

Abstract
BRICHOS is a domain found in several proteins consisting of ∼ 100 amino acids with sequence and structural similarities. Mutations in BRICHOS domain have been associated with both degenerative and proliferative diseases in several nonpulmonary organs, although the pathogenic mechanisms are largely undefined. Recently, several mutations in surfactant protein C (SP-C) mapping to the BRICHOS domain located within the proprotein (proSP-C) have been linked to interstitial lung diseases. In vitro expression of one of these BRICHOS mutants, the exon 4 deletion (hSP-CΔexon4), promotes a dominant-negative perinuclear aggregation of the protein. The present study characterizes the trafficking behavior and pathogenic consequences resulting from hSP-CΔexon4 expression. Time-lapse and co-localization microscopy studies demonstrated enhanced green fluorescent protein (EGFP)/hSP-CΔexon4 expression in calnexin-positive (endoplasmic reticulum [ER]) compartment with subsequent time- and concentration-dependent development of ubiquitinated perinuclear inclusion bodies followed by apoptosis. Compared with controls, EGFP/hSP-CΔexon4 promoted upregulation of multiple ER stress species, activated caspase 3, and induced annexin V binding. Furthermore, in GFP-u cells, hSP-CΔexon4 directly inhibited proteasome activity. These results support a model whereby proSP-C BRICHOS mutations induce a dynamic toxic gain-of-function, causing apoptotic cell death both by early ER accumulation leading to an exaggerated unfolded protein response and by enhanced deposition of cellular aggregates associated with proteasome dysfunction.
Keywords: conformational disease, apoptosis, unfolded protein response, epithelial cells, protein aggregation
Surfactant protein C (SP-C) is a small hydrophobic protein, found in pulmonary surfactant, initially identified upon isolation from bronchoalveolar lavage of a variety of mammalian species. SP-C is synthesized in alveolar type II cells as a 21-kD integral membrane precursor (proSP-C), which undergoes proteolytic C- and N-terminal cleavages as it is trafficked through the biosynthetic pathway from the Golgi complex, by way of small vesicles and multivesicular bodies, to the lumen of lamellar bodies, yielding a 3.7-kD mature peptide (1, 2). Together with other surfactant proteins and phospholipids contained in the lamellar body, SP-C is released into the alveolar space, via regulated secretion (1, 3), where it functions to modulate lung surface tension during the respiratory cycle.
The C-terminus of proSP-C shares a highly conserved region with several other proteins that were previously thought to be functionally and structurally unrelated. Proteins that comprise the British and Danish dementia (BRI) family of proteins, CA11, Chondromodulin-1 (ChM-I), and SP-C have a homologous conserved region of ∼ 100 amino acids known as the BRICHOS Domain (4). Many of these proteins are produced via proteolytic processing from a larger proprotein in which the BRICHOS domain is located within the propeptide region.
An intriguing similarity among the BRICHOS domain–containing proteins is their association with degenerative or proliferative diseases affecting one or more organ systems. Expression of BRI2 (a mutant from BRI gene family) is associated with a central nervous system syndrome whose pathologic, morphologic, and histologic features resemble that of Alzheimer's disease (5, 6). Deficiency of CA11, which is specifically expressed in the stomach, is linked to gastric cancer (7). Mutation in the gene encoding ChM-I is related to chondrosarcoma, a malignant neoplasm of cartilage and bone (8).
A remarkable structural homology can be found among BRICHOS domain proteins. They exist as integral transmembrane proteins with a type II (Ncytosol/Clumen) orientation. In addition, they mainly contain a pair of conserved cysteine residues within the BRICHOS domain that are predicted to form disulfide bridges. Based on available experimental data, it has been proposed that the BRICHOS domain may play a role in the intricate post-translational processing of these proteins (4).
The disulfide bridge-forming cysteine residues in the BRICHOS domain of proSP-C play a key role in the proprotein's trafficking and processing. Substitution mutation studies have demonstrated that the absence or replacement of these residues results in protein misfolding leading to aberrant trafficking, cytosolic aggregation, aggresome formation, and complete abrogation of post-translational processing of the proprotein (9, 10). Moreover, due to the homomeric associative characteristic of SP-C, the consequences of these mutations are exasperated by a dominant-negative effect (10).
Several proliferative and degenerative interstitial lung diseases have been linked to mutations in the SP-C gene (SFTPC) (11–16). All five exons have been targets; however, the great majority of these mutations were found within the BRICHOS domain (exons 4 and 5) of the propeptide where mutations are believed to affect propeptide folding and conformation. The first reported human SFTPC mutation, c.460 + 1 G → A identified on one allele, produced alternative splicing of the SP-C mRNA and exclusion of the fourth exon. This mutation not only produced a proSP-C peptide foreshortened by 37 amino acids (SP-CΔexon4), but also eliminated a consensus BRICHOS cysteine residue in the COOH flanking region of the propeptide (Figure 1A). Likewise, a heterozygous exon 5 + 128 T → A transversion mutation was reported that substitutes a glutamine for leucine at amino acid position 188 (SP-CL188Q) of the propeptide, which is adjacent to the disulfide bridge-forming cysteine 189. Other BRICHOS domain mutations linked to chronic lung disease include substitution mutations (SP-CL188R, SP-CI126R, SP-CP115L, SP-CY104H, and SP-CG100V) (12) (Figure 1A).
Figure 1.


Wild-type SP-C is trafficked to vesicles distal to the Golgi independent of cell type. (A) Amino acid sequence of the human SP-C pro-protein showing cytosolic, transmembrane, luminal, and BRICHOS domains. The secreted mature SP-C, the BRICHOS domain (hatched oval circle), and mutations found in association with ILD are shown. The Δexon 4 BRICHOS domain mutation and a non–BRICHOS domain mutation analyzed in the present study are highlighted in gray. The BRICHOS domain flanks the C-terminal starting at phenylalanine 94. Amino acid nomenclature is based on the published SP-C sequence (31). (B) A549, MLE, and HEK cells transiently transfected with EGFP/SP-CWT (top row) show similarities in vesicular co-localization (bottom row) with Texas Red–conjugated CD63 (middle row) antibody. Bar, 5 μm.
In vitro studies have begun to elucidate the impact of SP-C BRICHOS domain mutant expression on cellular homeostasis. In A549 cells, wild type SP-C (SP-CWT) is normally trafficked to subcellular vesicles distal to the Golgi (Figure 1B). In contrast, transiently transfected SP-CΔexon4 accumulates in ubiquitinated perinuclear inclusion bodies resulting in aggresome formation (17). Furthermore, when SP-CWT is co-transfected with SP-CΔexon4, trafficking to distal vesicles is abrogated and SP-CWT co-localizes with the mutant form in aggresomes (17) implying a dominant negative effect of SP-CΔexon4 via heteromeric association. Another BRICHOS domain mutant, SP-CL188Q, induces lactate dehydrogenase (LDH) release in transiently transfected MLE cell lines (13). Finally, induction of an endoplasmic reticulum (ER) stress element by SP-CΔexon4 in HEK cell lines has been reported suggesting an activation of the ER-associated degradation (ERAD) system (18). To date, no mechanistic links between BRICHOS domain mutants, ER stress response, and cell death have been described.
The present study examines the cellular response to mutant SP-C expression to further elucidate the pathologic mechanism of BRICHOS domain mutants. We hypothesized that BRICHOS domain mutants cause apoptotic cell death via both induction of ER stress and deposition of toxic cellular aggregates. We demonstrate that in vitro expression of, and cellular reaction to, SP-CΔexon4 is cell type–independent and results in sequential ER accumulation and cytosolic aggregate formation of the mutant protein, followed by apoptotic cell death. Concomitant to these observations, we found an upregulation of the unfolded protein response (UPR) species signifying ER stress and an inhibition of proteasome activity by SP-CΔexon4 indicating disruption of the ubiquitin/proteasome system (UPS). These results support a model in which misfolded proteins can cause cell injury and ultimately cell death through combined disruption of at least two cellular quality control systems.
MATERIALS AND METHODS
SP-C cDNA Expression
Human wild-type proSP-C (amino acids 1–197) in pEGFPC1 and pcDNA3 vectors have been previously described, as have EGFP/hSP-CΔexon4 and EGFP/hSP-CI73T (10, 17). To create HA-tagged and untagged species of the SP-C isoforms, a single PCR reaction with two primers was used. The forward and reverse primers contained restriction sites at the 5′ and 3′ ends of human proSP-C, respectively, for insertion into a pcDNA vector, as previously described (10, 17). The primer used to create HA-tagged SP-C isoforms also contained the HA (YPYDVPDYA)-coding sequence (10, 17).
Cell Lines
Human lung epithelial (A549), human embryonic kidney (HEK 293), and GFPu –1 cell lines were obtained from ATCC (Manassas, VA). The mouse line epithelial (MLE 15) cell line was a generous gift from Dr. Jeffrey Whitsett (Cincinnati, OH).
Immunocytochemistry
Cells grown to 70% confluence on glass coverslips in 35-mm plastic dishes were transiently transfected with EGFP- or HA-tagged or untagged proSP-C constructs (4 μg/dish) using FuGene 6 (Roche Diagnostics, Indianapolis, IN). For time-lapse microscopy, the same procedure described above was followed with the exception of using glass-bottom 35-mm petri dishes with 0.16- to 0.19-mm coverglasses (MatTek Corporation, Ashland, MA) for live microscopic observation. Cells were maintained for up to 48 h after introduction of plasmid DNA (unless otherwise indicated). Colocalization of GFP tagged fusion proteins with subcellular organelles was performed by immunostaining plated cells that were fixed by immersion of coverslips in 4% paraformaldehyde. After permeabilization, cells were immunolabeled with primary antibodies for 1 h at room temperature at the following dilutions: anti-NPro (polyclonal antisera raised against the Met [10]–Glu [23] domain of rat proSP-C recognizes the N-terminal flanking peptide of human proSP-C), 1:200; anti-calnexin (Stressgene, Victoria, BC, Canada), 1:200; anti-CD63, (Immunotech, Marseilles, France), 1:100; anti-ubiquitin (Chemicon, Temecula, CA), 1:100; anti-HA (Roche Molecular Biochemicals, Indianapolis, IN) 1:1,000; anti-activated caspase 3 (Sigma, St. Louis, MO) 1:200. Texas Red–conjugated secondary goat anti-mouse IgG monoclonal or secondary goat anti-rabbit IgG polyclonal antibodies (Jackson Immunoresearch Laboratories, West Grove, PA) at 1:200 dilution were used for visualization. Fluorescence images of air-dried and Mowiol mounted slides were viewed on an Olympus I-70 inverted fluorescent microscope (Olympus, Melville, NY). Fluorescence and phase images were captured using a Hamamatsu 12-bit coupled-charged device camera (Hamamatsu, Hamamatsu, Japan). Image processing and overlay analysis were performed using IMAGE 1 Metamorph software (Universal Imaging, West Chester, PA).
Necrotic and Apoptotic Cell Death Assays
Cell necrosis was monitored by nuclear labeling of propidium iodide (10 μg/ml of culture medium) in live cells and analyzed by direct fluorescence microscope at 48 h and 72 h after transfection. For apoptosis, two assays were used: (1) activated caspase 3 labeling in permeablized cells (see Immunocytochemistry for method), and (2) annexin V binding. The assay for annexin V binding was performed according to the manufacturer's recommendation with minor modifications. A549 cells grown to 70% confluence in 35-mm glass-bottom dishes were transiently transfected with EGFP/proSP-C constructs (4 μg/dish) using FuGene 6. For positive control, cells in separate 35-mm glass-bottom dishes were treated with tumor necrosis factor (TNF)-α (BD Biosciences, Bedford, MA) 16 h before assay. Forty-eight hours after the introduction of plasmid DNA, cells were washed twice (5 min each) with phosphate-buffered saline (137 mM NaCl, 10 mM Na2HPO4, 2.7 mM KCl, 1.8 mM KH2PO4, pH 7.2), incubated with incubation buffer (10 mM Hepes/NaOH, pH 7.2, 140 mM NaCl, 5 mM CaCl2) for 10 min, and incubated with annexin V–Alexa 647 labeling solution (incubation buffer + 10 μl annexin V–Alexa647/1 ml buffer [Roche Diagnostics, Penzberg, Germany]) for 15 min at room temperature. Cells were visualized with a confocal fluorescence microscope.
Time-Lapse Microscopy
Time-lapse fluorescence imaging of A549 cells transfected with various isoforms of EGFP-tagged SP-C in 35-mm glass-bottom dishes was performed using the Nikon Eclipse TE2000-E (Melville, NY) with an automated stage and Evolution QEi MediaCybernetics camera (Silver Springs, MD). Multifield images were selected, programmed, acquired, and processed by Phase 3 Imaging Systems software (Glen Mills, PA). During imaging, cells were maintained at 37°C and 5% CO2 up to 5 h using a temperature controller with an open perfusion micro-incubator, Model PDMI-2 (Medical System Corporation, Greenvale, NY).
Immunoblotting
Approximately 100 μg of protein sample in lysis buffer (50 mM Tris, 190 mM NaCl, 6 mM EDTA, 2% Triton X-100, pH 7.2) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride [PMSF], leupeptin [1 μg/ml], pepstatin A [1 μg/ml], and aprotinin [1.5 μg/ml]) and sonicated on ice three times in 20-s bursts at 50 W, were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membrane. Immunoblotting was performed using successive incubations with primary polyclonal GFP antisera (Molecular Probes, Eugene, OR) 1:5,000; anti-activated caspase 3 (Sigma) 1:200; anti–XBP-1 (M186) and anti–GRP 78 (H-129; Santa Cruz Biotechnologies, Santa Cruz, CA) both at 1:200; anti–HDJ-2 (Lab Vision, Fremont, CA) 1:400; or anti–β-actin (Sigma) 1:2,000, for 1 h followed by goat anti-rabbit horseradish peroxidase–conjugated secondary antibody (1:10,000) for another hour at room temperature. To induce ER stress, Tunicamycin Streptomyces sp. (Sigma) was added at the indicated concentrations 12 h before analysis. Bands were visualized by enhanced chemiluminescence using a commercially available kit, ECL Western Blotting Detection Reagents (Amersham Inc., Arlington Heights, IL). Fluorescence images were produced either by exposure to film or by direct acquisition using the Kodak 440 Imaging System (New Haven, CT).
In Vitro Proteasome Inhibition Assay
Proteasome inibition assays were performed using GFPu –1 cells (ATCC, Manassas, VA) as described previously (19). Cells grown to 70% confluence in 35 mm polylysine-coated dishes were transiently transfected with untagged proSP-C constructs (4 μg/dish) using FuGene 6. Forty-eight hours after transfection, cells were prepared for immuncytochemistry or immunoblot analysis according to the methods described above. As a positive control, nontransfected cells were treated with 10 μM clastro-lactasystin β-lactone (lactacystin) (Calbiochem, San Diego, CA) 16 h before analysis.
Statistical Analyses
Experimental data were analyzed by one-way ANOVA with the Tukey-Kramer post hoc test using GraphPad InStat software, version 3.0 for Windows (GraphPad Software Inc., San Diego, CA). All values were expressed as mean ± SE.
RESULTS
Trafficking of the SP-C BRICHOS Domain Mutant Is Cell Type– and Tag-Independent
We have previously reported that 48 h after transfection, EGFP/SP-CΔexon4 forms cytosolic aggregates in A549 cells (17). To determine whether trafficking of the mutant isoform is dependent upon cell type, we performed subcellular localization studies in A549, HEK, and MLE cell lines. Forty-eight hours after transfection in each of the three cell lines, EGFP/SP-CWT exhibited homogenous sorting to punctate vesicles positive for CD63 (Figure 1B), a marker antigen associated with lysosomes and lysosomal-like organelles including lamellar bodies and multivesicular bodies of alveolar type II cells. Rapid trafficking of EGFP/SP-CWT to punctate organelles was observed as early as 6 h and until monitoring was stopped 72 h after transfection with no apparent ER retention or subcellular compartment localization other than to vesicles (data not shown).
In contrast, a heterogeneous pattern of trafficking was observed in cells transfected with EGFP/SP-CΔexon4 transfected cells. The EGFP/SP-CΔexon4 formed cytosolic aggregates with no significant differences in observed expression between the three cell lines (Figure 2) 48 h after transfection. Consistent with previous reports, many of these aggregates are ubiquitin-positive (Figure 2A), a characteristic of proteins directed to ubiquitin/proteasome degradation pathways and aggresome formation (9, 17). In addition, expression of mutant protein was not limited to cytosolic aggregates, but also exhibited a reticular pattern suggesting ER localization. Calnexin, an ER-specific marker, partially colocalized with EGFP/SP-CΔexon4 confirming retention of a portion of mutant SP-C in the ER (Figure 2B). Comparison of EGFP/SP-CΔexon4 expression patterns between cell lines showed that whereas A549 and MLE cells exhibited comparable localization with predominant cytosolic aggregation, HEK cells showed more ER retention.
Figure 2.
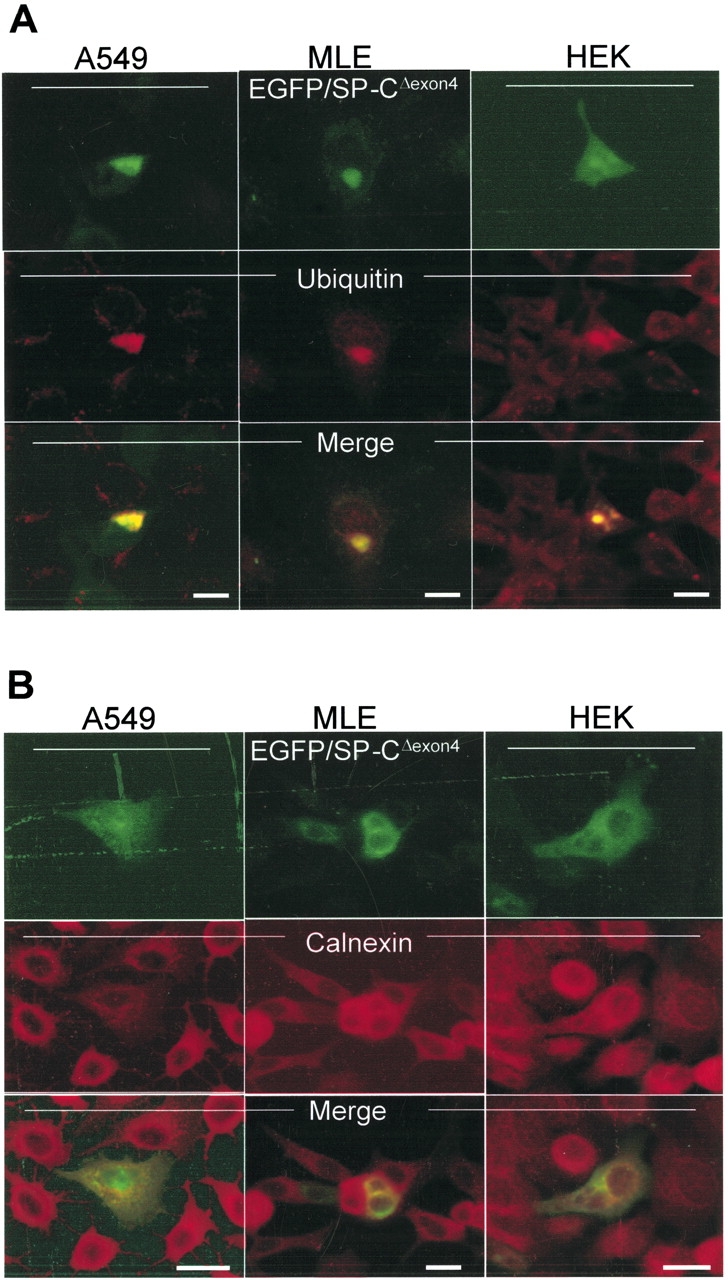

EGFP/SP-CΔexon4 accumulates in the ER and forms cellular aggregates independent of conjugated marker tags. A549, MLE, and HEK cells transiently transfected with EGFP/SP-CΔexon4 (top rows) were immunostained with ubiquitin (A, middle row) and calnexin (B, middle row). In all three cell lines, both markers co-localize with the mutant (merged images). (C) Fluorescent images of A549 cells transfected with different plasmid constructs containing either EGFP-tagged (top row), HA-tagged (middle row), or untagged (bottom row) SP-CWT (left column) and SP-CΔexon4 (right two columns) isoforms immunostained with Texas Red–conjugated anti-HA (middle row) and anti-NPro (bottom row) antibodies. Bar, 5 μm.
Previous studies showing cytosolic aggregation of SP-CΔexon4 mutant were performed using an EGFP tag (17). To establish whether or not EGFP affects trafficking of SP-C isoforms, tagged and untagged SP-CWT and SP-CΔexon4 were transiently transfected in A549 cells. Fluorescence analysis of cells 48 h after transfection showed no variation in trafficking whether the wild-type (Figure 2C, left column) or the mutant (Figure 2C, right two columns) SP-C isoforms were EGFP-tagged (Figure 2C, top row), HA-tagged (Figure 2C, middle row), or untagged (Figure 2C, bottom row). Similar findings were observed in MLE and HEK cells (data not shown).
Time Dependence of Expression Patterns
We hypothesized that the morphologic heterogeneity seen in transient transfection was due to the dynamics between ER retention and retrotranslocation of the mutant protein to the cytosol. To investigate the sequential subcellular trafficking events of mutant protein, expression patterns of A549 cells transfected with EGFP/SP-CΔexon4 were determined at intervals following introduction of cDNA. At 12 h after transfection, very faint expression of mutant proteins was apparent, which was entirely ER localized (Figure 3A, a and b). Rising fluorescence intensity of ER localized proteins (Figure 3A, c–e) and increasing formation of cytosolic aggregates (Figure 3A, f–h) occurred from 24–48 h after transfection, suggesting ER accumulation and subsequent retrotranslocation of the mutant isoform to the cytosol. This shift from total to partial ER localization continued with a subset of the cells exhibiting progressive aggregation in the cytosol. By 72 h after transfection, cytosolic aggregates (mostly perinuclear) were the predominant feature of transfected cells (Figure 3A, i–l). In contrast, SP-CWT was not retained in the ER but was rapidly transferred to distal vesicles where it reached steady-state expression by 24 h (see online supplement video1.mov).
Figure 3.
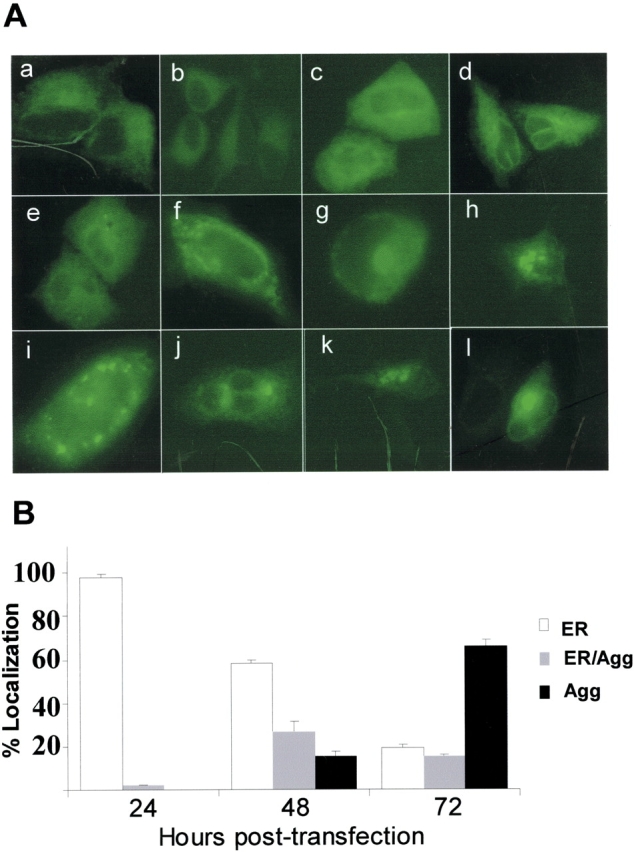
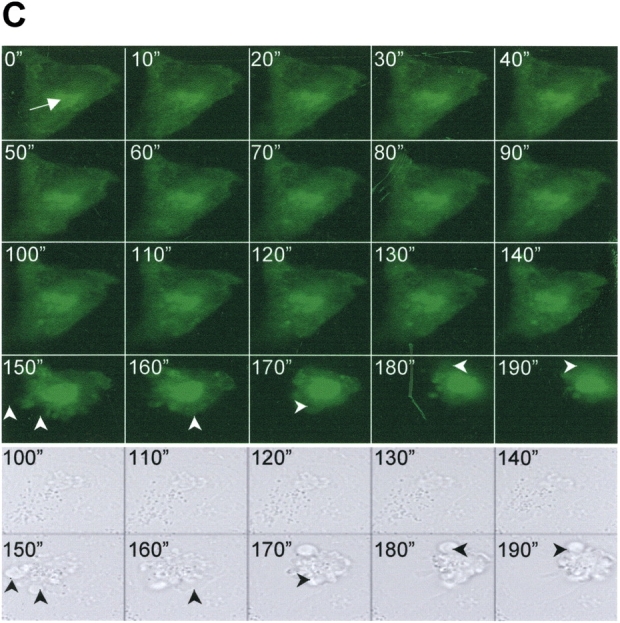
Time-dependent trafficking and cell injury associated with SP-CΔexon4 expression. (A) Representative live fluorescent images of A549 cells at different time intervals following introduction of EGFP/SP-CΔexon4. Times are post-transfection: (a, b) 12 h with 2 min exposure time; (c, d) 24 h with 100 ms exposure time; (e–h) 36–48 h with 100 ms exposure time; (i–l) 48–72 h with 100 ms exposure time. Heterogeneous expression patterns of mutant proteins included ER localization (a–e), aggregation (h, j, k, and l), or combined ER localization and aggregation (f, g, and i). (B) Subcellular localization of EGFP/SP-CΔexon4 at 24, 48, and 72 h after transfection in A549 cells. Data were obtained from three separate experiments, each with counts of at least 300 transfected cells. (C) A representative time-lapse fluorescent imaging series of cellular response to the expression of EGFP/SP-CΔexon4 showing time-dependent development of aggregates and subsequent cell death (10-min intervals). Imaging was initiated at 47 h after transfection (t = 0 min) with apparent ER localized expression and initial formation of juxtanuclear aggregates (arrow). Onset of cell recoiling at t = 140 min (49 h and 20 min after transfection). Within the next hour, complete recoiling with concurrent dish-surface detachment and cell blebbing (arrowheads) (bottom rows of both fluorescent and phase images). Cell death following transfection of EGFP/SP- CΔexon4 ranged from 48–72 h.
The time-dependent subcellular localization was quantified in transfected cells by cell counting. At 24 h after transfection, nearly 98% of transfected cells exhibited ER localization of the protein, while less than 3% showed some cytosolic aggregates in the presence of ER localization. At this time point, isolated aggregate formation, without ER expression, was not observed. By 48 h after transfection, the proportion of transfected cells with only ER localized proteins was reduced to 56% with the emergence of more cells containing both ER-localized proteins and cytosolic aggregates (27%) and cells exhibiting only cytosolic aggregates (15%). By 72 h after transfection, 66% of the cells showed isolated cytosolic aggregates, whereas cells showing both ER and cytosol localization were reduced to 15%, suggesting that ER localization of the mutant protein is transient. Although 21% of transfected cells still displayed proteins localized in ER without any detectable cytosolic aggregates, the level of mutant expression in these cells was very low, similar to that observed at 12 h after transfection.
Time-Dependent Formation of Perinuclear Aggregates and Subsequent Cell Death
Time-lapse microscopy of individual A549 cells transfected with EGFP/SP-CΔexon4 confirmed time-dependent aggregate formation of the mutant protein (Figure 3C, video2.mov). This process starts with the formation of small aggregates and a display of retrograde-like movement toward the nucleus resulting in the creation of larger, and frequently single, perinuclear aggregates. In many cases, as the size and fluorescence intensity of the perinuclear aggregates increased, a proportional decrease in fluorescence intensity of the ER was observed. This perinuclear aggregation induced sequential events consisting of cell recoiling, loss of surface adherence, and cell blebbing (Figure 3C, 150” to 190”, video3A.mov and video3B.mov) suggestive of cell death by apoptosis.
SP-CΔexon4 Induces Apoptotic Cell Death via Activation of Caspase 3
Propidium iodide nuclear labeling in cells with mutant EGFP/SP-CΔexon4 aggregates (Figure 4A, right column) confirmed cell death observed during the time-lapse microscopy. The plasma membrane of cells containing mutant aggregates frequently labeled with annexin V (Figure 4A, middle column), an agent that binds translocated (inner to outer leaflet) plasma membrane phophatidylserine during apoptosis.
Figure 4.
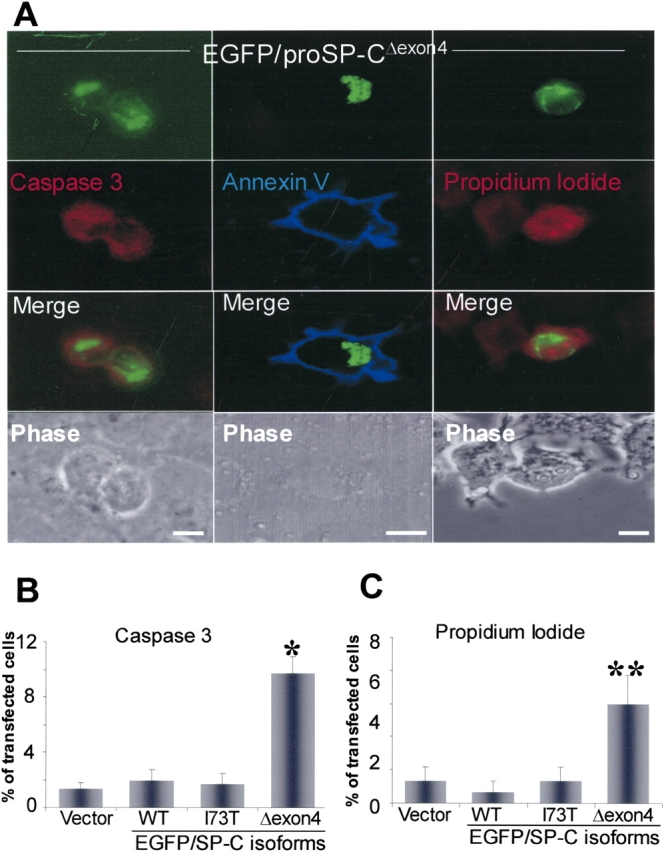
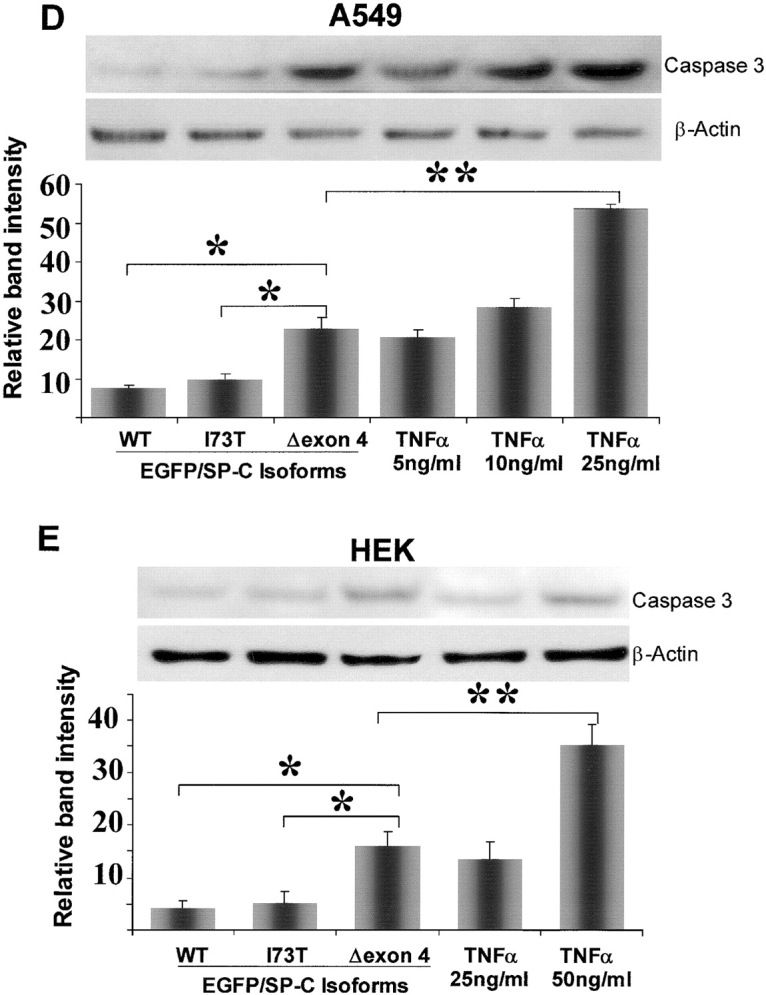
Apoptotic cell death caused by EGFP/SP-CΔexon4 induction of Caspase 3. (A) EGFP/SP-CΔexon4–transfected A549 cells (top row) were immunostained with Texas Red–conjugated anti-activated caspase 3 (left column), Alexa 647–tagged annexin V (middle column), or nuclear-stained with propidium iodide (right column). Activated caspase 3 was present in cells expressing the mutant protein (left merged image). Plasma membrane phospatidylserine binding to annexin V was also observed in mutant protein-containing cells (middle merged image). Similarly, propidium iodide labeled nuclei of cells with mutant protein (right merged image). Phase contrasts of fluorescent images are shown (bottom row). Images represent results from at least three separate experiments. Bar, 5 μm. (B and C) Percentage of cell count showing significant immunostaining of activated caspase 3 (B) and nuclear labeling of propidium iodide (C) in cells transfected with EGFP/SP-CΔexon4 as compared with those transfected with vector (EGFPC1), EGFP/SP-CWT, or EGFP/SP-CI73T. Cells were counted 48 h and 72 h after transfection for caspase 3 and propidium iodide, respectively. Data were obtained from at least 250 transfected cells per construct over at least three separate experiments. *P < 0.001, **P < 0.005. Immunoblots of actived caspase 3 in lysates of A549 (D) and HEK (E) cells 48 h after transfection with various isoforms of EGFP/SP-C. Band intensity was quantified and results from at least three separate experiments are shown. Representative immunoblots appear above each graph. TNF-α was used as a positive control. For normalization, the band value of vector-transfected cells and the value of background intensity from the same experiment were subtracted from the measured intensity values for each construct or TNF-α. β-Actin bands were used to normalize for equal loading. *P < 0.005, **P < 0.001.
Although the above observations (including the time-lapse analyses and propidium iodide and annexin V labeling) suggest apoptosis, they do not exclude other mechanisms of cell death such as necrosis. To substantiate whether EGFP/SP-CΔexon4–transfected cells die via apoptosis, we used detection assays for caspase 3 (an effector caspase that induce apoptosis). Labeling of activated caspase 3 was observed more frequently in cells transfected with EGFP/SP-CΔexon4 aggregate-containing cells (Figure 4A, left column) than those transfected with SP-CWT or SP-CI73T (another ILD-associated SP-C mutation that is located outside the BRICHOS domain [Figure 1A]) (data not shown). Moreover, cell counts were performed in cells transfected with EGFP/SP-CΔexon4, SP-CWT, and SP-CI73T that were labeled with either anti-activated caspase 3 (Figure 4B) or propidium iodide (Figure 4C). In both cases, significantly more labeling was seen in cells transfected with the BRICHOS domain mutant than in cells transfected with either SP-CWT or SP-CI73T, suggesting a marked aberrant effect by SP-CΔexon4 expression.
Caspase 3 activation by SP-CΔexon4 was also assessed using whole cell lysate immunoblot analysis. A549 and HEK cells were transiently transfected with EGFP-tagged SP-CWT, SP-CΔexon4, or SP-CI73T 48 h before analysis. Caspase 3 activation was significantly increased in SP-CΔexon4–transfected cells in both A549 (Figure 4D) and HEK (Figure 4E) cells compared with cells transfected with either SP-CWT or the non–BRICHOS domain mutant (SP-CI73T). Cells treated with TNF-α, a known inducer of apoptosis, show caspase 3 activation in a dose-dependent manner in both A549 (Figure 4D, right three bars) and HEK (Figure 4E, right two bars) cells; however, the TNF-α concentration required to activate caspase 3 appeared to be cell type–dependent.
SP-CΔexon4 Expression Elicits an ER Stress Response
Cellular response to the stress of misfolded protein accumulation in the ER is regulated by various transcription factors. One of the upstream transcription factors, XBP-1, has been shown to regulate ER resident chaperone genes, including BiP/GRP78, HEDJ, EDEM, and HDJ-2/HSP40 (20). To investigate whether SP-CΔexon4 accumulation in the ER elicits an upregulation of XBP-1 and subsequent activation of chaperone genes, A549 cells were transiently transfected with various isoforms of SP-C. Immunoblot of whole cell lysates of transfected cells (Figure 5A) showed that XBP1 is significantly upregulated with transfection of SP-CΔexon4, as compared with SP-CWT or SP-CI73T (Figure 5B). Moreover, expression of the chaperone genes GRP78 (Figure 5C) and HDJ-2 (Figure 5D) were significantly enhanced by the presence of this misfolded protein. Tunicamycin (TM), an ER stress–inducing agent used as a control, considerably increased the expression of all three species of the UPR in a dose-dependent manner (Figure 5, right two columns of data).
Figure 5.
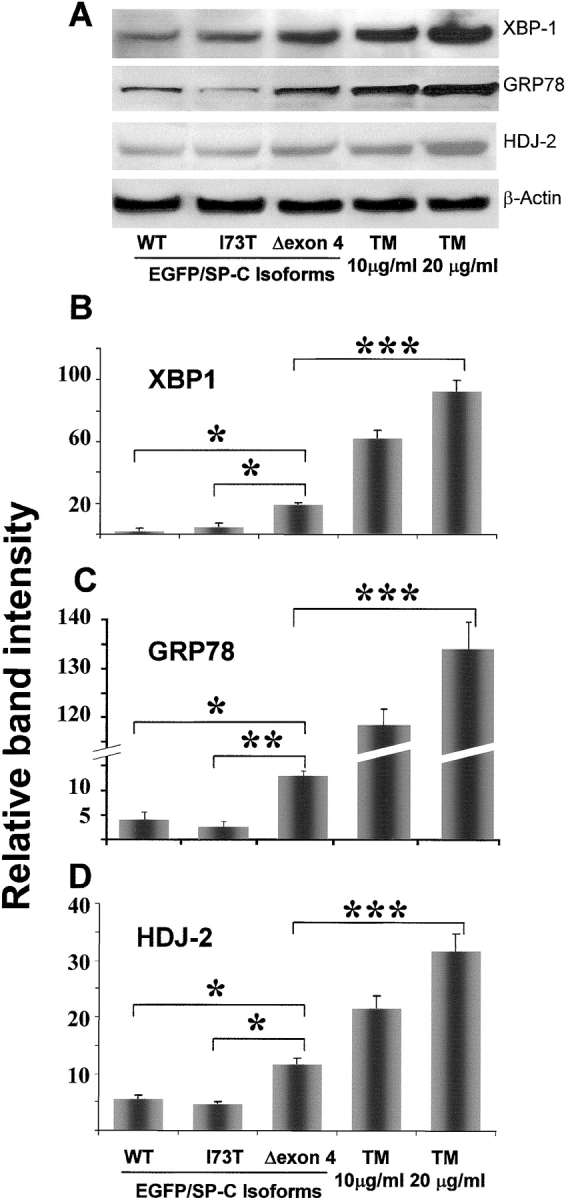
Induction of ER stress species by SP-CΔexon4. (A) Representative immunoblot bands of samples from A549 whole cell lysates showing increased transcription of XBP-1, GRP78, and HDJ-2 48 h after transfection in cells with EGFP/SP-CΔexon4 compared with those with either EGFP/SP-CWT or EGFP/SP-CI73T. TM-treated A549 cells were used as a positive control. Relative band intensity from at least three experiments is shown for XBP-1 (B), GRP78 (C), and HDJ-2 (D) expression in cells transfected with EGFP/SP-CΔexon4, SP-CWT, and SP-CI73T. Variable intensity of ER stress species in response to positive control (TM-treated cells) is shown. For normalization, the band value of vector-transfected cells and the value of background intensity from the same experiment were subtracted from the measured intensity values for each construct or TM. β-Actin bands were used to normalize for equal loading. *P < 0.005, **P < 0.001, ***P < 0.0005.
SP-CΔexon4 Inhibits the Ubiquitin/Proteasome System
The ubiquitination of cytosolic aggregates suggests that the BRICHOS domain mutant may affect the UPS in a similar manner to that previously reported in other proteins prone to aggregation (19). GFPu-1 cells were used to investigate the impact of aggregates formed by SP-CΔexon4 on the UPS. GFPu-1 is an HEK-derived, stably transfected cell line expressing a short degron (CL1) tagged to GFP (21). The stability of GFP can be used as a marker to determine relationships between protein aggregation and UPS dysfunction (19). Under normal conditions, the GFP/degron expression is short-lived as the efficient UPS of HEK cells rapidly degrades the product soon after translation, before there is any detectable green fluorescence (Figure 6C, top). However, conditions that impair the UPS, thereby inhibiting protein degradation, will promote the accumulation of the GFP-tagged peptide. Control treatment with lactacystin, a proteasome inhibitor, resulted in GFP expression across the entire cell culture population (Figure 6C, middle). Cells transiently transfected with untagged SP-CWT (Figure 6A, top), identified by Texas Red–labeled anti–SP-C, exhibited no GFP-fluorescence (Figure 6A, middle), indicating that the UPS was not affected by the presence of SP-CWT. In contrast, cells transfected with SP-CΔexon4, similarly identified by Texas Red–labeled anti–SP-C (Figure 6B, top), showed GFP-fluorescence (Figure 6B, middle). Moreover, immunoblot of GFPu-1 whole cell lysates using anti-GFP shows a significant increase in GFP protein levels in samples where cells were either treated with the control reagent lactacystin (Lac) or transfected with SP-CΔexon4, as compared with vector, SP-CWT, and SP-CI73T (Figure 6D). Collectively, these results demonstrate increased GFP expression by BRICHOS mutant aggregates, indicating UPS inhibition.
Figure 6.
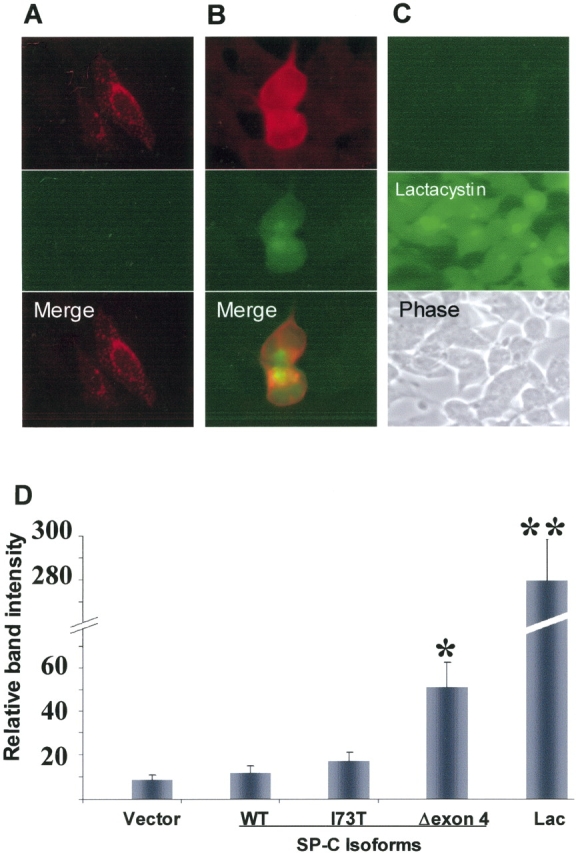
Proteasome inhibition by SP-CΔexon4. Representative GFPu-1 cells transfected with either SP-CWT (A) or SP-CΔexon4 (B) untagged SP-C isoforms. Immunostaining with anti-NPro (A and B, top row) antibody 48 h following plasmid introduction. Control GFPu-1 cell population before (C, top) and after (C, middle) treatment with lactacystin (Lac) with GFP expression only after treatment. Phase contrast image is shown (C, bottom). (D) Histogram of quantified immunoblot (using anti-GFP) from whole lysate samples of GFPu-1 cells transfected with either pcDNA3 (vector), or untagged isoforms of SP-C. Significant GFP expression is apparent in SP-CΔexon4–transfected cells compared with SP-CWT, SP-CI73T, or vector. Expression of large amounts of GFP in control cells treated with lactacystin (Lac) (right bar) is consistent with the fluorescent imaging results (C, middle). Data were obtained from three separate experiments, each consisting of at least two culture dishes per construct. *P < 0.05, **P < 0.001.
DISCUSSION
This study demonstrates a previously undescribed mechanism of cellular dysfunction in pulmonary epithelium based on the direct destabilization, by a misfolded protein, of two major cellular pathways leading to cell injury and death. Previously we had shown that the SP-C BRICHOS domain mutant, SP-CΔexon4, co-localizes in the microtubule organizing center to form inclusion bodies called aggresomes (17). In addition, evidence of cytotoxicity in cells expressing another mutant isoform of the SP-C BRICHOS domain has been reported (13, 18). However, the mechanism linking mutation, aggregation, and cellular dysfunction has not been fully elucidated. The data from the present study demonstrate that the SP-CΔexon4 BRICHOS domain mutant not only forms aggresomes, inhibits proteasome function, and causes apoptosis but may also induce ER stress. More importantly, aggregation of SP-CΔexon4 is independent of cell type. These results suggest that at least two subcellular systems are directly and negatively affected by this mutation: (1) the UPR, which is activated by ER overload of accumulated misfolded proteins; and (2) the UPS, which is weakened and ultimately inhibited by prolonged retrotranslocation and cytosolic accumulation of aberrantly folded proteins.
Under normal conditions, native isoforms of secretory or transmembrane proteins are rapidly processed and translocated from the ER to the Golgi. In contrast, misfolded proteins are either retained in the ER or rapidly retrotranslocated to the cytoplasm. In the cytosol, these misfolded proteins either undergo proteasome-mediated degradation or form aggregates. We found that SP-C BRICHOS mutants showed early retention in the ER and induction of the UPR in three epithelial cell lines (Figures 3 and 6). Similar ER accumulation of SP-CΔexon4 without formation of cytosolic aggregates was shown to induce an ER stress response element, BiP, in stably transfected human embryonic kidney cells (HEK293) (18). However, the capacity for ER accumulation seems to have a retention threshold beyond which the ER no longer accommodates mutant protein, resulting in protein retrotranslocation to the cytosol and subsequent aggregation. This threshold may vary depending on the cell type, as suggested by the presence of significantly more ER localized and less aggregated SP-CΔexon4 in HEK cells compared with either A549 or MLE cells at each analyzed time point. At 72 h after transfection in A549 cells, faint ER labeling of the EGFP-tagged SP-CΔexon4 was observed without aggregate formation in 21% of cells. This finding is likely secondary to either transfection of low copy numbers of the mutant protein in these cells, resulting in low levels of expression, or these cells may be able to suppress mutant aggregation by balancing synthesis with proteasome-mediated degradation. Similar results have been shown in HEK cells (which appear to exhibit a robust ERAD pathway) expressing the CFTRΔF508 mutant (22).
Using immunocytochemical, immunoblot, and direct fluorescence analyses, we have convincingly demonstrated the marked differences in cellular response between the expression of SP-CΔexon4 and other isoforms of SP-C. These results indicate that the responses observed in cells expressing SP-CΔexon4 are not consequences of overexpression but rather a characteristic cellular response to a particular misfolded mutant protein.
The accumulation of other mutant proteins in the ER has previously been shown to have detrimental effects on cell function in organs other than the lung. Deficiency of α1-AT causes chronic lung disease in some patients and inflammatory liver diseases in others. Polymerization of mutant α1-ATZ is retained in the ER of hepatocytes (23) and activates the UPR in vitro (24). In a mouse model of diabetes due to a mutant form of insulin, ER accumulation of the mutant isoform has been shown to induce ER stress and apoptosis (25). Mutant forms of peripheral myelin protein-22 (PMP22) associated with Charcot-Marie-Tooth disease and Dejerine-Sottas syndrome are ER-retained and closely associate with calnexin (an ER chaperone) to cause dominant gain-of-function disease (26, 27).
In addition to ER signaling, cytosolic aggregation of mutant proteins has also been shown to disrupt cellular function. In this article we demonstrate that cytosolic aggregation of mutant SP-C is associated with cell toxicity. Conditions such as Alzheimer's disease, Parkinson's disease, Huntington disease (HD), Machado-Joseph disease, and cystic fibrosis represent a large class of conformational diseases associated with intra- and/or extracellular accumulation of abnormal protein aggregates. Recent evidence indicates that in some of these diseases, including Machado-Joseph disease, aggregates localized in the nucleus or cytoplasm activate the UPR and promote cell death (19, 28). The polyglutamine repeat-associated spinocerebrocellular atrophy protein (SCA3) of Machado-Joseph disease aggregates in the cytosol, inhibits proteasome function, and induces activation of the UPR and caspase 12 (19, 28).
The induction of ER stress ultimately results in apoptotic cell death as indicated by caspase 3 activation in cell expressing mutant SP-C. These results were confirmed by Western blotting of transfected cell lysates. Lower levels of activated caspase 3 (Figure 4) and UPR species (Figure 5) were observed in cells transfected with the SP-C BRICHOS domain mutant when compared with cells treated with TNF or tunicamycin. This is likely related to the transfection system used. The FuGene 6 transfection method was chosen for SP-C cDNA delivery due to its ability to give the maximum transfection efficiency with minimum toxicity in these particular cell lines. Nevertheless, transfection efficiency with this method was only ∼ 30%. When TNF-α or tunicamycin was used as a positive control, 100% of the cells were exposed to an apoptotic signal or ER stress, respectively. Given the low transfection efficiency, the relative increase of activated caspase 3 and UPR species in cells transfected with the BRICHOS domain mutant is highly significant compared with those transfected with other isoforms of SP-C.
GFPu-1 cells were used to demonstrate SP-CΔexon4 aggregates also inhibit proteasome function (Figure 6) suggesting a direct cause-and-effect relationship between this mutant and disruption of cellular function as proposed for other aggregate-prone mutant proteins. The misfolded mutant protein of the cystic fibrosis transmembrane conductance regulator (CFTRΔF508), and the mutant polyglutamine repeat of the Huntington protein form distinct perinuclear inclusion bodies that are structurally similar (29). The ΔF508 isoform is normally retrotranslocated and degraded via the ERAD and UPS pathways (30). Studies in HEK cells have shown that at low levels of expression, the UPS is capable of degrading the mutant protein and suppressing aggregation. In contrast, overexpression of ΔF508 results in the formation aggresomes (22). Comparable studies using mutant HD protein have shown that protein accumulation is significantly increased by the inhibition of the 26S proteasome (29). Such studies, together with the results from our study, support the model that cytosolic aggregates are the direct consequence of an overwhelmed UPS that has exceeded its degradation capacity due to accumulation of aggregate-prone misfolded proteins (22).
Using SP-CΔexon4 as a model molecule, this study provides evidence suggesting the concept that misfolded BRICHOS mutant isoforms could induce cell injury via disruption of two separate but mechanistically linked subcellular systems, the UPR and the UPS (Figure 7). Unlike previously described disease paradigms in which mutations result in either ER retention and accumulation of mutant protein without cytosolic aggregates formation or cytosolic aggregation of mutant protein without ER retention, the SP-C BRICHOS domain mutant appears to exhibit both characteristics and affects cellular function accordingly. As summarized in Figure 7, the misfolded protein is initially retained in the ER, where it accumulates, thereby promoting the upregulation of UPR species including the transcription factor XPB-1 and chaperone proteins GRP78 and HSP40, suggestive of ER stress, and eventually inducing caspase 3 activation, leading to apoptotic cell death. Within those same cells, prolonged retrotranslocation of the misfolded protein to the cytosol by the ERAD system overwhelms the UPS, which is no longer capable of adequately degrading the protein, resulting in the accumulation of cytosolic aggregates. These aggregates may interact with a wide range of cellular targets and interrupt normal processes. These interactions include dominant-negative heteromeric association of SP-CΔexon4 and SP-CWT and entrapment of ubiquitin within aggresomes leading to inhibition of ubiquitin-mediated signaling function. These responses of subcellular systems collectively disrupt cellular function to ultimately cause cell injury and apoptotic cell death.
Figure 7.

Dual effect of the SP-C BRICHOS domain mutant leading to cellular dysfunction. At least two separate subcellular systems are affected by BRICHOS domain mutant expression: the UPR and UPS pathways. UPR: Mutant expression induces ER stress as demonstrated by ER retention of misfolded proteins, increased expression of the transcription factor XBP-1, and upregulation of chaperone genes GRP78 and HDJ2. Consequently, through undefined intermediates, caspase 3 is activated with subsequent apoptotic cell death. UPS: Persistent production of misfolded proteins overwhelms the UPS and prevents normal homeostatic degradation of proteins resulting in cellular accumulation and aggregation of proteins. Aggregation is enhanced in the case of proteins in which heteromeric association between wild type and mutant isoforms results in a dominant negative effect, as seen with SP-CΔexon4. Inhibition of UPS not only promotes the accumulation of degradation-destined proteins, but also impedes other signaling pathways including those mediated by ubiquitin. Such subcellular system dysfunction is likely to cause cell injury and, ultimately, cell death. Hatched arrows indicate undefined intermediates.
Although expression of many disease-causing misfolded proteins results in cell death, differences in cellular responses and pathologic features observed in the presence of various mutations indicate that not all subcellular organelles or pathways are affected equally. The nature of the protein and its misfolding is likely to dictate which pathway and/or organelle is impacted directly. While misfolded proteins that are ER retained, such as α1-ATZ, may have a direct effect on the ER and ER associated responses, other proteins that form cytosolic aggregates, such as SCA3, may primarily target cytosolic entities including the UPS. Alternatively, cellular response to SP-CΔexon4 may represent a hybrid that shows both ER retention and cytosolic aggregation with a concomitant disturbance of both the UPR and the UPS. Understanding the nature of cellular response to different types of misfolded proteins should assist in the development of new therapeutic strategies tailored to specific subcellular targets for degenerative disorders caused by aberrantly folded proteins.
Supplementary Material
Acknowledgments
The authors thank Dr. Jeffrey Whitsett of the University of Cincinnati (Cincinnati, OH) for his generous gift of the MLE 15 cell line, and Colleen D. Campbell of The Children Hospital of Philadelphia (Philadelphia, PA) for her editorial expertise. This work was supported by the National Institutes of Health Grants HL-19737, HL074064, and P50-HL56401 (M.F.B.) and the American Lung Association Dalsemer Research Grant DA-188-N (S.M.) and the Parker B. Francis Foundation Fellowship Grant (S.M.).
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Conflict of Interest Statement: S.M. has no declared conflicts of intetrest; V.N. has no declared conflicts of interest; S.J.R. has no declared conflicts of interest; M.M. has no declared conflicts of interest; and M.F.B. has no declared conflicts of interest.
References
- 1.Beers M. Molecular processing and cellular metabolism of surfactant protein C. In: Rooney SA, editor. Lung surfactant: cellular and molecular processing. Austin: R.G. Landes; 1998. pp. 93–124.
- 2.Johansson J. Structure and properties of surfactant protein C. Biochim Biophys Acta 1998;1408:161–172. [DOI] [PubMed] [Google Scholar]
- 3.Weaver TE. Synthesis, processing and secretion of surfactant proteins B and C. Biochim Biophys Acta 1998;1408:173–179. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez-Pulido L, Devos D, Valencia A. BRICHOS: a conserved domain in proteins associated with dementia, respiratory distress and cancer. Trends Biochem Sci 2002;27:329–332. [DOI] [PubMed] [Google Scholar]
- 5.Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, Ghiso J. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature 1999;399:776–781. [DOI] [PubMed] [Google Scholar]
- 6.Vidal R, Revesz T, Rostagno A, Kim E, Holton JL, Bek T, Bojsen-Moller M, Braendgaard H, Plant G, Ghiso J, et al. A decamer duplication in the 3′ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc Natl Acad Sci USA 2000;97:4920–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shiozaki K, Nakamori S, Tsujie M, Okami J, Yamamoto H, Nagano H, Dono K, Umeshita K, Sakon M, Furukawa H, et al. Human stomach-specific gene, CA11, is down-regulated in gastric cancer. Int J Oncol 2001;19:701–707. [DOI] [PubMed] [Google Scholar]
- 8.Hiraki Y, Inoue H, Iyama K, Kamizono A, Ochiai M, Shukunami C, Iijima S, Suzuki F, Kondo J. Identification of chondromodulin I as a novel endothelial cell growth inhibitor. Purification and its localization in the avascular zone of epiphyseal cartilage. J Biol Chem 1997;272:32419–32426. [DOI] [PubMed] [Google Scholar]
- 9.Kabore AF, Wang WJ, Russo SJ, Beers MF. Biosynthesis of surfactant protein C: characterization of aggresome formation by EGFP chimeras containing propeptide mutants lacking conserved cysteine residues. J Cell Sci 2001;114:293–302. [DOI] [PubMed] [Google Scholar]
- 10.Wang WJ, Russo SJ, Mulugeta S, Beers MF. Biosynthesis of surfactant protein C (SP-C): sorting of SP-C proprotein involves homomeric association via a signal anchor domain. J Biol Chem 2002;277:19929–19937. [DOI] [PubMed] [Google Scholar]
- 11.Nogee LM, Dunbar AE III, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med 2001;344:573–579. [DOI] [PubMed] [Google Scholar]
- 12.Nogee LM. Abnormal expression of surfactant protein C and lung disease. Am J Respir Cell Mol Biol 2002;26:641–644. [DOI] [PubMed] [Google Scholar]
- 13.Thomas AQ, Lane K, Phillips J III, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 2002;165:1322–1328. [DOI] [PubMed] [Google Scholar]
- 14.Chibbar R, Shih F, Baga M, Torlakovic E, Ramlall K, Skomro R, Cockcroft DW, Lemire EG. Nonspecific interstitial pneumonia and usual interstitial pneumonia with mutation in surfactant protein C in familial pulmonary fibrosis. Mod Pathol 2004;17:973–980. [DOI] [PubMed] [Google Scholar]
- 15.Brasch F, Griese M, Tredano M, Johnen G, Ochs M, Rieger C, Mulugeta S, Muller KM, Bahuau M, Beers MF. Interstitial lung disease in a baby with a de novo mutation in the SFTPC gene. Eur Respir J 2004;24:30–39. [DOI] [PubMed] [Google Scholar]
- 16.Beers MF, Mulugeta S. Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu Rev Physiol 2005;67:663–696. [DOI] [PubMed] [Google Scholar]
- 17.Wang WJ, Mulugeta S, Russo SJ, Beers MF. Deletion of exon 4 from human surfactant protein C results in aggresome formation and generation of a dominant negative. J Cell Sci 2003;116:683–692. [DOI] [PubMed] [Google Scholar]
- 18.Bridges JP, Wert SE, Nogee LM, Weaver TE. Expression of a human surfactant protein C mutation associated with interstitial lung disease disrupts lung development in transgenic mice. J Biol Chem 2003;278:52739–52746. [DOI] [PubMed] [Google Scholar]
- 19.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001;292:1552–1555. [DOI] [PubMed] [Google Scholar]
- 20.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 2003;23:7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilon T, Chomsky O, Kulka RG. Degradation signals for ubiquitin system proteolysis in Saccharomyces cerevisiae. EMBO J 1998;17:2759–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol 1998;143:1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lomas DA, Mahadeva R. Alpha1-antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest 2002;110:1585–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawless MW, Greene CM, Mulgrew A, Taggart CC, O'Neill SJ, McElvaney NG. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J Immunol 2004;172:5722–5726. [DOI] [PubMed] [Google Scholar]
- 25.Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 2002;109:525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickson KM, Bergeron JJ, Shames I, Colby J, Nguyen DT, Chevet E, Thomas DY, Snipes GJ. Association of calnexin with mutant peripheral myelin protein-22 ex vivo: a basis for “gain-of-function” ER diseases. Proc Natl Acad Sci USA 2002;99:9852–9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snipes GJ, Orfali W, Fraser A, Dickson K, Colby J. The anatomy and cell biology of peripheral myelin protein-22. Ann N Y Acad Sci 1999;883:143–151. [PubMed] [Google Scholar]
- 28.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 2002;16:1345–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 2001;12:1393–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kopito RR. Biosynthesis and degradation of CFTR. Physiol Rev 1999;79:S167–S173. [DOI] [PubMed] [Google Scholar]
- 31.Warr RG, Hawgood S, Buckley DI, Crisp TM, Schilling J, Benson BJ, Ballard PL, Clements JA, White RT. Low molecular weight human pulmonary surfactant protein (SP5): isolation, characterization, and cDNA and amino acid sequences. Proc Natl Acad Sci USA 1987;84:7915–7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.