

Abstract
This study demonstrates that the two biomarkers, MUC5AC/ Muc5ac and hCLCA1/Gob5, which are frequently associated with surface mucous/goblet cells in asthmatic airways, are differentially regulated. Intratracheal instillation of IL-13 (0.5 μg/mouse lung) elicited 8- and 110-fold induction of Muc5ac and Gob5 messages, respectively, within 24 h in wild-type mouse lung, whereas these inductions were abrogated in Stat6 knockout mice. The induction of MUC5AC/Muc5ac message could not be duplicated in vitro with primary tracheobronchial epithelial (TBE) cells derived from wild-type mice or humans, despite significant inductions still seen for hCLCA1/Gob5. Further studies with JAK inhibitors and STAT6 signaling showed active signaling of the JAK/STAT6 pathway in these primary TBE cultures by IL-13 in the regulation of hCLCA1 expression. Dual immunofluorescent staining with antibodies specific to MUC5AC and hCLCA1 revealed a differential nature of the expression of these two biomarkers by distinct cell types of primary TBE cultures. Finally, MUC5AC expression could be elevated by a bacterial product, peptidoglycan, without any induction of hCLCA1. Thus, these results suggest that the two biomakers of the metaplastic airway mucous cell type are differentially regulated by JAK/STAT6-dependent and -independent pathways.
Keywords: airway epithelium, cytokine, JAK/STAT, mucin, Gob-5
Excessive secretion of mucus in the airways is an important cause of morbidity and mortality in diseases such as asthma, chronic obstructive pulmonary disease (COPD), and cystic fibrosis (CF) (1, 2). Understanding the mechanisms that lead to increased mucus secretion in these diseases will be important for developing improved therapeutics in the future. In patients and experimental animal models with these airway diseases, increased numbers of mucus-producing cells line the surface airway epithelium of the lung (3). These secretory cells vary in appearance with the cycle of secretory activity. At rest, these surface secretory cells, namely, the goblet cell type, have a goblet- like shape with a basally situated nucleus, and are filled with mucin containing electron-lucent secretory granules of varying sizes (4). Mucins are a family of large, heavily glycosylated proteins that comprise a significant portion of the secreted mucus and the secretory granules (5–7). Their heavily glycosylated structures are believed to contribute to the high viscoelastic property of secreted airway mucus. MUC genes encode the protein backbones of mucins. The expression of the mucin gene MUC5AC/Muc5ac is normally used as a marker for these airway surface mucous/goblet cells (8, 9), whereas MUC5B/Muc5b (10, 11) and the recently identified MUC19/Muc19 (12) are regarded as markers for the mucous cell type of the submucosal glands.
In mucous cell metaplasia, another gene, called calcium- dependent chloride channel 1 (CLCA1), or the mouse homolog, Gob5 (or mCLCA3), is also expressed at increased levels (13–15). This membrane-associated protein is expressed in the mucus secretory granules of goblet cells of airways, intestines, and uterus (16). In airways, it is believed to play a role in secreting chloride anions into the lumen and contributing to the salt and water composition of secreted mucus (17, 18). The expression of this gene has been shown to be increased in airway epithelial cells in patients with asthma (15) as well as in animal models (13) of asthma. In addition, studies have also showed that overexpression of hCLCA1 could increase the expression of MUC5AC in NCI-H292 airway cells (14), whereas its downregulation by antisense Gob5 RNA led to a corresponding decrease in Muc5ac (13). These results suggest that hCLCA1 is not only a strong marker for goblet cells, but may play a functional role in upregulating the expression of the MUC5AC gene in airway epithelial cells.
Because mucous cell metaplasia in airway diseases is frequently accompanied by significant airway inflammation, it is widely believed that inflammatory cells and their secreted mediators directly act on airway epithelial cells to induce goblet cell formation. In asthma, T helper type 2 cytokines, such as IL-4, IL-9, and IL-13 are believed to play important roles in directly causing MUC5AC expression in airway cells. Among these cytokines, IL-13 seems to have the most prominent role in causing the asthma phenotype (19, 20). IL-4 and IL-9 have been shown to induce mucin expression in vitro and in vivo (21–24), but several studies have shown their presence is not critical to mucous cell hyperplasia (25–27). The ability of IL-13 to directly increase MUC5AC expression in airway epithelial cells is also not entirely clear. At least one group has found that IL-13 can increase MUC5AC expression in airway epithelial cells (25), but others have reported either no change (28, 29) or a decrease in its expression (30) after IL-13 treatment. It is difficult to determine what the true effect of IL-13 is on MUC5AC in airway epithelial cells, as many of these studies varied widely in terms of the cells used (cell lines versus primary cells), the culture conditions (immersed versus air–liquid interface (ALI) culture conditions, serum-free defined medium versus serum-supplemented medium), and duration of treatment (1 d to 2 wk).
Our recent work on primary human tracheobronchial epithelial (TBE) cells cultured under an ALI condition demonstrated no induction of either MUC5B or MUC5AC expression by IL-13 after 24 h of treatment (28). In the present study, we sought to characterize the expression of both MUC5AC/Muc5ac and hCLCA1/Gob5 by airway epithelial cells to determine whether these genes are coordinately or differentially regulated. We demonstrate that at 24 h, IL-13 induced the expression of both Muc5ac and Gob5 in mouse lungs after an intratracheal administration. However, a similar 24-h treatment by IL-13 on primary mouse airway cell cultures lead to only Gob5 induction, but not Muc5ac. A similar induction of hCLCA1, but not MUC5AC, by IL-13 was also demonstrated in primary human TBE cells. Interestingly, the bacteria product, peptidoglycan, was a potent inducer of MUC5AC, but not hCLCA1, expression in primary human TBE cells. These results suggest that the two biomarkers, MUC5AC/Muc5ac and hCLCA1/Gob5, of surface mucous/goblet epithelial cells are differentially regulated.
MATERIALS AND METHODS
Culturing of Human and Mouse Primary Airway Epithelial Cells
For human cells, discarded trachea and bronchii with consent from the local hospital, University of California at Davis Medical Center, and the National Institutes of Health–sponsored nationwide organization, the National Disease Research Interchange (Philadelphia, PA), were minced and washed with Minimum Essential Medium (MEM; Sigma, St. Louis, MO) and then digested overnight with 0.2% protease (Type XIV; Sigma) in MEM. The protocol for human tissue procurement was periodically reviewed and approved by University Human Subject Research Review Committee and consent forms for these tissues were obtained. The protease-dissociated TBE cells were dislodged from tissues and pelleted by centrifugation as described previously (31). Cell pellets were suspended in a serum-free hormone-supplemented medium and plated onto 100-mm tissue culture dishes, incubated in a 5% CO2 incubator until confluency (within 7–10 d). The serum-free hormone-supplemented medium used for establishing primary cultures was Clonetics' small airway basal medium (Cambrex BioScience, Walkersville, MD)/Dulbecco's modified Eagle's medium at 1:1 and supplemented with insulin (5 μg/ml), transferrin (5 μg/ml), epidermal growth factor (10 ng/ml), dexamethasone (0.1 μM), cholera toxin (10 ng/ml), bovine hypothalamus extract (15 μg/ml), and bovine serum albumin (0.5 mg/ml). The confluent primary cultures were further passaged onto a Transwell (Corning Costar, Corning, NY) chamber (25 mm diameter) at 1–2 × 104 cells/cm2 in the above-mentioned serum-free hormone-supplemented medium with the addition of 30 nM all-trans-retinoic acid, as described previously (31). After 1 wk in an immersed condition, the passage-1 cultures were transferred from the immersed condition to an ALI to allow for differentiation into a mucociliary epithelium (Days 14–21 after cell plating). Experiments were performed mostly on these passage-1, well-differentiated TBE cultures. For mouse TBE cell cultures, both wild-type (Charles River Laboratory, Wilmington, MA) and Stat6-deficient (Jackson Laboratories, Bar Harbor, ME) Balb/c female mice at 10–12 wk of age were killed and their trachea were sterilely removed. TBE cells were isolated and plated on collagen gel–coated Transwell chambers as described previously (31). The culture medium used was the same as that for primary human TBE cells with the supplement of 30 nM all-trans-retinoic acid. Primary TBE cells were maintained initially under an immersed condition and then shifted to the ALI as described for human cells. Cultures used in the study were incubated for 14–21 d after the initial plating.
Intratracheal Instillation of IL-13 in Mouse
Balb/C mice were lightly anesthetized with inhaled isoflurane and their tracheas were surgically exposed. A sterile 26-gauge needle was then used to instill either PBS (for control) or IL-13 (R&D Systems, Minneapolis, MN) at 0.5 μg in 50 μl of PBS. The skin was then sutured, and the mice placed back into their cages.
Treatment of Cells with IL-13 and Peptidoglycan
Primary TBE cells were grown and plated onto Transwell chamber. After 1 wk in immersed conditions, the cells were switched to an ALI for 14 d to promote mucociliary differentiation. During this time, medium was changed every other day. After 14 d in ALI conditions, the cells were exposed to IL-13 (2–50 ng/ml) or peptidoglycan (2.5 μg/ml, InvivoGen, San Diego, CA) on both the apical and basolateral surfaces. Medium was changed on the apical side of the ALI culture by briefly exposing it to 0.5 ml media with or without IL-13 and peptidoglycan. For the basal side of ALI culture, 2 ml of media with or without IL-13 and peptidoglycan were added. After removing the apical media, cultures were continuously maintained in ALI in a humidified CO2 incubator. Cultures were harvested 24 h (for gene expression studies), and 24, 48, and 96 h (for Western blot studies) after treatment. For inhibitor studies, JAK inhibitor I and AG490 (EMD Biosciences, San Diego, CA) were added to the media on both the apical and basolateral surfaces at indicated concentrations and the cells pretreated for 30 min. After the pretreatment, the cells were further treated with or without IL-13 along with the inhibitors for another 24 h before the cells were harvested.
RNA Isolation
For intratracheally instilled mice, mice were anesthetized and then killed by exsanguination by severing the axillary arteries. The lung and the trachea were then dissected free, minced, and placed in 3 ml of Trizol (Invitrogen, Carlsbad, CA). The tissue was macerated with a blender, and the RNA was then extracted according to the manufacturer's protocol. For each Transwell chamber, culture chambers were washed twice with PBS, and 1 ml of Trizol was added for RNA extraction. The RNA pellet was then dissolved in RNase-free water and stored at −80°C until used. Each mouse lung yielded a total of 100–150 μg RNA, a typical 25-mm Transwell culture yielded ∼ 30–50 μg of total RNA.
Real-Time RT-PCR
An aliquot of 3 μg of total RNA was used to anneal to 1 μM Oligo dT (Promega, Madison, WI) at 70°C for 10 min. The following chemicals were then added to the reaction mixture: 1 μM dNTP, 1X RT buffer, 1 U/μl of RNasin (Promega), and 20 U/μl MMLV reverse transcriptase (Promega). The reaction proceeded for 1 h at 420°C. After the reverse transcription reaction, the first-stranded cDNA was then diluted 1:5, and 10 μl of reaction was used in each subsequent PCR reaction. For the real-time PCR, 10 μl of cDNA was used with SYBR Green and a 5,700 real-time thermocycler (Applied Biosystems, Foster City, CA), according to the manufacturer's protocol. After the PCR, the relative expression of each gene was normalized to GAPDH to give a relative expression level. Mouse Muc5ac primers: forward 5′CCATGCAGAGTCCTCA GAACAA 3′; reverse 5′ TTACTGGAAAGGCCCAAGCA 3′. Mouse Gob5/mCLCA3 primers: forward 5′ ACTAAGGTGGCCTACCTC CAA 3′; reverse 5′ GGAGGTGACAGTCAAGGTGAGA 3′. Mouse GAPDH primers: forward 5′ TGTGTCCGTCGTGGATCTGA 3′; reverse 5′ CCTGCTTCACCACCTTCTTGAT 3′. Human MUC5AC primers: forward 5′ GCTCAGCTGTTCTCTGGATGAG 3′; reverse 5′ TTACTGGAAAGGCCCAAGCA 3′. Human hCLCA1 primers: forward 5′ ACCTCGTTAAAGCACCCACTTA 3′; reverse 5′ CGAT CGGCATTTACTGTGATTA 3′. Human GAPDH primers: forward 5′ CAATGACCCCTTCATTGACC 3′; reverse 5′GACAAGCTTCC CGTTCTCAG 3′.
Western Blotting
Primary TBE cells treated with or without IL-13 were lysed with RIPA buffer and sonicated. The extracts were quantified by the BioRad DC protein assay (BioRad, Hercules, CA). Equal amounts of protein (30 μg/lane) were then loaded and run on a 10% SDS-PAGE gel, followed by blotting to a PVDF membrane. Western blot analysis was performed as described previously (32) with anti–phosphorylated STAT6 monoclonal antibody (1:2,000 dilution; Cell Signaling, Beverly, MA), anti–total STAT6 (1:2,000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) or anti-hCLCA1 rabbit polyclonal antibody (1:250 dilution; a kind gift from Dr. M. Levitt, Genaera Corporation, Philadelphia, PA). Even loading of the gel was ascertained with anti–b-tubulin antibody (Sigma, St. Louis, MO) staining.
Dual Immunofluorescent Staining of IL-13–Treated Primary TBE Cells
Primary TBE cells were grown in ALI conditions for 2 wk and then treated with IL-13 for 1 wk. The cells were fixed in 4% paraformaldehyde for 24 h, and then paraffin embedded. Sections of the cultures were made on glass slides and deparaffinized with xylene, followed by rehydration by immersing in 95, 80, and 50% ethanol, then water. The cells were then incubated in blocking buffer composed of PBST (PBS with Tween-20 at 0.02%) plus 2% BSA for 1 h at room temperature. The sections were then incubated with a rabbit polyclonal antibody to hCLCA1 in blocking buffer at 1:250 dilution for 24 h at 4°C, followed immediately by incubation of a mouse monoclonal antibody to MUC5AC at 1:1,000 dilution (45M1 clone; Santa Cruz Biochemicals, Santa Cruz, CA) for 2 h at 37°C. These sections were washed 3 times with PBST, and then incubated with Alexa Fluor 568 (Molecular Probes, Eugene, OR) goat anti-rabbit (for hCLCA1), and FITC (Antibodies Incorporated, Davis, CA) goat anti-mouse (for MUC5AC) antibodies at 1:500 dilution in blocking buffer for 1 h at 37°C. The sections were again washed 3 times in PBST and then covered with 20 μl of Vectashield fluorescent mounting media (Vector Laboratories, Burlingame, CA) and a cover slip. Dual immunofluorescent stains were examined by a Carl Zeiss confocal fluorescent microscope with appropriate filters (Carl Zeiss Surgical, Inc., Thornwood, NY).
Statistical Analysis
Two sample t test was used to calculate P values when there were only two groups of treatment. When three groups or more of treatment were present, single factor ANOVA was used to calculate P values followed by post hoc testing with student test for individual group differences. The results were considered significant when P values were < 0.05.
RESULTS
In Vivo/In Vitro Mouse Studies
Because IL-13 is one of the major mediators that have been implicated in mouse asthmatic goblet cell metaplasia, we initially performed the intratracheal instillation studies of this cytokine on both wild-type and Stat6-null mice. A single intratracheal instillation of IL-13 elevated both Muc5ac and Gob5 messages in wild-type mouse lungs (Figure 1). For Muc5ac, the stimulation was 8.5-fold (Figure 1A), whereas IL-13 elevated Gob-5 expression 110-fold (Figure 1B). However, these stimulations were abrogated in Stat6-deficient mice. These results have been repeated twice with three animals for each data point, and data were quite reproducible. These in vivo intratracheal instillation studies are consistent with previously published results (33–35), and suggest the involvement of IL-13–Stat6 signaling in the regulation of both Muc5ac and Gob5 expression in vivo (36). However, these experiments cannot rule out the possibility that an indirect action of IL-13, such as through the recruitment of inflammatory cells and secondary mediators upon the instillation, is involved in this in vivo phenomenon. To address this concern, experiments were performed in vitro using primary cultures of mouse TBE cells, which were long-term cultured under ALI conditions. To our surprise, IL-13 treatment for 24 h had no stimulatory effect on mouse Muc5ac expression, whereas the stimulation of Gob5 message was increased more than 500-fold (Figure 2). Consistent with the in vivo data, the stimulation of Gob5 in vitro by IL-13 was also Stat6-dependent because no increase of Gob5 message was observed in IL-13–treated primary TBE cells derived from Stat6 knockout mouse.
Figure 1.
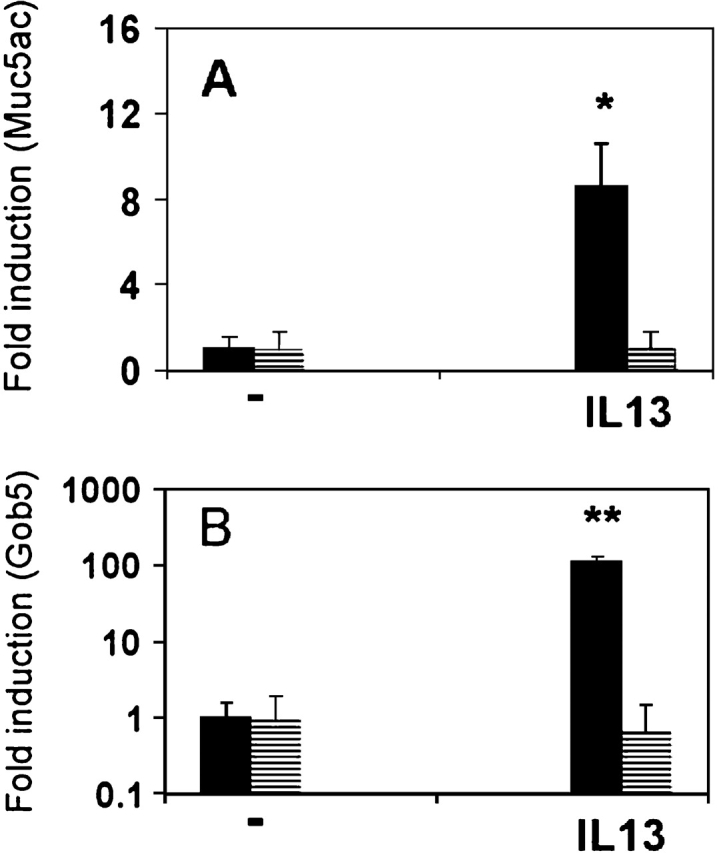
Expression of Muc5ac and Gob5 in mouse lungs after intratracheal instillation of IL-13. Balb/c female mice were given intratracheal injections of IL-13 at 500 ng/50 μl. The lungs were isolated 24 h later for total RNA isolation. A two-step real-time RT-PCR protocol was then performed with primers specific for Muc5ac and Gob5. Copy numbers of each gene were normalized to GAPDH. (A) Fold of induction of Muc5ac as normalized to GAPDH in wild-type mice (filled bar) and Stat6-deficient (striped bar) Balb/c mice after intratracheal instillation of PBS and IL-13. (B) Fold of induction of Gob5 expression as normalized to GAPDH of Gob5 in wild-type (filled bar) and Stat6-deficient (striped bar) Balb/c mice after intratracheal instillation. Y axis is in logarithmic scale. n = 3; *P = 0.002; **P = 0.003. Data are presented as mean ± 1 SEM.
Figure 2.
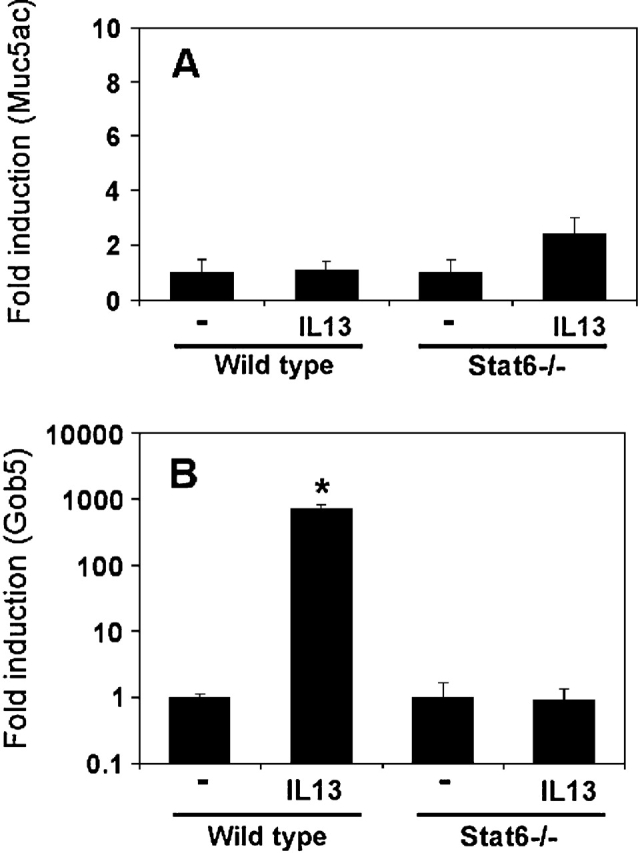
In vitro effects of IL-13 on Muc5ac and Gob5 gene expression in primary mouse TBE cells. Mouse TBE cells were obtained from wild-type and Stat6 knockout Balb/c mice, as described in the text. TBE cells were cultured in an air-liquid interface (ALI) condition, with either vehicle (PBS) or IL-13 at 20 ng/ml for 24 h, and then total RNA was isolated and real-time RT-PCR was performed as described in Figure 1. (A) Fold induction of Muc5ac (normalized with GAPDH) in these mouse lungs after IL-13 instillation. (B) Fold induction of Gob5 expression (normalized with GAPDH) in mouse TBE cultures after IL-13 treatment. n = 3; *P = 0.069; **P = 0.029.
In Vitro Studies with Primary Human TBE Cells
One of the problems associated with primary mouse TBE cultures is that goblet cell differentiation is very low in culture, even when those cells are maintained under ALI culture conditions. We could routinely find ciliated cells in these primary mouse TBE cultures, but very few “mucous” cells can be found, a situation quite different from the human primary TBE cultures (unpublished data). We noticed that the level of Muc5ac expression in mouse primary TBE cultures is extremely low. It is possible that the lack of Muc5ac expression in response to IL-13 in mouse cells is due to its low expression in nature. For this reason, we extended the above study to human primary TBE cultures, which express high levels of MUC5AC and MUC5B (28). Consistent with mouse data and our recent publication (28), IL-13 treatment had no stimulatory effect on MUC5AC message (Figure 3A), whereas the stimulatory effect on hCLCA1 could be demonstrated (Figure 3B). A dose-dependent study of IL-13, up to 50 ng/ml, also failed to show the stimulation of MUC5AC expression, but the stimulation of hCLCA1 was demonstrated. A time-course study showed that significant induction of hCLCA1 by IL-13 occurred after 3 h treatment, and was maximal 24–48 h later (data not included). There was a slight increase of MUC5AC message in human TBE cultures after a 1 wk treatment with IL-13, but the induction was not statistically significant (data not shown).
Figure 3.
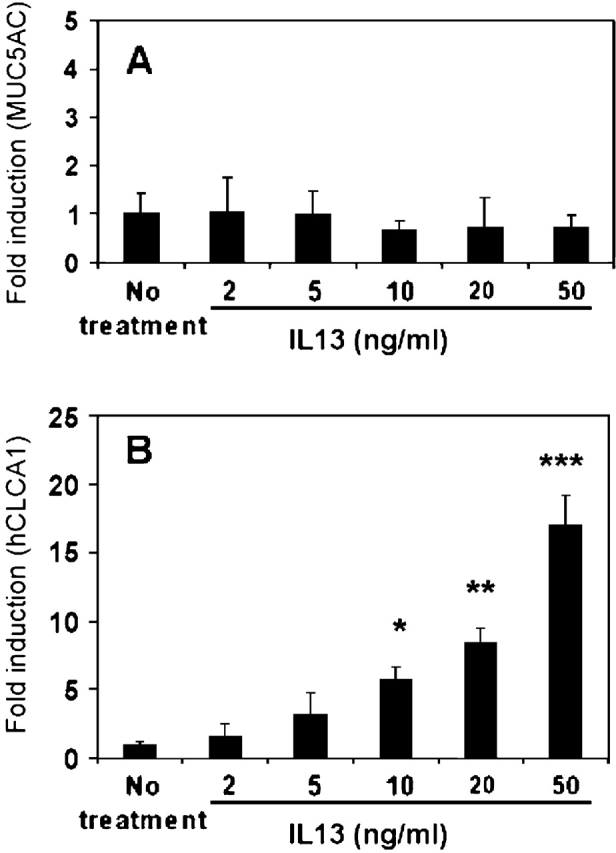
Dose-dependent effects of IL-13 on MUC5AC and hCLCA1 expression by primary human TBE cells in vitro. Well differentiated primary human TBE cells were prepared as described in the text, and treated with IL-13 at doses from 2 to 50 ng/ml for 24 h as shown. (A) Effects of IL-13 doses on MUC5AC expression by primary human TBE cells. (B) Effects of IL-13 doses on hCLCA1 expression by primary human TBE cells. n = 4; *P < 0.018; **P < 0.001; ***P < 0.007.
Inhibitor Studies
Because IL-13 mediates many of its effects through the activation of JAK/STAT signaling pathways (37), we wanted to test whether IL-13's ability to upregulate hCLCA1/Gob5 could be inhibited by JAK inhibitor 1 (a nonspecific inhibitor of JAK1, JAK2, JAK3, and TYK2) and a JAK2-specific inhibitor, AG490. As can be seen, both JAK inhibitor 1 at 100 nM and AG490 at 50 μM significantly decreased the upregulation of hCLCA1 by IL-13 (Figure 4). Cell viability was tested and confirmed to be > 95% throughout this study. A similar response to these inhibitors for the inhibition of IL-13–induced Gob5 was also demonstrated in mouse primary TBE cultures (Figure 5).
Figure 4.
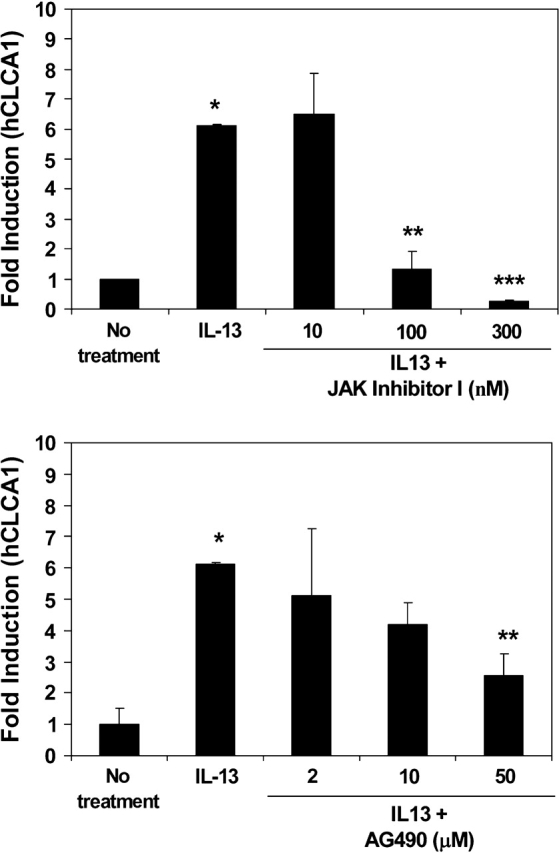
Effect of JAK inhibitors on IL-13–induced hCLCA1 expression by human primary TBE cells. The experiment was performed the same as described in Figure 2. Before IL-13 treatment, cells were treated with different amounts of JAK inhibitors; JAK inhibitor I (A) and AG490 (B) for 30 min. The doses of JAK Inhibitor I were 10, 100, and 300 nM. The doses of AG490 were 2, 10, and 50 μM. Twenty-four hrs after IL-13 treatment, the cells were harvested for measuring the relative fold induction of hCLCA1 expression (normalized with GAPDH). The control study, treated with an equal amount of these inhibitors showed no quantitative change of hCLCA1 message in these cultures. For this reason, only one data point with “no treatment” was used in the figure. n = 3; *P = 0.005; **P = 0.009; ***P = 0.014.
Figure 5.

Effects of JAK inhibitors on IL-13–induced Gob5 expression by mouse primary TBE cells in culture. Mouse TBE cell cultures were performed as described in Figure 2. Before IL-13 (20 ng/ml) treatment, cultured cells were pretreated for 30 min with the JAK inhibitors, JAK inhibitor I (100 nM) and AG490 (10 μM), and RNA was isolated 24 h after IL-13 treatment. Relative fold increase of Gob5 message that is normalized with GAPDH is shown in the figure. n = 3; *P = 0.029; **P = 0.047.
Because IL-13 can induce hCLCA1 significantly in airway epithelial cells in culture and its effects appear to depend on JAK signaling, we tested further to see whether the cellular signaling mechanisms would involve STAT6. To do this, we first tested the ability of IL-13 to induce STAT6 phosphorylation in primary human TBE cells. Primary cells were treated with IL-13, and then cell lysates were prepared from these cultures at 1, 6, and 24 h after the treatment. Western blot analysis of these cell lysates using an antibody specific to phosphorylated STAT6 was then performed (Figure 6). It was observed that IL-13 induced the phosphorylation of STAT6 within 1 h of treatment, and its effects persisted at 24 h. The control experiments showed that the total STAT6 protein loading in this gel was the same for all cell lysates.
Figure 6.

Western blot analysis of STAT6 phosphorylation in primary humanTBE cells after IL-13 treatment. Primary human TBE cells were treated with IL-13 at 20 ng/ml, as described in Figure 2. At various times after the treatment, cultures were harvested for protein extraction and Western blot analysis with both anti–phosphorylated STAT6 (upper blot) and anti–total STAT6 protein (lower blot) antibodies. A similar result has been repeated in another primary human TBE culture derived from a different donor (data not shown).
Stimulation of MUC5AC but Not hCLCA1 Expression by Peptidoglycan
Because IL-13 appeared to increase the expression of hCLCA1, but did not increase the MUC5AC level in primary TBE cell cultures, we set out to look for other mediators that could increase the expression of MUC5AC, and then determine if those same mediators had any effect on hCLCA1 expression. Through preliminary studies with various bacterial products on airway epithelial cells, we saw that Staphylococcus Aureus lysates appeared to increase the expression of MUC5AC in primary TBE cells (data not shown). Therefore, we tested whether some of the bacterial components could also stimulate MUC5AC. We found that peptidoglycan, when given in concentrations of ∼ 2.5 μg/ml, could significantly stimulate the expression of MUC5AC (Figure 7). This same amount of peptidoglycan did not induce the expression of hCLCA1. Thus, peptidoglycan appeared to stimulate selectively the expression of MUC5AC, but not hCLCA1, in these primary human TBE cultures.
Figure 7.
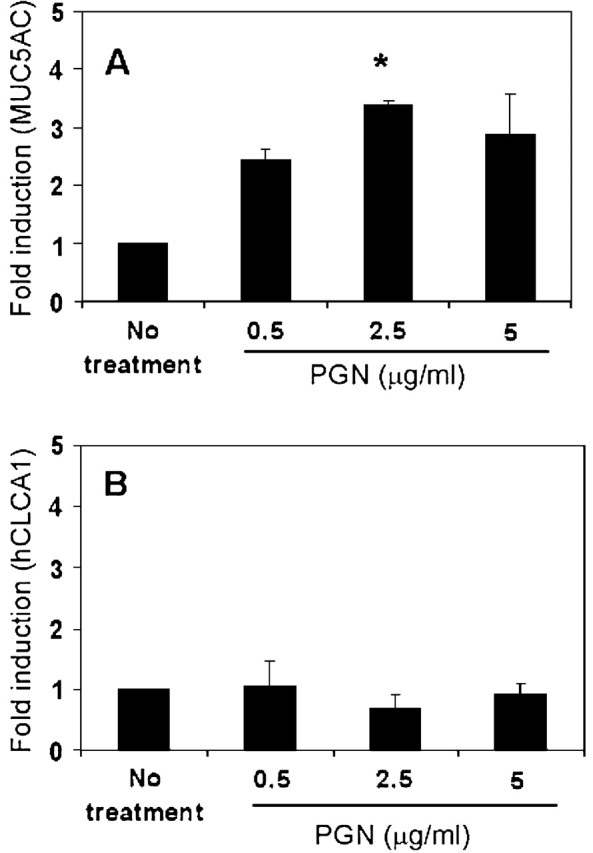
Effects of peptidoglycan on the expression of MUC5AC and hCLCA1 by primary human TBE cells. Well differentiated primary human TBE cells were treated with different amounts of peptidoglycan (PGN) from Staphylococcus Aureus (Invivogen) for 24 h. (A) Fold induction of MUC5AC (normalized with GAPDH). (B) Fold induction of hCLCA1 (normalized with GAPDH). n = 2; *P = 0.041.
Western Blot Analysis and Immunofluorescent Staining of hCLCA1 and MUC5AC in Primary TBE Cells
Previous data have shown that both gene products of hCLCA1/Gob5 and MUC5AC/Muc5ac are expressed in goblet cells in both human and mouse airways (13, 15). Gob5 expression, as analyzed at an electron microscopic level, was shown to be localized at the membranes of mucus-secreting granules (16). To assess whether the observed IL-13 effects on hCLCA1 and MUC5AC messages also occur at the protein level, both Western blot analysis and dual immunofluorescent staining were performed. As shown in a time-course study (Figure 8), hCLCA1 protein was significantly elevated within 24 h of treatment with IL-13. This elevation persisted in 48 and 96 h samples. In contrast to this finding, no significant change in MUC5AC, as detected by 45M1 antibody, was seen in these samples (data not shown). To further elaborate on the nature of the differential expression of hCLCA1 and MUC5AC, paraffin-embedded sections of IL-13–treated primary TBE cultures were stained with the two antibodies specific to these molecules. As seen in Figure 9, dual immunofluorescent staining supported some of the claims of colocalization of these two biomarkers, whereas the figure also displays the differential nature of the expression by different TBE cells. The dual fluorescent staining showed a colocalization of anti-hCLCA1 (Alex Fluor–stained red) and 45M1-specific (FITC–stained green) stains in the same cell (Figure 9A, arrow) within 0.5 μm thick, as scanned by a Carl Zeiss confocal fluorescent microscope. However, in a different area of the same paraffin section, the 0.5 μm thick section, also scanned by confocal fluorescent microscope, revealed different locations and different cells for both anti-hCLCA1 and 45M1-specific stains (Figure 9B). In a separate paraffin section, none of the dual-stained cells could be found (Figures 9C and 9D). Roughly 3% of all cells stained for hCLCA1 and 20% of cells stained for MUC5AC. Costaining of hCLCA1 and MUC5AC was seen infrequently (< 0.5% of the cells) in primary human TBE cultures 1 wk after IL-13 treatment (data not shown). These results further support the notion that the expression of these two biomarkers of surface mucous/goblet cells are differentially regulated in airway cells.
Figure 8.

Western blot analysis of hCLCA1 expression in human TBE cells after IL-13 treatment. Well-differentiated primary TBE cultures (at Day 21) were treated with IL-13 (10 ng/ml) as described in Figure 3. At 24, 48, and 96 h after IL-13 treatment, both untreated and treated cultures were harvested for protein preparations. Equal amount of proteins (30 μg/lane) were loaded on to the gel. Immunoblot preparation and analysis were performed as described in the text. Anti–β-tubulin staining was used to further verify the equal protein loading (data not shown). This figure is taken from one of three experiments, all of which showed similar findings.
Figure 9.

Double immunofluorescent staining of primary human TBE cells treated with IL-13. Paraffin blocks were prepared from primary TBE cultures treated with IL-13 (10 ng/ml) for 1 wk on both the basolateral and apical areas, and paraffin sections (5 μm) were prepared. After deparaffinization, double antibodies specific for hCLCA1 and MUC5AC were used to stain these sections, which were further stained with Alexa Fluor (red)-conjugated or FITC (green)-conjugated secondary antibody, respectively. Nuclei were further stained with DAPI, yielding a blue color. The slides were examined using a Zeiss Confocal microscope under the ×63 lens (A, B) and a Zeiss fluorescent microscope (C, D). Z-Stacking was performed at 0.5-μm cuts and 10–12 images were obtained through the thickness of the sections. The images in (A) and (B) are single 0.5-μm slices from the Z-Stacked cuts. (A) A 0.5-μm slice showing the colocalization of hCLCA1 and MUC5AC. Arrow indicates a cell that was stained by both antibodies, and the merged color gives off a yellowish glow. Blue fluorescent stains are nuclei stained with DAPI. (B) Another region of the same slide that showed different cells stained for hCLCA1 and MUC5AC with no overlap seen. (C and D) Fluorescent views of another paraffin section that was examined with a Zeiss fluorescent microscope under the filter specific for FITC (green [C], 45M1) or Alexa Fluor (red, (D), anti-hCLCA1), with a 40× lens.
DISCUSSION
This study demonstrates that the expression of two goblet cell–associated biomarkers, MUC5AC/Muc5ac and hCLCA1/Gob5, are differentially regulated by IL-13, depending on whether the study is performed in vivo or in vitro. When IL-13 was intratracheally instilled, both Muc5ac and Gob5 messages were elevated in mouse lungs after 24 h, and were presumably expressed by surface goblet cells of mouse airways. This elevated expression was abrogated by Stat6-null mutation, indicating a Stat6-dependent event for both. However, when isolated primary airway epithelial cells from mice and humans were treated with IL-13 in vitro for 24 h, only hCLCA1/Gob5A was found to be elevated, but not MUC5AC/Muc5ac. The lack of stimulation of MUC5AC/Muc5ac in vitro by IL-13 is not related to the activation of STAT6, the transcription factor known to be the main downstream signaling molecule for IL-4 and IL-13. In this study, we showed that STAT6 was readily phosphorylated in human airway epithelial cells by IL-13 within 1 h (Figure 6). Subsequently, dual immunofluorescent staining with antibodies specific to these two biomarkers revealed the differential nature of the expression of these proteins by different airway cells (Figure 9).
MUC5AC/Muc5ac and hCLCA1/Gob5 are well known markers for surface goblet cells in the airway epithelium (8, 9, 15, 16,). IL-13 is the T helper type 2 cytokine that is believed to affect the mucous cell metaplasia on airway epithelium present in allergic-mediated asthma models (20, 27). The logical conclusion to draw would be that IL-13 acts on airway epithelial cells to induce expression of MUC5AC/Muc5ac and hCLCA1/Gob-5, which then leads to the metaplastic change of cells into a mucous type. But if this model is correct, how can we explain the discrepancy between the ability of IL-13 to induce both Muc5ac and Gob-5 expression in vivo, but only Gob-5 in vitro? The first factor that we considered was whether the in vitro cultures that we used are an acceptable model for airway epithelial cells in vivo. The primary cell culture system used in this study has a heterogeneous cell population that, under ALI, develops into well differentiated cells with morphologies very close to that of the native airway epithelium (31). Although there are limitations in all in vitro systems, and our cultures cannot totally mimic the cells in the intact animal, we believe that our system is among the best for studying biological mechanisms, and provides conditions that are as close as possible to those in vivo. Although our mouse and human cultured TBE cells did not increase MUC5AC expression in response to IL-13, as they did in vivo, they were still able to respond to this cytokine by increasing hCLCA1/Gob-5 expression and the activation of STAT6 signaling. These results suggest that our primary TBE cells have a preserved ability to respond to IL-13 and mediate its intracellular signaling cascades, but that the regulation of hCLCA1/Gob5 and MUC5AC/Muc5ac are different. Our hypothesis is that shortly after ligand binding by IL-13 to its receptor on airway epithelial cells, there is an activation of a signaling cascade of the JAK/STAT pathway. The Gob5/hCLCA1 gene is the target of this activated JAK/STAT signaling cascade. However, for MUC5AC/Muc5ac induction in vivo, the increased expression may depend on more than simple IL-13/epithelial interaction. Additional factors or signaling pathways that are present in vivo, but missing in vitro, may be required. These factors/signalings may lead to either the stabilization of the message (38) or the activation of transcriptional factors, such as NF-κB, activator protein 1, surfactant protein 1, etc., that are needed for the transcription of MUC5AC/Muc5ac gene in airway cells (39). Other investigators have found the NF-κB pathway to be important in the regulation of MUC5AC (39). Our results with peptidoglycan support the possibility that the NF-κB pathway may play a role in the regulation of the MUC5AC/Muc5ac gene because signaling through TLR2 by the peptidoglycan could activate NF-κB through the MyD88/TIRAP/TRAF6 pathways (40). Interestingly, one research group has found that IL-13's ability to induce Muc5ac in vivo may depend on its cooperation with cysteinyl leukotrienes and chemokines (35), further suggesting the significance of coordinated signaling in the regulation of gene expression. This possibility can be tested, but is beyond the scope of this study.
Other investigators have shown that IL-13 is capable of inducing the formation of goblet cells under in vitro conditions in primary cell cultures similar to ours (25, 41). Although these studies did not always look specifically at MUC5AC/Muc5ac (41), it is very likely that its induction occurred in their systems, as MUC5AC/Muc5ac is a well-established marker for mucous cell metaplasia (7–9). The likely reason for the discrepancy between our results and those of other investigators is the differences in culture conditions and/or doses and time of treatment. Our dose treatment of IL-13 is similar to what other investigators have used, although one group did find that typical doses of IL-13, such as 10 ng/ml, actually inhibited goblet cell formation, but small doses, such as 1 ng/ml, promoted it (41). Although we did not include doses as low as 1 ng/ml, we did go as low as 2 ng/ml, and did not see any induction of MUC5AC/Muc5ac. Our treatment course (24 h) was much shorter than that used by most other investigators (1–2 wk) in the treatment of primary cells to induce goblet cell formation and/or MUC5AC/Muc5ac induction (25, 41). However, we chose our time based on our observation that intratracheal induction of MUC5AC/Muc5ac by IL-13 can occur at 24 h. This time course is consistent with that found by other investigators after a single intratracheal injection of IL-13. If IL-13 can induce MUC5AC/Muc5ac within 24–48 h in vivo (33–35), we believe that it is reasonable for our in vitro treatments to parallel the same time course.
Several studies have suggested that hCLCA1/Gob5 may play a regulatory role in stimulating MUC5AC/Muc5ac expression (13, 15). Our data do not directly address the issue of whether MUC5AC expression is dependent on hCLCA1/Gob5 expression. Instead, our focus was aimed at understanding the relationship of these two genes in their responses to IL-13. Nevertheless, we suspect that if MUC5AC/Muc5ac expression was critically dependent on hCLCA1/Gob-5, there would not be a discrepancy seen between their coordinated upregulation by IL-13 in vivo compared with the isolated upregulation of hCLCA1/Gob5 in vitro. The ability to induce MUC5AC without the induction of hCLCA1 by peptidoglycan, as demonstrated in this study, further supports this notion.
One group of investigators has found a critical importance of Stat6 expression in airway epithelial cells for the induction of Muc5ac in vivo (36). Using an ingenious method, whereby mice that were Stat6-deficient but had it reconstituted, only on airway epithelial cells, through a CC10 promoter, this group showed that overexpression of IL-13 could induce mucous cell metaplasia only in mice with reconstituted epithelial Stat6. These data demonstrate convincingly that IL-13's ability to induce Muc5ac and Gob5 depends critically on Stat6. Because no other mediator than IL-4/IL-13 has been identified that can activate STAT6 (42), it is reasonable to conclude that IL-13's induction of Muc5ac, Gob-5, and mucous cell metaplasia occurs through its direct effects on airway epithelial cells. Our in vivo data involving the intratracheal injection of IL-13 are consistent with those of this study (36). Although we did not see an induction of MUC5AC/Muc5ac by IL-13 in vitro, this does not contradict the notion that IL-13 can induce MUC5AC/Muc5ac and mucous cell metaplasia directly in vivo. But our data do suggest, as we hypothesized previously here, that IL-13 may require additional factors or signals that are present in vivo to induce Muc5ac. These factors and signals could still be present in the reconstituted epithelial STAT6 of mice noted previously here.
What are the roles for these two gene products in mucous cell metaplasia and mucus hypersecretion in the airways? The expression of both genes are increased in goblet cells (8, 9, 13–15), and both gene products have been localized to mucin granules on goblet cells (16). However, our data, with the double immunostaining for hCLCA1 and MUC5AC in primary TBE cultures, did not often show colocalization of these proteins. Most cells that expressed either of these proteins appeared to express either one or the other, but not both. Cells that did express both proteins were fairly uncommon in our sections. What can explain this apparent discrepancy? One possibility is that these proteins are expressed by cells of different lineage. We believe this is unlikely, as both proteins have well-established associations with goblet cells. A more likely possibility is that cells stained for hCLCA1 and MUC5AC are cells of the same lineage but along different time-points of differentiation. It is possible that early on, cells destined for differentiation into goblet cells may express hCLCA1, but then express mucin genes later on as they mature. The different staining that we see representing cells at different time points of this maturation process with the rare costained cells are in a transition phase period.
In summary, through an intratracheal instillation, we demonstrated that both Muc5ac and Gob5 were elevated by IL-13 in mouse lungs. These elevations are Stat6-dependent. However, the stimulatory phenomena could not be reproduced for MUC5AC/Muc5ac in primary TBE cultures, including those from human tissues. This negative result is not due to the impairment of STAT6 signaling in these primary TBE cultures, because STAT6 phosphorylation still occurs and JAK inhibitors could still partially block hCLCA1/Gob5 expression. In addition, we have shown that it is possible to selectively stimulate MUC5AC, but not hCLCA1, expression by a bacterial product, peptidyloglycan, in vitro. This suggest to us that, in IL-13 induction of mucous cell metaplasia, the induction of the two biomarkers MUC5AC/Muc5ac and hCLCA1/Gob5 may be differently regulated, with additional factors required for the former compared with the latter.
Acknowledgments
The authors thank Yu Hua Zhao for her work with the cell culture experimental animals. They also thank Dr. Suzette Smiley-Jewell for her extensive efforts in reviewing this manuscript.
This work was supported in part by National Institutes of Health (NIH) grants (HL35635, HL077315, HL077902, HL073160). P.T. was supported by an NIH training grant (T32 HL07013).
Originally Published in Press as DOI: 10.1165/rcmb.2004-0220RC on September 8, 2005
Conflict of Interest Statement: None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Jeffery PK. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care Med 2001;164(Suppl. 10 Pt 2):S28–S38. [DOI] [PubMed] [Google Scholar]
- 2.Puchelle E, Bajolet O, Abely M. Airway mucus in cystic fibrosis. Paediatr Respir Rev 2002;3:115–119. [DOI] [PubMed] [Google Scholar]
- 3.Voynow JA. What does mucin have to do with lung disease? Paediatr Respir Rev 2002;3:98–103. [DOI] [PubMed] [Google Scholar]
- 4.Rogers DF. Airway goblet cells: responsive and adaptable front-line defenders. Eur Respir J 1994;7:1690–1706. [PubMed] [Google Scholar]
- 5.Davies JR, Svitacheva N, Lannefors L, Kornfalt R, Carlstedt I. Identification of MUC5B, MUC5AC and small amounts of MUC2 mucins in cystic fibrosis airway secretions. Biochem J 1999;344:321–330. [PMC free article] [PubMed] [Google Scholar]
- 6.Wickstrom C, Davies JR, Eriksen GV, Veerman EC, Carlstedt I. MUC5B is a major gel-forming, oligomeric mucin from human salivary gland, respiratory tract and endocervix: identification of glycoforms and C-terminal cleavage. Biochem J 1998;334:685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hovenberg HW, Davies JR, Herrmann A, Linden CJ, Carlstedt I. MUC5AC, but not MUC2, is a prominent mucin in respiratory secretions. Glycoconj J 1996;13:839–847. [DOI] [PubMed] [Google Scholar]
- 8.Hovenberg HW, Davis JR, Carlstedt I. Different mucins are produced by the surface epithelium and the submucos in human trachea: identification of MUC5AC as a major mucin from the goblet cells. Biochem J 1996;318:319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zuhdi AM, Piazza FM, Selby DM, Letwin N, Huang L, Rose MC. Muc5/5ac mucin messenger RNA and protein expression is a marker of goblet cell metaplasia in murine airways. Am J Respir Cell Mol Biol. 2000;22:253–260. [DOI] [PubMed] [Google Scholar]
- 10.Sharma P, Dudus L, Nielsen PA, Clausen H, Yankaskas JR, Hollingsworth MA, Engelhardt JF. MUC5B and MUC7 are differentially expressed in mucous and serous cells of submucosal glands in human bronchial airways. Am J Respir Cell Mol Biol 1998;19:30–37. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Zhao YH, Wu R. 2001. In silico cloning of mouse Muc5b gene and upregulation of its expression in mouse asthma model. Am J Respir Crit Care Med 164:1059–1066. [DOI] [PubMed]
- 12.Chen Y, Zhao YH, Kalaslavadi TB, Hamati E, Nehrke K, Le AD, Ann DK, Wu R. Genome-wide search and identification of a novel gel-forming mucin MUC19/Muc19 in glandular tissues. Am J Respir Cell Mol Biol 2004;30:155–165. [DOI] [PubMed] [Google Scholar]
- 13.Nakanishi A, Morita S, Iwashita H, Sagiya Y, Ashida Y, Shirafuji H, Fujisawa Y, Nishimura O, Fujino M. Role of gob5 in mucus overproduction and airway hyperresponsiveness in asthma. Proc Natl Acad Sci USA 2001;98:5175–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Y, Dong Q, Louahed J, Dragwa C, Savio D, Huang M, Weiss C, Tomer Y, McLane MP, Nicolaides NC, et al. Characterization of a calcium-activated chloride channel as a shared target of Th2 cytokine pathways and its potential involvement in asthma. Am J Respir Cell Mol Biol 2001;25:486–491. [DOI] [PubMed] [Google Scholar]
- 15.Hoshino M, Morita S, Iwashita H, Sagiya Y, Nagi T, Nakanishi A, Ashida Y, Nishimura O, Fujisawa Y, Fujino M. Increased expression of the human Ca2+-activated Cl− channel 1 (CaCC1) gene in the asthmatic airway. Am J Respir Crit Care Med 2002;165:1132–1136. [DOI] [PubMed] [Google Scholar]
- 16.Leverkoehne I, Gruber AD. The murine mCLCA3 (alias gob-5) protein is located in the mucin granule membranes of intestinal, respiratory, and uterine goblet cells. J Histochem Cytochem 2002;50:829–838. [DOI] [PubMed] [Google Scholar]
- 17.Gruber AD, Elble RC, Ji HL, Schreur KD, Fuller CM, Pauli BU. Genomic cloning, molecular characterization, and functional analysis of human CLCA1, the first human member of the family of Ca2+-activated Cl− channel proteins. Genomics 1998;54:200–214. [DOI] [PubMed] [Google Scholar]
- 18.Loewen ME, Gabriel SE, Forsyth GW. The calcium-dependent chloride conductance mediator pCLCA1. Am J Physiol Cell Physiol 2002;283:C412–C421. [DOI] [PubMed] [Google Scholar]
- 19.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol 2004;22:789–815. [DOI] [PubMed] [Google Scholar]
- 20.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science 1998;282:2258–2261. [DOI] [PubMed] [Google Scholar]
- 21.Temann UA, Prasad B, Gallup MW, Basbaum C, Ho SB, Flavell RA, Rankin JA. A novel role for murine IL-4 in vivo: induction of MUC5AC gene expression and mucin hypersecretion. Am J Respir Cell Mol Biol 1997;16:471–478. [DOI] [PubMed] [Google Scholar]
- 22.Dabbagh K, Takeyama K, Lee HM, Ueki IF, Lausier JA, Nadel JA. IL4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J Immunol 1999;162:6233–6237. [PubMed] [Google Scholar]
- 23.Reader JR, Hyde DM, Schelegle ES, Aldrich MC, Stoddard AM, McLane MP, Levitt RC, Tepper JS. Interleukin-9 induces mucous cell metaplasia independent of inflammation. Am J Respir Cell Mol Biol 2003;28:664–672. [DOI] [PubMed] [Google Scholar]
- 24.Longphre M, Li D, Gallup M, Drori E, Ordonez CL, Redman T, Wenzel S, Bice DE, Fahy JV, Basbaum C. Allergen-induced IL9 directly stimulates mucin transcription in respiratory epithelial cells. J Clin Invest 1999;104:1375–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kondo M, Tamaoki J, Takeyama K, Nakata J, Nagai A. Interleukin-13 induces goblet cell differentiation in primary cell culture from Guinea pig tracheal epithelium. Am J Respir Cell Mol Biol 2002;27:536–541. [DOI] [PubMed] [Google Scholar]
- 26.McMillan SJ, Bishop B, Townsend MJ, McKenzie AN, Lloyd CM. The absence of interleukin 9 does not affect the development of allergen-induced pulmonary inflammation nor airway hyperreactivity. J Exp Med 2002;195:51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whittaker L, Niu N, Temann UA, Stoddard A, Flavell RA, Ray A, Homer RJ, Cohn L. Interleukin-13 mediates a fundamental pathway for airway epithelial mucus induced by CD4 T cells and interleukin-9. Am J Respir Cell Mol Biol 2002;27:593–602. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem 2003;278:17036–17043. [DOI] [PubMed] [Google Scholar]
- 29.Rose MC, Piazza FM, Chen YA, Alimam MZ, Bautista MV, Letwin N, Rajput B. Model systems for investigating mucin gene expression in airway diseases. J Aerosol Med 2000;13:245–261. [DOI] [PubMed] [Google Scholar]
- 30.Kim CH, Song KS, Koo JS, Kim HU, Cho JY, Kim HJ, Yoon JH. IL-13 suppresses MUC5AC gene expression and mucin secretion in nasal epithelial cells. Acta Otolaryngol 2002;122:638–643. [DOI] [PubMed] [Google Scholar]
- 31.Wu R. Growth and differentiation of tracheobronchial epithelium. In: McDonald JA, editor. Growth and development of the lung. New York: Marcel Dekker; 1997. pp. 211–241.
- 32.Di YP, Harper R, Zhao Y, Pahlavan N, Finkbeiner W, Wu R. Molecular cloning and characterization of spurt, a human novel gene that is retinoic acid–inducible and encodes a secretory protein specific in upper respiratory tracts. J Biol Chem 2003;278:1165–1173. [DOI] [PubMed] [Google Scholar]
- 33.Shahzeidi S, Aujla PK, Nickola TJ, Chen Y, Alimam MZ, Rose MC. Temporal analysis of goblet cells and mucin gene expression in murine models of allergic asthma. Exp Lung Res 2003;29:549–565. [DOI] [PubMed] [Google Scholar]
- 34.Shim JJ, Dabbagh K, Ueki IF, Dao-Pick T, Burgel PR, Takeyama K, Tam DC, Nadel JA. IL-13 induces mucin production by stimulating epidermal growth factor receptors and by activating neutrophils. Am J Physiol Lung Cell Mol Physiol 2001;280:L134–L140. [DOI] [PubMed] [Google Scholar]
- 35.Vargaftig BB, Singer M. Leukotrienes, IL-13, and chemokines cooperate to induce BHR and mucus in allergic mouse lungs. Am J Physiol Lung Cell Mol Physiol 2003;284:L260–L269. [DOI] [PubMed] [Google Scholar]
- 36.Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, Elias JA, Sheppard D, Erle DJ. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med 2002;8:885–889. [DOI] [PubMed] [Google Scholar]
- 37.Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD. Interleukin-4 and interleukin-13 signaling connections maps. Science 2003;300:1527–1528. [DOI] [PubMed] [Google Scholar]
- 38.Fischer BM, Voynow JA. Neutrophil elastase induces MUC6AC gene expression in airway epithelium via a pathway involving reactive oxygen species. Am J Respir Cell Mol Biol 2002;26:447–452. [DOI] [PubMed] [Google Scholar]
- 39.Chen R, Lim JH, Jono H, Gu XX, Kim YS, Basbaum CB, Murphy TF, Li JD. Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1–dependent p38 MAPK–AP1 and IKKbeta–IkappaBalpha–NF-kappaB signaling pathways. Biochem Biophys Res Commun. 2004;324:1087–1094. [DOI] [PubMed] [Google Scholar]
- 40.Kirschning CJ, Schumann RR. TLR2: cellular sensor for microbial and endogenous molecular patterns. Curr Top Microbiol Immunol 2002;270:121–144. [DOI] [PubMed] [Google Scholar]
- 41.Atherton HC, Jones G, Danahay H. IL-13–induced changes in the goblet cell density of human bronchial epithelial cell cultures: MAP kinase and phosphatidylinositol 3-kinase regulation. Am J Physiol Lung Cell Mol Physiol 2003;285:L730–L739. [DOI] [PubMed] [Google Scholar]
- 42.Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol 2001;13:211–217. [DOI] [PubMed] [Google Scholar]