

Abstract
Hexavalent chromium (Cr[VI]) is classified by the International Agency for Research on Cancer as a group I carcinogen. Although the U.S. Occupational Safety and Health Administration was obliged to reduce the permissible exposure limit (PEL), it was reported that U.S. workers continue to be exposed to dangerously high Cr(VI) levels. In this study, we examined the role of p53 and target genes in a bronchoalveolar carcinoma isogenic cell line system and in primary human bronchial epithelial cells. p53-Negative parental H358 cell line, the same line in which the wild-type p53 expression vector (pC53-SN3) was introduced, and cells obtained from biopsies of human bronchus were exposed to chromate. Induction of DNA strand breaks were evaluated by alkaline elution assay, and apoptosis was analyzed by gel ladder, annexin V-PI staining, and ELISA, whereas p53 and target genes were evaluated by Western blots. Although Cr(VI) induced DNA strand breaks in both H358 cell clones, apoptosis was present only in the p53-transfected cells (H358p53+/+). In these cells, Cr(VI)-induced apoptosis is mediated by p53 upregulation of p53-upregulated modulator of apoptosis (PUMA), BAX translocation to mitochondria, cytochrome c release, and caspase-3 activation. In primary human bronchial epithelial cells expressing functional p53, Cr(VI) induced expression of PUMA and Noxa, which promote apoptosis through BAX. This result establishes p53 as the “necessary” player in Cr(VI)-induced apoptosis. To the best of our knowledge, this is the first report indicating strict correlation of Cr(VI) apoptosis to PUMA induction on primary human bronchoalveolar cells in short-term cultures.
Keywords: apoptosis, bronchial epithelial cells, hexavalent chromium, p53, PUMA
Hexavalent chromium (Cr[VI]) is classified by the International Agency for Research on Cancer (IARC) as a group I carcinogen (1). Cr(VI)-containing compounds are widespread in cigarette smoke, automobile emissions, and in the environment (e.g., Cr[VI]-contaminated water). These compounds are commonly used in the chemical industry, artistic paints, anticorrosion paints, electroplating, and stainless steel welding (1–4). Although the U.S. OSHA reduced the permissible exposure limit (PEL), a recent study (5) reported that U.S. workers continue to be exposed to dangerously high Cr(VI) levels.
The lower respiratory tract is the target organ of Cr(VI) exposure, and its accumulation in lung tissue is found in workers with occupational exposure and in cigarette smokers (1–4). Epidemiologic studies have consistently shown that occupational exposure is strongly associated with a higher incidence of lung cancer (1, 6, 7). Exposure increases the incidence of lung cancer in cigarette smokers (1, 6, 8), thus the cancer morbidity rate for smokers who were formerly K2CrO4 workers, with 9 or more years of exposure, is 21.6 times higher than that of nonsmokers (1, 6).
Intracellular reduction of Cr(VI) to Cr(III) leads to the extensive formation of DNA-phosphate–based adducts that cause a variety of genetic damage (1, 9–16), which in turn determines the activation of the p53 signaling pathway and cell cycle arrest or apoptosis (9–16). Moreover, Cr(VI) induces the ataxia–telangiectasia mutated (ATM)-dependent DNA damage response pathway, which is paradoxically required for both apoptosis and survival after Cr(VI) insult (16, 17). Nucleotide excision repair (NER) is the principal repair mechanism for Cr(VI)-DNA adducts in human cells (18). However, Cr(VI) exposure causes the inhibition of NER, which enhances carcinogen-induced mutagenicity and cytotoxicity (11, 16). Although the inhibition of NER is an important mechanism for Cr(VI)-induced human carcinogenesis, the underlying molecular mechanisms of Cr(VI)-induced carcinogenesis remain unclear.
In this study, we examined the role of p53 in a bronchoalveolar carcinoma isogenic cell line system consisting in the p53-negative parental H358 cell line and parental line in which the wild-type p53 expression vector (pC53-SN3) was introduced. Because the only difference between these two cell lines is the absence or presence of a single gene, the interpretation of results is particularly straightforward and uncomplicated by the overexpression of exogenous genetic elements. Although Cr(VI) induced DNA strand breaks in both cell clones, the induction of apoptosis, mediated by p53 upregulated modulator of apoptosis (PUMA), Bax translocation to mitochondria, cytochrome c release, and caspase-3 activation, is present only in the p53+/+ clones. This result establishes p53 as the “necessary” player in Cr(VI)-induced apoptosis. To characterize the signaling pathways activated by Cr(VI) in primary human bronchial epithelial cells, we examined its induced effects in short-term primary cultures obtained from biopsies of human bronchus.
MATERIALS AND METHODS
Cell Lines and Primary Cell Cultures
Human bronchoalveolar carcinoma H358 cell line, obtained from the American Type Culture Collection (Rockville, MD), were grown in RPMI 1640 (Gibco BRL, Grand Island, NY) supplemented with 10% FBS (Gibco BRL).
To obtain primary cultures of human bronchus epithelium, samples of bronchial tissue were excised from surgical specimens of resected non–small cell lung cancer from patients presenting normal lung function. The procedure was approved by the ethics committee. Biopsies were assessed by conventional H&E staining. After excision, the bronchial samples were washed and incubated overnight at 4°C with 0.38 mg/ml hyaluronidase, 0.75 mg/ml collagenase, 1 mg/ml protease, and 0.3 mg/ml DNase in RPMI 1640 medium, and then filtered through a 70-mm mesh nylon strainer. After centrifugation, epithelial cells were resuspended in small airway epithelium basal medium (Clonetics; BioWhittaker, San Diego, CA) supplemented with 0.5 μg/ml human recombinant epidermal growth factors, 7.5 mg/ml bovine pituitary extract, 0.5 mg/ml epinephrine, 10 mg/ml transferrin, 5 mg/ml insulin, 0.1 μg/ml retinoic acid, 6.5 μg/ml triiodothyronine, 50 mg/ml gentamicin, 50 μg/ml amphotericin B, and 50 mg/ml BSA/fatty acid–free. Cells were cultured on plates precoated with coating media containing: 29 μg/ml collagen (vitrogen; Collagen Corp., Palo Alto, CA), 10 μg/ml BSA (Biofluids, Inc., Rockville, MD), and 10 μg/ml fibronectin (Calbiochem, La Jolla, CA) for 5 min. Fresh complete medium was replaced every 2–3 d until cells were confluent. Upon confluence, the cells were lifted by 1× trypsin-EDTA (Life Technologies, Inc., Gaithersburg, MD) and subcultured at a 1:2 dilution. Third-fourth passage confluent cultures were used for all the experiments. For in vitro testing of nontumorigenicity of each primary cell line, the anchorage-independent assay was performed using a soft-agar clonogenic method, as described previously (19). The cells were identified as bronchoalveolar by immunocytochemical staining.
Cell Proliferation Assay
Cells were treated as reported by Sugiyama and colleagues (20). Colonies were scored after 10 d.
Immunostaining for Flow Cytometry
p53 and p21waf-1 induction were analyzed in FacsCalibur (BD Biosciences, San Jose, CA).
DNA Damage
DNA damage was evaluated by the alkaline filter elution assay under deproteinizing conditions, as described previously (21).
Apoptosis
Apoptosis was detected using different methods: (1) internucleosomal DNA fragmentation; (2) annexin V-PI staining; and (3) cellular DNA fragmentation ELISA assay, as described previously (21, 22).
Western Blotting
Western blotting was performed as described previously (21, 22). Antibodies used were: monoclonal human p53 (DO-1), tubulin, caspase-3, human cleaved caspase-3 (Asp 175), and BCL-XL (Cell Signaling Technology, Beverly, MA); human p21 (polyclonal), PUMA, NOXA, and BAX (Oncogene; Dako Cytomation, Carpinteria, CA); human cytochrome oxidase IV (clone 1A12; Molecular Probes, Inc., Eugene, OR, and Cell Signaling Technology).
Cellular Fractionation
Cytosolic fractions (supernatant) and mitochondrial fractions (pellet) were obtained as described previously (23).
Determination of Caspase Activity
A total of 5 × 105 cells were incubated in the presence or absence of 200 μM chromate (K2CrO4) together with the PE-labeled affinity-purified anti-caspase-3 Abs (Pharmingen, San Diego, CA), that binds irreversibly to activated caspases-3, and then analyzed by flow cytometry using FacsCalibur. Alternatively, the enzymatic activity of caspase-8 and caspase-3 was assayed fluorometrically.
Immunofluorescence Analysis
Immunofluorescence analysis was performed as described previously (19).
RESULTS
Cell Growth Inhibition, Induction of DNA Single-Strand Breaks, and P53 Activation in a Bronchoalveolar Carcinoma Isogenic H358 Cell Line System
The bronchoalveolar carcinoma isogenic cell line system consisted of the p53-negative H358 parental cell line and the parental cell line in which the wild-type p53 expression vector (pC53-SN3) was introduced, as described previously (24) (parental cell line courtesy of the Medicine Branch (National Cancer Institute, National Institutes of Health, Bethesda, MD).
A different dose–response relationship was seen between parental H358p53−/− cells and transfected H358p53+/+ clones (Figure 1A). Cells transfected with wild-type p53 were sensitive to K2CrO4 treatment with an inhibitory concentration producing a 50% suppression (IC50) value of 200 ± 3.4 μM. On the contrary, in the parental cell line, H358, the IC50 was not reached; treatment with 500 μM K2CrO4 induced a survival fraction of 64%, suggesting that K2CrO4-induced cytotoxicity is p53-dependent (Figure 1A).
Figure 1.

Effects of K2CrO4 on a bronchoalveolar carcinoma isogenic H358 cell line system. (A) Clonogenic assay. Cells were plated at 1,000 cells/well in 60-mm dishes in complete medium for 24 h, then rinsed twice with salts-glucose medium (SGM: 50 mM Hepes [pH 7.2], 10 mM NaCl, 5 mM KCl, 2 mM Cacl2, 5 mM glucose), and treated for 2 h with different concentrations of K2CrO4 diluted in SGM medium. After drug removal (three washes in SGM), cells were cultured in complete medium. Colonies were scored after 10 days. Data (median ± SE) are representative of three replicate experiments yielding similar results. (B) DNA-SSB induction. H358 p53−/− and H358p3+/+ cells (2.5 × 106 cells/150 cm2) were labeled for 24 h with [3H]thymidine (0.2 μCi/ml) (New England Nuclear, Boston, MA) and, after 4 h incubation in a label-free medium, treated with 200 μM of K2CrO4 in SGM. Samples were withdrawn immediately after damage induction and analyzed by the alkaline filter elution assay under deproteinizing conditions (proteinase K 0.5 mg/ml). Data (median ± SE) are representative of three replicate experiments yielding similar results. (C) Effects of p53 or p21waf1 protein levels. Cells were treated with 200 μM of K2CrO4 in SGM for 2 h, washed, incubated in drug-free medium for a range of times, and then collected. Equal amounts of total protein were resolved on 12% SDS-PAGE gels, transferred onto nitrocellulose membranes (ECL; Amersham Biosciences, Piscataway, NJ), and immunoblotted. Blots were developed with ECL chemiluminescence (Amersham) in this and in all Western blotting experiments. Lanes: 1: control; 2: 1 h; 3: 2 h; 4: 4 h of recovery. Data are representative of three replicate experiments yielding similar results.
When both cell lines were incubated for 2 h at the equimolar concentration of 200 μM K2CrO4, a large amount of protein-associated DNA single-strand breaks (SSB) (Figure 1B) was generated, both in the parental cell line (p53−/−) and in the transfected clones, suggesting that p53 is not involved in DNA-SSB formation induced by Cr(VI). In these cells, p53 protein levels started to increase after 1 h of recovery, and were maximum after 4 h of recovery. It was observed only in H358p53+/+ clones (Figure 1C). Interestingly no p21waf-1, the expression of which is largely regulated at the transcriptional level by p53-dependent mechanism (25), was induced (Figure 1C).
Induction of Apoptosis
All the following experiments were performed using the concentrations of K2CrO4 needed to reach the IC50 value in H358p53+/+ clones and considered equimolar for both the parental and the transfected cells. Cells were treated with 200 μM K2CrO4 for 2 h, then the K2CrO4 was washed out and cells were incubated until 24 h of recovery in K2CrO4-free medium.
DNA ladder formation, characteristic of apoptosis, was detectable after 4 h of recovery and increased significantly until 24 h of recovery in H358p53+/++/+ clones; low levels of DNA ladder was detectable after 24 h of recovery in the parental cell line H358p53−/− (Figure 2A). Annexin–V-PI assay, performed after 24 h of recovery (Figure 2B), confirmed these observations.
Figure 2.

Induction of apoptosis by K2CrO4 on a bronchoalveolar carcinoma isogenic H358 cell line system. (A) Internucleosomal DNA fragmentation; cells were treated with 200 μM of K2CrO4 in SGM for 2 h, washed, and then incubated in drug-free medium for additional time periods. Finally, cells, including those floating and adherent, were collected. Lanes: M: marker; C: control. Data are representative of three replicate experiments yielding similar results. (B) Evidence of apoptosis was identified by staining with annexin V (Roche Diagnostic, Indianapolis, IN) (x axis) and propidium iodide (PI) (y axis). Cells were treated as in (A) and analyzed after 24 h of recovery. Cells were washed in PBS and resuspended in 100 μl binding buffer containing annexin V. Cells were analyzed by flow cytometry after the addition of PI. Cells binding annexin V and retaining PI were apoptotic (lower right quadrant); double-positive cells underwent secondary necrosis (upper right quadrant). Data are representative of three replicate experiments yielding similar results.
Because these data suggested that Cr(VI) triggered apoptosis in H358p53+/++/+ clones via p53 signal, p53 target gene products (25), such as PUMA, NOXA, BAX, and BCL-XL, were analyzed. According to p53 induction, PUMA and NOXA proteins were induced in a time-dependent manner (Figure 3A), but only in H358p53+/++/+ clones.
Figure 3.

Induction of p53 related genes (PUMA, NOXA, BAX, Smac and BCL-XL). (A) Induction of PUMA and NOXA. Cells were treated with 200 μM of K2CrO4 in SGM for 2 h, washed, and then incubated in drug-free medium for additional time periods. Data are representative of three replicate experiments yielding similar results. (B) Translocation of Bad and Smac. (Top) Cells were treated with 200 μM K2CrO4 for 2 h and proteins were extracted after 12 h recovery. Western blotting was performed both in the cytosolic (supernatant) and in the mitochondrial fractions (pellet). Cytochrome oxidase subunit IV (Cox IV) was the positive control for mitochondrial fraction. Tubulin, which is exclusively expressed in the cytosol, was used as a control for loading and for fractionation. Data are representative of three replicate experiments yielding similar results. (Bottom) cells were treated as in (B), and analyzed by immunofluorescence. For immunofluorescence analysis, cells were fixed with 2% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS, incubated with 25 nM Mitotracker Red CMXRos (Molecular Probes, Inc., Eugene, OR) for 30 min at room temperature, and then washed three times with PBS. Cells were then incubated with rabbit antihuman BAX diluted 1:200 in 5% FBS/PBS for 1 h at room temperature in a humidified chamber. Excess antibody was removed by washing the coverslips three times with PBS. Cells were then incubated with biotin-labeled goat antirabbit IgG (Zymed Laboratories Inc., San Francisco, CA) diluted 1:200 in 5% FBS/PBS, and for 1 h protected from light at room temperature. After washing three times with PBS, cells were incubated with FITC-streptavidin diluted 1:200 in 5% FBS/PBS for 4 h. After washing three times with PBS, coverslips were mounted onto microscope slides using ProLong antifade mounting reagent (Molecular Probes). The slides were analyzed by a confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany). Mitochondrial localization of BAX was defined by yellow spots indicating overlap of FITC and Mitotracker. Data are representative of three replicate experiments yielding similar results. (C) Induction of BCL-XL on a bronchoalveolar carcinoma isogenic H358 cell line system. Cells were treated with 200 μM of K2CrO4 in SGM for 2 h, washed, and then incubated in drug-free medium for additional time periods. Data are representative of three replicate experiments yielding similar results.◂
In these clones, a significant fraction of BAX translocated from the cytosol to the mitochondria (Figure 3B), and BCL-XL was downregulated (Figure 3C). As a result, some second mitochondria-derived activator of caspases, such as Smac/Diablo (Figure 3B), cytochrome c (Figure 4A), AIF, and endonuclease G, were released from mitochondria to the cytoplasm (Figure 4B). As a final consequence, caspase-3 was activated (Figures 5A–5D). When H358p53+/+ clones were incubated with 200 μM K2CrO4 in the presence of the specific inhibitors of caspase-3 z-DEVD.fmk 30 nM (26), no induction of apoptosis was observed (annexin V/PI assay) (Figure 5E) and, as expected, caspase-3 activation was inhibited (Figure 5D).
Figure 4.

Induction of p53 related genes (cytochrome c, AIF, and endonuclease G translocation). (A) Translocation of cytochrome c. Cells were treated with 200 μM of K2CrO4 in SGM for 2 h, washed, and then incubated in drug-free medium for 24 h. Western blotting was performed both in the cytosolic (supernatant) and in the mitochondrial fractions (pellet). Cytochrome oxidase subunit IV (Cox IV) was the positive control for mitochondrial fraction. Tubulin, which is exclusively expressed in the cytosol, was used as a control for loading and for fractionation. Data are representative of three replicate experiments yielding similar results. (B) Activity of AIF and endonuclease G. Cells were treated with 200 μM of K2CrO4 in SGM for 2 h, washed, and then incubated in drug-free medium for additional time periods. Western blotting was performed both in the cytosolic (supernatant) and in the mitochondrial fractions (pellet). Data are representative of three replicate experiments yielding similar results.
Figure 5.

Caspase-3 activation. (A) Cells were treated with 200 μM of K2CrO4 in SGM for 2 h, washed, and then incubated in drug-free medium for additional time periods. The enzymatic activity of caspase-3 was determined by incubation of 10 μg of total protein with 200 μM of its specific fluorogenic peptide substrate Ac-DEVD-MCA, in a 50-μl assay buffer at 37°C for 2 h and assayed fluorometrically. The release of AMC was measured with a spectrofluorometer (excitation/emission wavelengths: 360/460 nm). Data are expressed as the percent of controls that were nontreated. Data are representative of three replicate experiments yielding similar results. (B) Induction of active caspase-3 on H358 p53+/+ cells. Cells were treated as in (A). Active caspase-3 was evaluated by Western blot. Data are representative of three replicate experiments yielding similar results. (C) Cleavage of caspase-3 on a bronchoalveolar carcinoma isogenic H358 cell line system. Cells were treated as in (A); protein extractions were performed after 4 of recovery. Data are representative of three replicate experiments yielding similar results. (D) Caspase 3 activation on a bronchoalveolar carcinoma isogenic H358 cell line system. Experiments were performed in the same pool of cells examined in (C). Enzymatic activity of caspase-3–like proteases was determined by incubation of 10 μg of total protein with its specific fluorogenic substrate (Ac-DEVD-MCA) for 2 h at 37°C; cells were then analyzed by flow cytometry (FacsCalibur), and the increase of caspase activity was determined after proper gating in comparison with untreated cells. Illustrated plots are representative of three independent experiments, which yielded very comparable results. The caspase-3 inhibitor (zDEVD) impaired the activity of K2CrO4. (E) Inhibition of apoptosis by caspase-3 inhibitor. Experiments were performed as in Figure 2B (cells were treated with 200 μM of K2CrO4 in SGM for 2 h, washed, and then incubated in drug-free medium for 24 h), with the exception that H358p53+/+ cells were pretreated with 25 μM Z-DEVD-fmk (a caspase-3 inhibitor) for 1 h, followed by 23 hours of 200 μM of K2CrO4 in SGM. Data are representative of three replicate experiments yielding similar results.
In conclusion, K2CrO4 induced cell cytotoxicity and mitochondria-dependent caspase-3–mediated apoptosis via p53 induction.
Experiments on Primary Human Bronchoalveolar Cells in Short-Term Cultures
Evaluation of cellular characteristics.
Hexavalent chromium exposure experiments were performed in human bronchoalveolar cells freshly isolated from surgical specimens. Short-term culture cells were obtained from six human bronchial biopsies identified as non-neoplastic bronchoalveolar cells after H&E staining (Figure 6A). The failure of these cells to proliferate in soft agar (anchorage-independent assay) supported the nontumorigenicity of each primary cell line (Figure 6A). These primary cells were considered bronchial epithelial cells after immunohistochemistry analysis. These cells strongly stained with the mouse antihuman cytokeratin monoclonal antibodies 34βE12 and RCK102, while no significant immunostaining was found with mouse anti-human vimentin monoclonal antibodies, anti-CD3, or anti-CD68, thus excluding contamination of cultures by fibroblasts, lymphocytes, or macrophages (Figure 6B). These cells displayed normal p53 function, as p53 protein levels were increased after treatment with campthotecin, a positive p53-inducing agent (27), at 1.0 μM for 2 h (Figure 6C).
Figure 6.

Characterization of human bronchus epithelium in primary cultures. (A) (Left) Representative histopathology of human bronchial biopsy stained with H&E. All biopsies were fixed in 10% buffered formalin and embedded in paraffin wax. Paraffin blocks were sectioned according to a standard protocol at 5 μm, and serial sections were mounted in consecutive order on silane-coated glass slides (Superfrost-plus; Fisher Scientific, Pittsburgh, PA). A total of 12 slides containing 8 sections/slide were prepared from each paraffin block. Slides 4 and 9 were stained with H&E, and epithelial abnormalities were classified according to recent World Health Orgaization criteria as normal, reserve cell hyperplasia, squamous metaplasia, or dysplasia (low or high grade). The presence or absence of ASD, as defined subsequently, was noted in each biopsy. Two examiners evaluated the specimens, and joint review and consultation resolved diagnostic disagreements. (Right) Representative cloning in soft agar of one bronchoalveolar cell primary culture. H358p53+/+ cells were the positive control. (B) Representative immunocytochemical staining of primary cultures. Mouse antihuman cytokeratin monoclonal antibodies (mAbs) RCK102 (1:25; Organon Teknika, Boxtel, Holland), NCL5D3 (1:100; Organon Teknika), and 34βE12 (1:100; Dako Corp., Carpinteria, CA) were used to confirm epithelial phenotype. Immunostaining for vimentin using mouse antihuman vimentin mAbs (1:100; Organon Teknika) was performed to exclude contamination by fibroblasts; antihuman cluster of differentiation (CD) 3 mAbs (1:200; Dako, Glostrup, Denmark) to exclude lymphocyte contamination; and antihuman CD68 mAbs (1:100; Dako) to exclude macrophage contamination in cultures. (C) Effect on p53 protein levels. Cells in primary cultures were treated for 2 h with CPT 10 μM, then proteins were extracted, and p53 protein level increase was evaluated by Western blot. Lanes 1–6: control (untreated cells); 1a–6a: cells treated with CPT. Data are representative of three replicate experiments yielding similar results.
Cell growth inhibition and apoptosis induction.
A dose–response relationship was seen in primary cells treated with different concentrations of K2CrO4 (Figure 7). Primary human bronchoalveolar cells were more sensitive to K2CrO4 than was the bronchoalveolar carcinoma cell line (compare Figure 1A with Figure 7A). The IC50 values covered a range from 18 to 82 μM (Figure 7B), suggesting that primary cells are very sensitive to K2CrO4.
Figure 7.

Effects of K2CrO4 in primary cultures of human bronchus epithelium. (A) Clonogenic assay. Cells were plated at 1,000 cells/well in 60-mm dishes in complete medium for 24 h, then rinsed twice with SGM (50 mM Hepes [pH 7.2], 10 mM NaCl, 5 mM KCl, 2 mM Cacl2, 5 mM glucose), and treated for 2 h with different concentrations of K2CrO4 diluted in SGM medium. After drug removal (three washes in SGM), cells were cultured in complete medium. Colonies were scored after 10 d. Data are representative of three replicate experiments yielding similar results. (B) Values of 50% inhibitory concentration (IC50) (μM). The IC50 was calculated as the drug concentration that inhibits cell growth to 50% of the control cells. IC50 values were estimated by fitting the data with a nonlinear regression to the dose–effect model derived by Chou and Talalay (45, 46): fa/fu = (D/Dm)m, where D is the dose of the drug, Dm is the IC50, fa is the fraction affected by the dose, fu is the fraction unaffected, and m is a coefficient that determines the sigmoidicity of the curve.
When cells were incubated for 2 h with K2CrO4 at their respective IC50 values, followed by 2 h of recovery, p53, PUMA, and NOXA (Figures 8A, 8B, and 8D), but not p21waf-1, proteins were induced (Figure 8C). In the same pool of cells, caspase-3 was activated (Figure 9A). Apoptosis started 3 h after recovery, increased with time, and rose to the maximum after 9 h of recovery (Figures 9B–9C), as evaluated by ELISA and ladder assays.
Figure 8.

Effects on p53 protein levels and of target genes (p21waf-1, PUMA and NOXA) in primary cultures of human bronchus epithelium. (A and C) Cells were treated with K2CrO4 at their IC50 values for 2 h and analyzed after 2 h recovery, collected, washed with PBS, and finally resuspended in 500 μl of 4% paraformaldehyde in PBS for fixing. After 15 min at room temperature, 5 ml of 1% BSA in PBS-Tween (BPT) was added, followed by centrifugation at 300 × g for 5 min. Cells were resuspended in 0.2% Triton X-100 in PBS and incubated for 10 min on ice. After addition of 5 ml of BPT and centrifugation, the cells were incubated in primary antibody solution in 5% nonfat dry milk in PBS-Tween (0.05%). PAb 122, for p53 protein detection, and polyclonal anti-p21waf1 antibody (Abcam) were used. All antibody incubations were done in room temperature for 1 h. The cells were washed once with 5 ml BPT and incubated with the appropriate secondary antibody in BPT. After washing, the cells were analyzed in FacsCalibur (BD Biosystems). Data are representative of three replicate experiments yielding similar results. (B–D) In the same pool of cells as that of (A), p53 (B), PUMA and NOXA (D) protein increases were evaluated by Western blotting. Data are representative of three replicate experiments yielding similar results.
Figure 9.
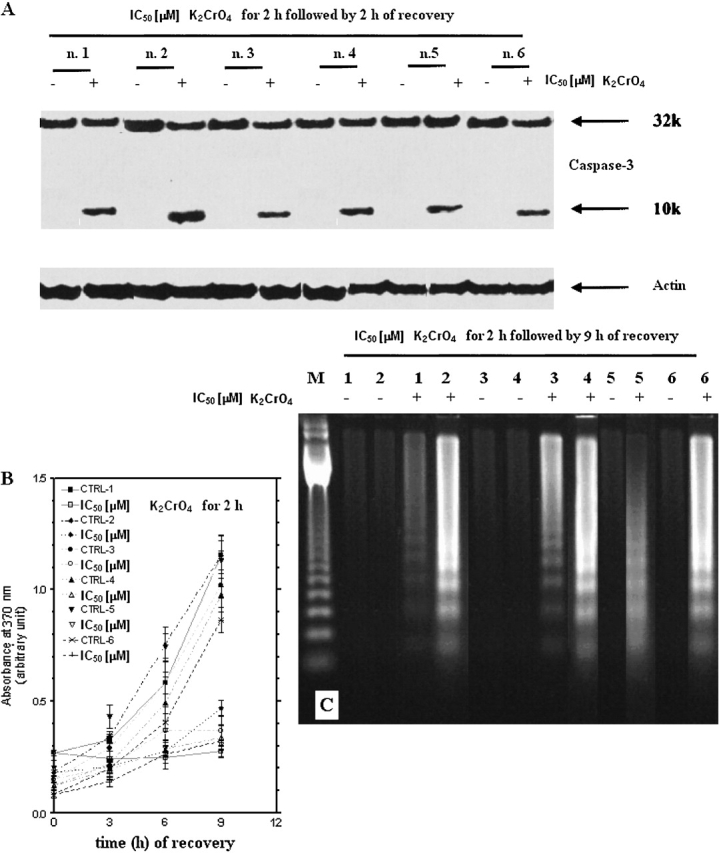
Induction of caspase-3 and apoptosis in primary human bronchoalveolar cells. (A) Induction of caspase-3 activation and apoptosis in primary cultures of human bronchus epithelium. Cells were treated with μM K2CrO4 at their IC50 values for 2 h and analyzed after 2 h recovery. Data are representative of three replicate experiments yielding similar results. (B) Kinetics of drug-induced apoptotic cell death evaluated by ELISA (Roche Diagnostics) according to manufacturer's instructions (catalog no. 1585 045). A total of 104 cells/well were incubated for 2 h with K2CrO4, their IC50 values, and then cells were incubated in drug-free medium for additional time periods. After the times indicated, 100 μl/well of supernatant plus 100 μl/well of lysates were removed and tested by ELISA. Data are expressed as mean ± SE of two independent experiments performed at least in duplicate and yielding similar results. (C) Internucleosomal DNA fragmentation. Cells were treated as in (A), and after 9 h of recovery, cells, including those floating and adherent, were collected. Data are representative of three replicate experiments yielding similar results.
When both H358 p53+/+ and primary human bronchoalveolar cells in short-term cultures were preincubated with PUMA antisense for 2 h, then treated with K2CrO4 for an additional 2 h at their respective IC50 values, followed by 2 h of recovery, no induction of PUMA proteins was observed (Figure 10A). Under these experimental conditions, no induction of apoptosis was observed (Figure 10B). These experiments showed that PUMA antisense inhibited apoptosis induced by Cr(VI).
Figure 10.

PUMA-dependence of apoptosis. (A) induction of PUMA in H358 p53+/+ (left) and in primary cultures of human bronchus epithelium (right). Cells were preincubated with PUMA HPLC-purified antisense phosphorothioate oligonucleotides (antisense Puma, 5′-CATCCTTCTCATACTTTC-3′; and control, CTTTCATACTCTTCCTAC-3′; GeneSet Oligos, Degussa, Germany) (100 nM) for 2 h and then treated with K2CrO4, at their respective IC50, for 2 h followed by 2 h of recovery. Data are representative of three replicate experiments yielding similar results. (B) Cells were treated as in panel A. (B) and, after different times of recovery, gel ladders were performed. Data are representative of three replicate experiments yielding similar results.
DISCUSSION
The analysis of the apoptosis induced by Cr(VI) in the isogenic human bronchoalveolar carcinoma cell system consisting of H358p53−/− parental cells and their derivative H358p53+/+, and in human primary bronchoalveolar cells displaying normal functional p53, revealed that p53 is necessary for Cr(VI)-induced expression of the BH3 domain–containing proteins, PUMA and NOXA. These proteins promote apoptosis through the multidomain Bcl-2 family member, BAX. When PUMA expression was inhibited by specific antisense, no apoptosis was observed.
The tumor suppressor protein, p53, mediates some parts of the response of mammalian cells to DNA damage, either by stimulating DNA repair or, beyond a certain threshold of DNA damage, by initiating apoptosis. The primary role of activated p53 is to act as a transcription factor to induce the expression of proteins involved in cell cycle arrest or apoptosis. The transcriptional activation of the cyclin-dependent kinase inhibitor CDKN1A (p21waf-1/cip1) is the key event for growth arrest after p53 activation (25). Alternatively, p53 transactivates a series of proapoptotic proteins from the BCL2 family; in particular, BAX, PUMA, and NOXA (25, 28, 29), which induce mitochondrial-membrane permeabilization and, therefore, release apoptogenic factors from the mitochondrial intermembrane space (25, 28, 29). p53 Can also induce apoptosis in a transcription-independent manner by acting directly at the mitochondrial level (25), although the extent of this for DNA damage–induced apoptosis is controversial. Whatever the mechanisms, p53 activates the intrinsic mitochondrial apoptotic pathways in response to DNA damage by inducing the expression of at least three BCL-2 proapoptotic family members (BAX, PUMA, and Noxa), shifting the balance toward proapoptotic effects. The formation of apoptosomes requires the release of cytochrome c and APAF-1 from mitochondria. The cytoplasm efflux of cytochrome c is the key event, with consequent mitochondrial release of Smac/DIABLO (second mitochondria-derived activator of caspases), which promotes the transactivation of caspase-9 and downstream effector caspase-3. Caspase-3 executes cell death (25).
In our experiments, Cr(VI) increased the expression of p53 protein levels, but did not induce p21waf-1, the principal mediator of G1 growth arrest, whereas p53 increases strongly promote apoptosis. It has been proposed (30) that the two outcomes induced by p53 (growth arrest or apoptosis) have a profound influence on the tumorigenic effects of genotoxic damage. As an example, O'Reilly and colleagues (31) suggested that p21waf-1 protects the lung from oxidative stress, in part by inhibiting DNA replication and thereby allowing additional time to repair damaged DNA.
The proapoptotic and cell cycle–arresting functions of p53 have been attributed to distinct transcriptional profiles (e.g., increased transcription of BAX, as in the case of Cr[VI]-treated cells [32], for apoptosis induction versus transcription of p21waf-1 for cell-cycle arrest). These profiles correlate, to some extent, with the phosphorylation of p53 on serine (Ser) 15, 20, or 46 (30). Experiments in transgenic mice have indicated that lack of phosphorylation of residues 15 or 20 does not affect growth arrest, but reduces the apoptotic response (30). Cr(VI) induced phosphorylation of p53 at Ser15 (33). A further potential mechanism for determining apoptosis versus p21waf-1 induction is the coexpression of proteins that act to repress p21waf-1 expression; one example is c-jun (34), which is enhanced after Cr(VI) exposure (32).
In conclusion, the response to genotoxic stress mediated by p53 is a highly complex process, as it must be to maintain viability. Tumors are very complex, and their initiation and progression involve disruption of interacting mechanisms that normally act to maintain tissue homeostasis after genotoxic damage. Although apoptosis eliminates the majority of damaged cells, a stochastic fraction of cells may survive (Figure 11) with unrepaired DNA damage. These cells may exhibit a predisposition to genomic instability and neoplasia. Hirose and colleagues (35) reported that a longer period of K2CrO4 exposure was associated with a tendency toward a higher frequency of microsatellite instability (MSI). In their study, 30 (78.9%) out of 38 tumors present in the K2CrO4 lung cancer group exhibited MSI. In contrast, only 4 (15.4%) out of 26 tumors present in the non-K2CrO4 lung cancer group exhibited MSI. The authors suggested that MSI may play a role in chromium-induced carcinogenesis, and that the carcinogenic mechanism of K2CrO4 lung cancer may differ from that of non-K2CrO4 lung cancer.
Figure 11.
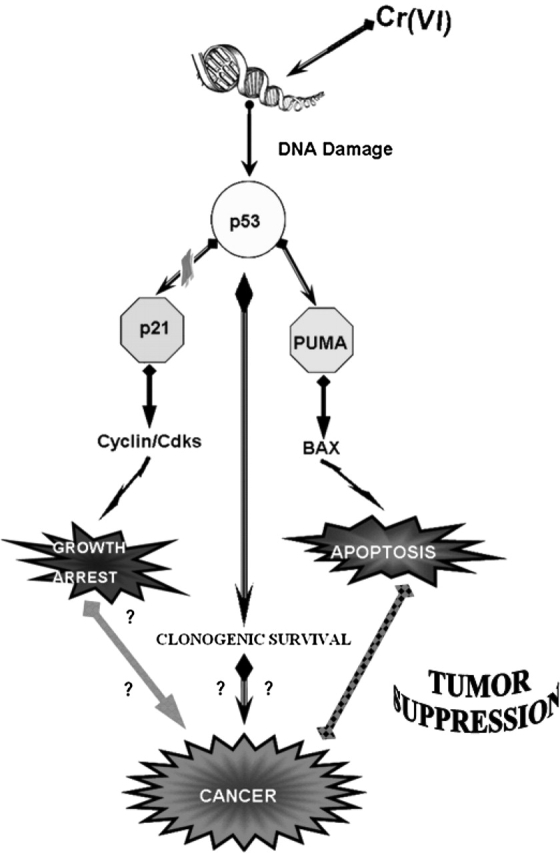
Mechanism of Cr(VI)-induced apoptosis and/or carcinogenicity in functionally p53 human bronchoalveolar cells.
Apoptosis is a survival mechanism aimed at maintaining good cellular events; however, adaptive responses to genotoxic stress may paradoxically generate mechanisms that can drive to carcinogenesis. In culture, a progressive change was observed in the phenotype of cells exposed repetitive times to Cr(VI); one such observation was of an increased resistance to apoptosis (36). The altered apoptotic susceptibility in these cells may be relevant to the response of Cr(VI) exposure in individuals with long-term repetitive exposures. Lung tissue that has undergone cellular turnover in response to recurrent cytotoxic Cr(VI) exposure may present an attenuated response to subsequent exposures, This implies that individuals with longer-term occupational exposure may be more susceptible to Cr(VI)-induced lung cancer due to selection for apoptosis-resistant cells.
Finally, considerable literature supports a role of apoptosis in the remodeling of lung tissue after acute lung injury (37–40). For example, Cr(VI)-related lung cancers are often associated with respiratory toxicity involving high levels of cell death, such as perforation of the nasal septum and/or respiratory tract ulcerations (1). In response to injury to the bronchial epithelium, there is an immediate requirement to initiate tissue repair and restore barrier function (41). When the degree of lung injury is mild, damaged tissue will normally be repaired, whereas excess cell death may lead to irreparable lung damage and pulmonary fibrosis. Pulmonary fibrosis is associated with an increased risk of lung cancer (40).
We have explored the possibility that pulmonary toxicity from Cr(VI) is due in part to apoptosis caused by DNA damage. Several studies have shown that intracellular reduction of Cr(VI) to Cr(III) leads to extensive formation of DNA phosphate–based adducts that cause different genetic damage, such as chromosomal aberration, sister-chromatid exchange, DNA strand breaks, and DNA-DNA and DNA-protein cross-links (9, 14). Using isogenic human bronchoalveolar carcinoma cells (H358 cell system) that differ only in their p53 status, we have shown that the induction of DNA strand breaks is independent of the status of p53, but, importantly, the resolution of these breaks is p53-dependent. In p53 wild-type cells (transfected cell clone and primary human bronchoalveolar cells), PUMA induction is essential for Cr(VI)-induced apoptosis, as demonstrated by experiments in the presence of PUMA antisense (Figure 11). To the best of our knowledge, this is the first report indicating that Cr(VI)-apoptosis strictly correlates to PUMA induction on primary human bronchoalveolar cells. Moreover, Cr(VI) determined the release of Smac/DIABLO, as well as apoptosis-inducing factors, such as AIF and endoprotein G.
PUMA was initially identified as a gene activated by p53 in cells undergoing p53-induced apoptosis (28, 42, 43), and as a protein interacting with Bcl-2 (29). PUMA and another p53 target, Noxa, share homology with Bcl-2 family proteins. PUMA is extremely effective in inducing apoptosis: when expressed, it kills cancer cells within a few hours (28, 44). Importantly, gene knockouts in human colorectal cancer cells showed that PUMA was required for apoptosis induced by p53, hypoxia, and DNA-damaging agents (43). Finally, PUMA acts by modulating Bax activity to facilitate cytochrome c release from the mitochondria, thereby triggering the apoptotic cascade (44).
Overall, our results suggest that the mitochondrial death pathway mediating apoptosis is activated by Cr(VI), and that Cr(VI) is directly genotoxic to relevant lung target cells, such as bronchoalveolar cells. Cr(VI)-induced DNA damage in p53 wild- type cells triggers complex cellular signaling pathways that determine cell fate. These responses include increases in p53 protein levels and transcriptional activation of p53 targets genes, such as PUMA and NOXA, which, in turn, trigger mitochondrial apoptosis.
Originally Published in Press as DOI: 10.1165/rcmb.2005-0213OC on September 15, 2005
Conflict of Interest Statement: None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.IARC. Chromium, nickel and welding. IARC Monogr Eval Carcinog Risks Hum 1990;49:1–648. [PMC free article] [PubMed] [Google Scholar]
- 2.Hayes RB. Review of occupational epidemiology of chromium chemicals and respiratory cancer. Sci Total Environ 1998;71:331–339. [DOI] [PubMed] [Google Scholar]
- 3.Gibb HJ, Lees PS, Pinsky PF, Rooney BC. Lung cancer among workers in chromium chemical production. AM J Ind Med 2000;38:115–126. [DOI] [PubMed] [Google Scholar]
- 4.Luippold RS, Mundt KA, Austin RP, Liebig E, Panko J, Crump C, Crump K, Proctor D. Lung cancer mortality among chromate production workers. Occup Environ Med 2003;60:451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lurie P, Wolfe SM. Continuing exposure to hexavalent chromium, a known lung carcinogen: an analysis of OSHA compliance inspections, 1990–2000. Am J Ind Med 2002;42:378–383. [DOI] [PubMed] [Google Scholar]
- 6.Langard S. One hundred years of chromium and cancer: a review of epidemiological evidence and selected case reports. Am J Ind Med 1990;17:189–215. [DOI] [PubMed] [Google Scholar]
- 7.Nurminen M. On the carcinogenicity risk assessment of chromium compounds. Am J Ind Med 2004;45:308–309. [DOI] [PubMed] [Google Scholar]
- 8.Hu W, Feng Z, Tang MS. Chromium(VI) enhances (+/−) anti-7beta, 8alpha-dihydroxy-9alpha,10alpha-epoxy-7,8,9,10-tetrahydrobenzo[a] pyrene–induced cytotoxicity and mutagenicity in mammalian cells through its inhibitory effect on nucleotide excision repair. Biochemistry 2004;43:14282–14289. [DOI] [PubMed] [Google Scholar]
- 9.De Flora S, Bagnasco M, Serra D, Zanacchi P. Genotoxicity of chromium compounds. Mutat Res 1990;278:99–172. [DOI] [PubMed] [Google Scholar]
- 10.Costa M. Toxicity and carcinogenicity of Cr(VI) in animal models and humans. Crit Rev Toxicol 1997;27:431–442. [DOI] [PubMed] [Google Scholar]
- 11.O'Brien TJ, Ceryak S, Patierno SR. Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat Res 2003;533:3–36. [DOI] [PubMed] [Google Scholar]
- 12.Dayan AD, Paine AJ. Mechanisms of chromium toxicity, carcinogenicity and allerginicity: review of the literature from 1985 to 2000. Hum Exp Toxicol 2001;20:439–451. [DOI] [PubMed] [Google Scholar]
- 13.Petrilli FL, Camoirano A, Bennicelli C, Zanacchi P, Astengo M, De Flora S. Specificity and inducibility of the metabolic reduction of chromium(VI) mutagenicity by subcellular fractions of rat tissues. Cancer Res 1985;45:3179–3187. [PubMed] [Google Scholar]
- 14.Tsapakos MJ, Hampton TH, Wetterhahn KE. Chromium (VI)–induced DNA lesions and chromium distribution in rat kidney, liver and lung. Cancer Res 1983;43:5662–5667. [PubMed] [Google Scholar]
- 15.Voitkun V, Zhitkovich A, Costa M. Cr(III)-mediated cross-links of glutathione or amino acids to the DNA phosphate backbone are mutagenic in human cells. Nucleic Acids Res 1998;26:2024–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridgewater LC, Manning FC, Patierno SR. Arrest of replication by mammalian DNA polymerases alpha and beta caused by chromium-DNA lesions. Mol Carcinog 1998;23:201–206. [PubMed] [Google Scholar]
- 17.Wakeman TP, Kim WJ, Callens S, Chiu A, Brown KD, Xu B. The ATM-SMC1 pathway is essential for activation of the chromium[VI]-induced S-phase checkpoint. Mutat Res 2004;554:241–251. [DOI] [PubMed] [Google Scholar]
- 18.Sancar A. DNA excision repair. Annu Rev Biochem 1996;65:43–81. [DOI] [PubMed] [Google Scholar]
- 19.Trombino S, Cesario A, Margaritora S, Granone P, Motta G, Falugi C, Russo P. Alpha 7-nicotinic acetylcholine receptors affect growth regulation of human mesotelioma cells: role of mitogen-activated protein kinase pathway. Cancer Res 2004;64:135–145. [DOI] [PubMed] [Google Scholar]
- 20.Sugiyama M, Tsuzuki K, Haramaki N. DNA single-strand breaks and cytotoxicity induced by sodium chromate(VI) in hydrogen peroxide–resistant cell lines. Mutat Res 1993;299:95–102. [DOI] [PubMed] [Google Scholar]
- 21.Russo P, Catassi A, Malacarne D, Margaritora S, Cesario A, Festi L, Mule A, Ferri L, Granone P. Tumor necrosis factor enhances SN38-mediated apoptosis in mesothelioma cells. Cancer 2005;103:1503–1518. [DOI] [PubMed] [Google Scholar]
- 22.Russo P, Arzani D, Trombino S, Falugi C. c-myc Down-regulation induces apoptosis in human cancer cell lines exposed to RPR-115135 (C31H29NO4), a non-peptidomimetic farnesyltransferase inhibitor. J Pharmacol Exp Ther 2003;304:37–47. [DOI] [PubMed] [Google Scholar]
- 23.Vikhanskaya F, Falugi C, Valente P, Russo P. Human papillomavirus type 16 E6–enhanced susceptibility to apoptosis induced by TNF in A2780 human ovarian cancer cell line. Int J Cancer 2002;97:732–739. [DOI] [PubMed] [Google Scholar]
- 24.Goldsmith ME, Gudas JM, Schneider E, Cowan KH. Wild type p53 stimulates expression from the human multidrug resistance promoter in a p53-negative cell line. J Biol Chem 1995;270:1894–1898. [PubMed] [Google Scholar]
- 25.Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW. Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene 2004;23:2934–2949. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Ladrech S, Pujol R, Brabet P, Van De Water TR, Puel JL. Caspase inhibitors, but not c-Jun NH2-terminal kinase inhibitor treatment, prevent cisplatin-induced hearing loss. Cancer Res 2004;64:9217–9224. [DOI] [PubMed] [Google Scholar]
- 27.Ullmannova V, Haskovec C. Gene expression during camptothecin-induced apoptosis in human myeloid leukemia cell line ML-2. Neoplasma 2004;51:175–180. [PubMed] [Google Scholar]
- 28.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001;7:683–694. [DOI] [PubMed] [Google Scholar]
- 29.Han J, Flemington C, Houghton AB, Gu Z, Zambetti GP, Lutz RJ, Zhu L, Chittenden T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc Natl Acad Sci USA 2001;98:11318–11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coates PJ, Lorimore SA, Wright EG. Cell and tissue responses to genotoxic stress. J Pathol 2005;205:221–235. [DOI] [PubMed] [Google Scholar]
- 31.O'Reilly MA, Staversky RJ, Watkins RH, Reed CK, de Mesy Jensen KL, Finkelstein JN, Keng PC. The Cyclin-dependent kinase inhibitor p21 protects the lung from oxidative stress. Am J Respir Cell Mol Biol 2001;24:703–710. [DOI] [PubMed] [Google Scholar]
- 32.D'Agostini F, Izzotti A, Bennicelli C, Camoirano A, Tampa E, De Flora S. Induction of apoptosis in the lung but not in the liver of rats receiving intra-tracheal instillations of chromium(VI). Carcinogenesis 2002;23:587–593. [DOI] [PubMed] [Google Scholar]
- 33.Wang S, Shi X. Mechanisms of Cr(VI)-induced p53 activation: the role of phosphorylation, mdm2 and ERK. Carcinogenesis 2001;22:757–762. [DOI] [PubMed] [Google Scholar]
- 34.Wang CH, Tsao YP, Chen HJ, Chen HL, Wang HW, Chen SL. Transcriptional repression of p21 ((Waf1/Cip1/Sdi1)) gene by c-jun through Sp1 site. Biochem Biophys Res Commun 2000;270:303–310. [DOI] [PubMed] [Google Scholar]
- 35.Hirose T, Kondo K, Takahashi Y, Ishikura H, Fujino H, Tsuyuguchi M, Hashimoto M, Yokose T, Mukai K, Kodama T, et al. Frequent microsatellite instability in lung cancer from chromate-exposed workers. Mol Carcinog 2002;33:172–180. [DOI] [PubMed] [Google Scholar]
- 36.Carlisle DL, Pritchard DE, Singh J, Owens BM, Blankenship LJ, Orenstein JM, Patierno SR. Apoptosis and P53 induction in human lung fibroblasts exposed to chromium (VI): effect of ascorbate and tocopherol. Toxicol Sci 2000;55:60–68. [DOI] [PubMed] [Google Scholar]
- 37.Higenbottam T, Kuwano K, Nemery B, Fujita Y. Understanding the mechanisms of drug-associated interstitial lung disease. Br J Cancer 2004;91:S31–S37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer 2005;5:231–237. [DOI] [PubMed] [Google Scholar]
- 39.Bardales RH, Xie SS, Schaefer RF, Hsu SM. Apoptosis is a major pathway responsible for the resolution of type II pneumocytes in acute lung injury. Am J Pathol 1996;149:845–852. [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar P, Goldstraw P, Yamada K, Nicholson AG, Wells AU, Hansell DM, Dubois RM, Ladas G. Pulmonary fibrosis and lung cancer: risk and benefit analysis of pulmonary resection. J Thorac Cardiovasc Surg 2003;125:1321–1327. [DOI] [PubMed] [Google Scholar]
- 41.Matthay MA. Function of the alveolar epithelial barrier under pathologic conditions. Chest 1994;105:67S–74S. [DOI] [PubMed] [Google Scholar]
- 42.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001;7:673–682. [DOI] [PubMed] [Google Scholar]
- 43.Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci USA 2003;100:1931–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erster S, Mihara M, Kim RH, Petrenko O, Moll UM. In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol 2004;24:6728–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chow TC, Talalay P. A simple generalized equation for the analysis of multipleinhibitions of Michaelis-Menten kinetic systems. J Biol Chem 1977;252:6438–6442. [PubMed] [Google Scholar]
- 46.Chow TC, Talalay P. Generalized equations for the analysis of inhibitors of Michaelis-Menten and higher order kinetic systems with two or more mutually exclusive and nonexclusive inhibitors. Eur J Biochem 1981;115:207–216. [DOI] [PubMed] [Google Scholar]