

In 2005, the American Thoracic Society marks its 100th year of existence. For over a third of this span, since 1967, clinicians and investigators have struggled with a common, often lethal condition originally termed the adult respiratory distress syndrome (ARDS) (1).* While the syndrome almost certainly occurred in earlier times well before the advent of intensive care units, this date establishes its modern era. Subsequently, ARDS has more correctly come to indicate the acute respiratory distress syndrome because it occurs in children as well as in adults (2). ARDS causes severe acute respiratory failure with dynamic impairment in oxygen and carbon dioxide transfer, with the need for high levels of supplementary oxygen and a high minute ventilation (3, 4). Efforts to understand the pathophysiologic events underlying ARDS, a constellation that is now generally termed acute lung injury (ALI), have been substantial and remain a priority of the National Institutes of Health (5). The expectation has been that basic, translational, and clinical studies will result in new strategies for management of ALI/ARDS based on clear definition of the cellular and molecular events requisite in lung injury and repair. While important discoveries have been made, the goal of fundamental characterization of ALI/ARDS in a way that results in highly effective prevention and treatment remains incomplete and elusive. One major advance in supportive care, a strategy of lung protective mechanical ventilation, has substantially reduced mortality in ALI/ARDS (6, 7). This therapeutic intervention resulted from both experimental and clinical studies that evaluated the effect of different ventilation strategies on the course of ALI, and continues a theme initiated in the original description of ALI/ARDS in which positive end-expiratory pressure was introduced as a management modality (1).
In spite of the advances in supportive care of patients with ALI/ARDS, unacceptable morbidity and mortality persist and formidable challenges remain. Our objective here is to review briefly what we knew in 1967 regarding pathogenesis and what we know in 2005, and to provide a perspective on how insights have evolved over the last four decades.
Pulmonary Edema and the Early Events in ALI/ARDS
Congestion, atelectasis, and pulmonary edema were features of the original description of the syndrome (1). Within a decade, clinical and experimental studies established the concept that increased permeability pulmonary edema is the primary physiologic abnormality in the early stages of ALI/ARDS. Large animal models with measurements of hemodynamics and lung lymph flow demonstrated that clinically relevant causes of ARDS, including live bacteria, endotoxin, and microemboli, induced an increase in lung vascular permeability that causes protein-rich lung edema (8–10). An important clinical study reported protein concentrations in the undiluted edema fluid of mechanically ventilated patients with acute respiratory failure from severe pulmonary edema. In all subjects with features consistent with ARDS, the ratio of the protein concentration in the edema fluid compared with than the simultaneously measured plasma sample was > 0.75, whereas patients with cardiogenic, or high pressure, pulmonary edema had an edema fluid to plasma protein ratio < 0.75 (11). More recent observations are consistent with this finding, showing a separation between patients with hydrostatic versus an increased permeability pulmonary edema (Figure 1). Thus, increased permeability edema, interpreted as accumulation of protein-rich edema fluid into the alveoli, has become a hallmark of ALI/ARDS (12) (Figure 2).
Figure 1.

Increased permeability pulmonary edema is a hallmark of ALI/ARDS. Edema fluid to plasma protein concentrations expressed as a ratio to determine whether the edema fluid is a transudate as in hydrostatic pulmonary edema (< 0.65) or an exudate (> 0.65) as in increased permeability pulmonary edema (ALI/ARDS). The pulmonary edema fluid samples were obtained from patients within the 15 min of endotracheal intubation for acute respiratory failure. These are representative data from previously published and ongoing studies by Matthay and coworkers, some of which are discussed in Ref. 18.
Figure 2.

Early events in ALI/ARDS. A variety of “direct” (lung infection, aspiration) and “indirect” (sepsis, multiple trauma with shock and large volume blood replacement) clinical insults lead to ALI. Initial clinical descriptions identified pulmonary edema as a major consequence. Subsequent investigations yielded evidence for inflammatory injury to the alveolar–capillary membrane as a central pathogenetic mechanism. The key effector cells, molecules, and mechanisms that lead to dysregulation of inflammatory and hemostatic pathways in ALI/ARDS remain incompletely defined. (Modified from Ref. 34.)
Subsequently many studies have been done to define mechanisms that account for the acute increase in lung vascular permeability (5). Earlier experiments concentrated mostly on large animal models in which pulmonary and systemic hemodynamics could be measured, whereas more recent studies have used mouse models, largely to exploit the opportunity to use specific gene deletions to define molecular events that may be pivotal in the development of altered lung vascular permeability. Novel in vitro models have used cultured endothelial cells. The progress of these investigations is outlined later in this review. In parallel with early attempts to understand the mechanisms that lead to lung edema factors that compound the physiologic derangements were considered. One hypothesis was that ALI is associated with surfactant dysfunction, either because of reduced production or neutralization of surfactant by the plasma proteins and fibrin that extravasate into the alveoli (1). A decrease in functional surfactant would contribute to alveolar instability and arterial hypoxemia, potentially increase lung edema formation (13), and perhaps impair innate immune defenses (14). Analysis of bronchoalveolar lavage samples indicates that the lipid and protein components of surface active material are altered in patients with ALI/ARDS (15). Nevertheless, replacement of surfactant has not reduced mortality in large clinical trials in adults with ALI (16, 17), perhaps because, unlike infant respiratory distress syndrome, the fundamental lesion in the acute respiratory distress syndrome is not lack of surfactant production by immature alveolar type II cells. In sharp contrast, surfactant replacement has substantially reduced mortality in infant respiratory distress syndrome in preterm infants.
The role of lung epithelial cell injury and dysfunction and the mechanisms that regulate removal of alveolar edema fluid, which were not recognized in early considerations of ALI/ARDS, emerged in experimental and clinical studies in the 1980s and '90s (18) (also see below). The concept of lung fluid balance incorporates both formation and removal and provides a more dynamic concept of the factors that contribute to the net quantity of edema in the lung in animals or humans with ALI (Figure 2). Edema formation and accumulation in the interstitium and airspaces of the lung may be substantial because of a marked increase in lung vascular permeability, but if alveolar epithelial fluid reabsorption balances the formation of alveolar edema, then a steady state can be established that may allow time for recovery from the fundamental cause of lung injury. Perhaps this is why patients who maintain higher rates of alveolar epithelial fluid transport in the face of ALI have a better survival (19, 20). In addition, although a primary event in ALI/ARDS is an increase in lung vascular permeability, it was apparent from experimental models (21) and clinical observations (22) that the magnitude of lung edema in ALI may be substantially increased when lung vascular pressures and volume are elevated, consistent with the effects of hydrostatic pressure on transvascular flux of fluid and protein. More recently, a large NHLBI-sponsored clinical trial is now testing the hypothesis that measures to lower lung vascular pressures in patients with ALI/ARDS can improve clinical outcomes by reducing the quantity of extravasated protein-rich edema fluid in the lung, thus reducing the severity of respiratory failure and ultimately decreasing mortality.
In addition to these investigations, the intimate relationship between alveolar edema formation and inflammatory and thrombotic effector mechanisms gradually emerged in the decades after the clinical and physiologic presentations of ALI/ARDS were described. These concepts were integral to the clinical studies that demonstrated the variety of “triggering” disorders that can be associated with the development of ALI, resulting in injury to the alveolar barrier (Figure 2).
More than Water: Inflammatory Mechanisms in ALI/ARDS
While it initially seemed clear that altered alveolar barrier function and pulmonary edema are central characteristics of ALI/ARDS, mechanisms that could account for the increase in permeability of the alveolar–capillary membrane were poorly understood. Even in early years, however, some investigators believed that inflammation might be involved. One stimulus for this view was the presence of leukocytes together with alveolar edema, hemorrhage, and hyaline membranes in some autopsy specimens (1) (Figure 3). Although subsequent pathologic studies were hampered by heterogeneity in predisposing conditions underlying ALI/ARDS (Figure 2) (12), the risk of lung biopsy in respiratory failure, and the timing and relative infrequency of autopsies, they still provided key information. One study reported purulent exudates in the alveoli of some patients, although the contributions of infection versus inflammatory injury were not dissected (23). Seminal ultrastructural studies of the lung in patients dying with ALI secondary to sepsis then demonstrated increased numbers of intravascular and extravascular neutrophils (PMNs), platelets, and fibrin (24, 25), and both endothelial and epithelial injury, findings that are still incorporated into modern concepts of inflammatory edema in ARDS (6, 12, 26) (Figure 3). Additional pivotal observations included the presence of PMNs and other leukocytes in bronchoalveolar lavage samples from subjects with ALI/ARDS (27), studies of PMN-dependent lung injury and edema in animal models (28), and evidence in cell biologic and in vivo analyses that PMN oxidants and proteases can injure cells of the alveolar–capillary membrane (28, 29). Subsequent observations are consistent with the concept that inflammation is a component of many, and perhaps all, causes of “direct” injury to the alveolar–capillary membrane as occurs with aspiration and many forms of infectious pneumonitis (12, 30, 31) (Figure 2).
Figure 3.
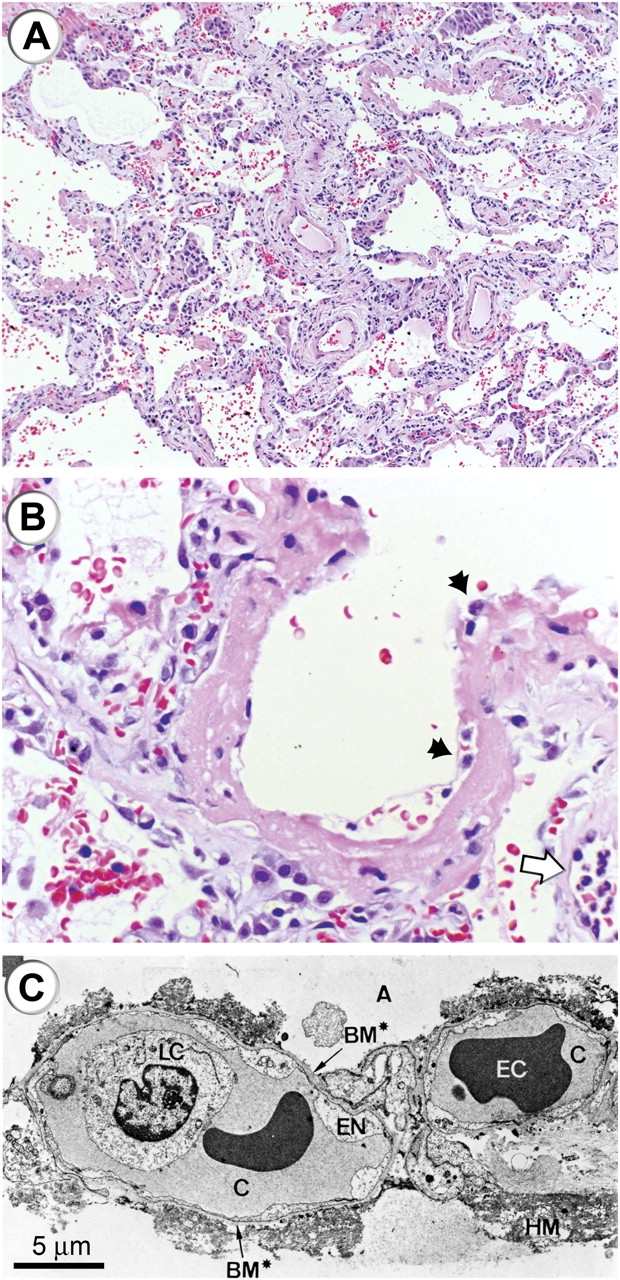
Histologic and ultrastructural analysis of the injured lung has been integral to current concepts of pathogenesis of ALI/ARDS. (A) A low-power light micrograph of a lung biopsy specimen collected 2 d after the onset of ALI/ARDS secondary to gram-negative sepsis demonstrates key features of diffuse alveolar damage, including hyaline membranes, inflammation, intraalveolar red cells and neutrophils, and thickening of the alveolar–capillary membrane. (B) A higher-power view of a different field illustrates a dense hyaline membrane and diffuse alveolar inflammation. Polymorphonuclear leukocytes are imbedded in the proteinaceous hyaline membrane structure (black arrows). The white arrow points to the edge of an adjacent alveolus, which contains myeloid leukocytes (thank you to K. Jones, M.D., UCSF Pathologist, for the histologic sections in A and B). (C) An electron micrograph from a classic analysis of ALI/ARDS showing injury to the capillary endothelium and the alveolar epithelium. LC refers to a leukocyte (a neutrophil) within the capillary lumen. EC designates enythrocytes. EN shows blebbing of the capillary endothelium. BM refers to exposed basement membrane where the epithelium has been denuded. C refers the capillary. A refers to the alveolar space. (Reprinted with permission from Ref. 24.)
Studies of complement activation, specifically generation of C5a, and consequent activation of PMNs contributed a new facet to concepts of the pathogenesis of ALI/ARDS (32, 33). Together with reports demonstrating endothelial injury by PMN proteases and oxidants, these observations provided a conceptual model in which generation of a circulating mediator (in this case, C5a) induces systemic activation of PMNs, resulting in aggregation and sequestration in pulmonary microvessels and consequent diffuse lung injury (33, 34). This suggested a mechanism by which “indirect” alveolar–capillary membrane injury is caused, as in systemic sepsis and nonthoracic trauma (12) (Figures 2 and 3). Leukocyte activation leading to intravascular sequestration, alone or involving interacting platelets, is consistent with findings in much more recent studies of the pathophysiology of ALI/ARDS (4, 34). These concepts of PMN activation and aggregation also contributed to early suggestions that glucocorticoids, inhibitors of PMN granular proteases, and antioxidants might be specific therapeutic agents (28, 33, 45)—a potential that, although mechanistically based, has not subsequently been realized (12).
Additional analyses of leukocytes in the blood and alveoli of subjects with ALI/ARDS (35–37) indicated that accumulation and activation of PMNs could not be completely explained by existing paradigms and known mediators such as C5a, and stimulated new investigators in the field to develop alternative models and hypotheses and to draw on previous and parallel investigations of inflammatory pathways in other systems. One outcome was identification of endothelial cell activation as a mechanism of leukocyte accumulation and signaling (38, 39). Thus, key mediators might not be acting directly on the leukocytes but instead on the lung endothelium, resulting in accumulation of PMNs, inflammatory injury, and, potentially, accumulation of other leukocyte types in the injured lung (40). A key concept in current pathobiology is that endothelial activation is central to inflammation in the lung and elsewhere (41). Subsequent studies have examined mechanisms that regulate leukocyte–endothelial interactions in the pulmonary versus systemic circulations, where there are both differences and similarities (40–43). These issues have additional complexity because ALI/ARDS can involve both pulmonary and systemic vascular beds (5, 34).
The concept of ALI mediated by PMNs and circulating or locally generated mediators (Figure 4) with origins outlined above, continues to be central to current concepts of the pathogenesis of ALI/ARDS and has been emphasized in reviews and symposia that span two decades (12, 44–51). Inflammation was identified as a key feature in the 1994 Consensus conference on ALI/ARDS (47). The concept has also evolved in complexity and detail. C5a, which remains topical (52, 53) but is not a final common pathway to ALI/ARDS, is now complemented by chemokines, cytokines, and lipid signaling molecules (54–58). PMNs are themselves sources of some of these factors (51). In some cases, identification of individual inflammatory mediators in cell biologic and preclinical models has generated candidate therapeutic strategies that have been tested in clinical trials. Intracellular signaling pathways that link PMN surface receptors to activation responses, including kinases and the NF-κB family of transcription factors, are implicated as potential points of intervention, and it is recognized that these pathways are modulated by surface integrins, selectins, and selectin ligands, which are adhesion molecules that also mediate tissue accumulation of leukocytes (40, 42, 49, 51). PMN activities besides acute generation of oxidants and release of proteases, such as signaling of endothelium (59), inflammatory gene expression (51), and apoptosis (60) are believed to be relevant. There is evidence that injurious versus protective responses of PMNs may influence outcomes (61, 62) and that there are time-dependent changes in PMN number and phenotype in ALI/ARDS (62, 63), although the significance of these findings remains incompletely explored.
Figure 4.

Multiple cellular responses and mediators contribute to alveolar–capillary membrane injury (right-hand side) and the transition from normal alveolar structure and function (left-hand side) in the acute phase of ALI/ARDS. Original investigations of the pathogenesis of ALI/ARDS searched for single mediators that provided final common pathways to inflammation and alveolar edema in ALI/ARDS. Current concepts of pathogenesis involve multiple molecular factors of several classes, a variety of responding cells, and imbalance between injurious and reparative signals and pathways. See text and Refs. 5 and 12 for details. (Reprinted from Ref. 12 with permission.)
It also seems unlikely that PMNs act alone in any of the phases of ALI/ARDS (Figure 4), although they may have particular roles at acute and subacute time points (62, 63). Lung macrophages, circulating monocyte subpopulations, and other leukocyte subtypes have been suggested as key effector cells and have been considered in a preliminary fashion in both early and later studies (27, 45, 64, 65), although little functional data exist. The roles of platelets, which were detected in the acutely injured lung in early studies (24, 25, 66), and molecular and cellular links between the thrombotic and inflammatory systems remain topical issues (5, 34). This is in part because of the potential therapeutic use of recombinant activated protein C (APC) in sepsis, a key triggering conditions for ALI/ARDS (5, 34, 67). APC interrupts both inflammatory and thrombotic events in experimental studies (68–70). Platelets release IL-1β, HMBG-1, and chemokines and also directly interact with PMNs and monocytes in addition to organizing fibrin clots; thus, they are active in innate immune cascades and may modify inflammatory injury in ALI/ARDS (34, 71). In parallel, however, platelets also release sphingosine-1-phosphate, which promotes endothelial barrier function in experimental models (72–74). This illustrates the well known fact that platelets have both defensive and reparative activities. Thus, it is not yet clear if their accumulation and activation in the lungs has a net positive or negative effect on the outcome of ALI/ARDS.
Animal and human studies demonstrate that mechanical factors and the pattern of ventilation induce or amplify alveolar inflammation and injury (12, 43, 75) and contribute to nonpulmonary organ injury (5, 12, 76, 77). Thus, unfavorable ventilation strategies can compound the severity of ALI, adding an iatrogenic variable to classic triggers of injury (5, 6, 12). Oxygen in high concentrations, which is vital in the support of patients with ALI/ARDS, can also be toxic to alveolar–capillary membrane cells of animals and humans and presents another mechanism of iatrogenic injury (44), although one recent clinical trial found no decrease in mortality when a lower fraction of inspired oxygen was used in patients with higher levels of positive end-expiratory pressure (7). In animal models, hyperoxia increases intrapulmonary retention of PMNs and induces dysregulation of other innate immune mechanisms (44, 78). Inflammatory cytokines can worsen or, conversely, ameliorate oxygen injury in surrogate models (79).
Resolution of Lung Injury
The natural history of ALI/ARDS has been progressively defined by clinical studies (12, 44, 47, 48) and follows a variable course. One outcome is resolution and repair (Figure 5). The successful resolution of pulmonary edema and lung inflammation is an important determinant of recovery from acute lung injury. Early studies demonstrated that lung lymphatics and the pulmonary microcirculation remove edema fluid from the interstitium of the lung (8, 9), and that fluid flow into the pleural space is an additional major pathway for edema translocation (80). A new mechanism was recognized in the 1980s when investigators demonstrated that removal of alveolar edema fluid depends on vectorial transport of salt and water across the alveolar epithelium in part through apically located epithelial sodium channels (ENaC), followed by extrusion into the lung interstitium via a basolaterally located Na-KATPase (18) (Figure 6). The transport of sodium creates a mini–osmotic gradient that absorbs water from the airspaces via water channels (aquaporins) and paracellular pathways (18). Net alveolar fluid clearance can be upregulated by cAMP agonists in many species, including the human lung (81). Recent evidence suggests that the cystic fibrosis chloride channel, CFTR, is required for effective cAMP stimulated fluid transport (82). Both alveolar type II and type I cells may be capable of driving sodium transport and net alveolar fluid clearance (83–85). The transport capacity of the alveolar epithelium is markedly diminished in ALI, a finding that correlates with higher mortality (19, 20). The mechanisms for the decrease in alveolar fluid clearance include frank injury to alveolar epithelial cells and their apoptosis or necrosis, resulting in loss of barrier and transport properties as well as more subtle defects in ion transport capacity. Oxidant injury, reactive nitrogen species, hypoxia, and direct effects of bacterial and viral pathogens alter the transport machinery in the epithelial cells (18), as do unfavorable ventilatory strategies (75, 86).
Figure 5.

The natural history of ALI/ARDS includes resolution and repair versus persistence and progression. Clinical and epidemiologic studies demonstrate that ALI/ARDS resolves with return of alveolar function to normal or near normal in some patients, whereas in others there is persistence and/or progression of injury. The outcomes of persistence and progression include multiple organ failure, fibrosing alveolitis, pulmonary vascular obliteration with pulmonary hypertension, and death. The genetic, cellular, molecular and iatrogenic factors that contribute to each of these outcomes remain largely unknown. In addition, rational mechanism-based strategies that favorably influence repair of the alveolar–capillary membrane are undefined. (Modified from Ref. 34.)
Figure 6.

Cellular and molecular pathways regulate the resolution of alveolar edema formation and resolution in ALI/ARDS. Histologic section from patients with a lung biopsy in the setting of lung injury from bacterial pneumonia and sepsis with protein rich alveolar edema. The insert refers to ENaC, the epithelial sodium channel, which is a major pathway for the uptake of sodium on the apical surface of alveolar epithelial type I and II cells as well as distal airway epithelia. NaKATPase refers to the sodium pump located in the basolateral surface of alveolar and airway epithelia that actively pumps sodium into the interstitial space, thus creating a mini–osmotic gradient for the absorption of edema fluid from the alveolar. Both ENaC and NaKATPase can be upregulated by several catecholamine dependent and independent mechanisms. See Refs. 18 and 19 for more details. (Reprinted with permission from Ref. 18.)
More precise understanding of how the alveolar endothelium and epithelium interact, and their coordinate responses to inflammatory cells and cytokines, could clarify mechanisms by which lung endothelial injury leads to alveolar edema (Figure 2) and how resolution of edema is modulated (Figures 5 and 6). Experimentally, alveolar endothelial injury from endotoxin can occur without accumulation of alveolar edema, apparently because epithelial injury is mild. In contrast, substitution of live pathogenic bacteria results in both endothelial and epithelial injury (87). Since direct or indirect infection accounts for more than 60% of clinical cases of ALI in most studies (88), a better understanding of how the bacterial and viral products alter these and other aspects of alveolar–capillary function is critical. Furthermore, it is essential to determine how well the alveolar epithelium recovers from apoptotic or necrotic lung injury and the basic mechanisms that regulate this process of epithelial repair (5).
It is also critical to understand more about how inflammatory cells and soluble and insoluble proteins are removed from the lung. Characterizations of molecular mechanisms that regulate macrophage-dependent removal of apoptotic leukocytes in inflammatory lung injury is in progress (89, 90). Soluble protein appears to be primarily removed by a slow process of paracellular diffusion across the alveolar epithelium although transcytosis can contribute as well (91). Removal of insoluble proteins and cellular debris and remodeling of hyaline membranes appears to be primarily driven by alveolar macrophages although details of how this process proceeds are sketchy at best.
One subset of patients with ALI/ARDS has severe lung injury that includes an extensive exudative process, profound endothelial and epithelial injury, and often fibrosing alveolitis (12, 24, 25) (Figure 5). Although multiple organ failure and death are common outcomes in this group, remodeling of the injured lung can occur in some subjects with restoration of a functional alveolar–capillary barrier without major permanent pathologic or physiologic changes. The mechanisms that regulate this recovery process remain largely unknown and need precise definition (5).
Four Decades of Discovery with Much Yet To Be Learned
In spite of elements of progress, much remains to be learned. This was apparent to an interdisciplinary group of investigators that met to discuss the state of the field and future research priorities in ALI/ARDS in 2002 (5, 12). The issues are broad and the unknowns complex.
In all of the facets of the inflammatory and hemostatic responses in ALI/ARDS (Figures 2 and 4) we still have little insight into what is cause and what is consequence (12, 35, 47). Although some progress has been made, we are little closer than we were in 1967 to understanding how key inflammatory events that are defensive and reparative when they are regulated in lung infection and limited injury become damaging and destructive when they are unregulated in ALI/ARDS, and we have very little insight into what the molecular mechanisms of dysregulation actually are. In bacterial pneumonia, innate immune responses mediated by activated neutrophils, monocytes, and alveolar macrophages play requisite roles. In parallel, a local shift to a procoagulant and antifibrinolytic environment may serve to wall off infection and airspace injury, yet these same effectors and responses appear to be pivotal in the early events in ALI/ARDS (Figure 2) (5). The concept of a balance between pro- and anti-inflammatory and pro- and anti-edematous factors has current traction in the field, in part borrowed from other diseases and mechanisms of injury (5, 55–57). Further, specific regulatory molecules may have essential activities that usually keep lung inflammation and edema in check (5, 35, 58). Nevertheless, while the concepts are appealing, the defining mechanisms remain obscure and many of the molecular players have a Janus face. As an example, transforming growth factor-β, a regulatory cytokine, has important activities that modulate inflammation and edema, but recent evidence suggests that it may also enhance endothelial and epithelial permeability and have a net deleterious effect in the acutely injured lung (89, 91–93).
The drive to understand initiation of ALI, the acute inflammatory components of ALI/ARDS, and their resolution (Figure 2) has been accompanied by focused intent to define cellular and molecular events that contribute to progressive alveolar–capillary injury and dysregulated repair (Figure 5), particularly in the last decade of molecular medicine (5, 12). It is clear from clinical and epidemiologic studies that specific biologic features prevent the resolution of ALI in some subjects, resulting in persistence and progression to severe complications including multiple organ failure, fibrosing alveolitis, and alveolar endothelial and epithelial cell destruction (Figures 3 and 5) (24, 25, 94, 95). Our understanding here is extremely limited. Further progress will likely require reductionist cell biologic experiments and models to establish molecular paradigms that can then be explored in animal models and, ultimately, in critically ill patients. In addition, genetic determinants of these responses and how they vary among individuals and interact with environmental influences are largely unexplored (5). Cellular, genomic, proteomic, and lipidomic patterns in the acutely injured lung and in lungs undergoing regulated repair versus dysregulated progression remain to be defined. Thus, the biologic basis required for clear understanding of the natural history of ALI/ARDS and for new strategies for intervention remains incompletely defined (5).
Animal models exploring mechanisms of lung edema, roles of PMNs and other inflammatory effectors, repair mechanisms, and other features of ALI/ARDS have increasingly used mice because of the power of genetic manipulation (31, 42, 51, 52, 73, 74, 78, 79, 96, 97). One recent example of this strategy proposed a novel role for angiotensin-converting enzyme 2 in protecting against experimental ALI (98). If confirmed in additional preclinical studies, this treatment could be translated to clinical testing in patients with lung injury. Studies in rats, dogs, sheep, and other surrogates have been indispensable in characterizing mechanisms of increased permeability edema and developing the preclinical evidence for revised approaches to ventilating patients with ALI/ARDS (99–101). However, the relationships of preclinical animal models to the clinical syndromes of ALI/ARDS are often uncertain for several reasons. Animal studies commonly do not model the usual medical and surgical risk factors for developing ALI/ARDS that occur in patients; animals are not ordinarily supported for days in an intensive care setting with nutrition, antibiotics, and other supportive care; and the time frame for developing lung injury is usually abbreviated in animal models (5). In spite of these limitations, animal models still provide a critical method for exploring mechanisms and potential new therapies in a proof-of-principle fashion. Nevertheless, the value of early testing of new therapeutic strategies in human studies is increasingly appreciated by both the NHLBI and private industry.
The success of a lung protective ventilatory strategy in reducing mortality in ALI/ARDS (6, 7) is a good example of how clinical trials are needed to test novel hypotheses that derive initially from preclinical studies. The marked decrease in mortality with lung protective ventilation has evolved, as outlined earlier in this review, from animal studies that suggested that the standard approach of ventilating patients with ALI/ARDS with higher tidal volumes could amplify the severity of lung injury. Knowledge of the responses of individual cell types to mechanical and inflammatory stimuli provided insight into how this might occur at a molecular level (5). Subsequently, the success of lung protective ventilatory strategies has stimulated new investigations into how mechanical stresses may adversely affect the lung. The mechanisms of benefit are still being worked out, but seem to involve a modest decrease in lung and systemic inflammation (102) and a reduction in the severity of alveolar epithelial injury (75, 103, 104). The reduction in mortality with the reduction in tidal volume and plateau airway pressure has been accompanied by a decrease in nonpulmonary organ failure (6), thus clearly linking the severity of the lung injury to the systemic consequences of ALI/ARDS.
ALI/ARDS threatens the lives and well being of ∼ 200,000 patients annually in the United States alone (105). Important discoveries relevant to the pathogenesis have been made since the original description of ARDS in 1967, but what we do not know still exceeds what we do. Further progress will require all of the tools of modern integrative biology (5) and a continued infusion of new investigators to apply them. It is perhaps this last requirement that is most tenuous and uncertain, but the one with the greatest potential (106).
Acknowledgments
The authors thank our many colleagues and collaborators for invaluable discussions and debates; Paolo M. Miranda and Adrienne Triplett for help with preparation of the manuscript; Diana Lim for consultation on graphic presentations; and Zachary Matthay, Gabe Mednick, Xiao Fang, and Diana Lim for preparation of the figures.
The references in this article are eclectic rather than comprehensive and span a time frame from early reports to much more recent observations. We regret that we could not include many other articles that have had an impact on current concepts and understanding of the pathogenesis of ALI/ARDS because of space limitations. Additional detailed reviews of specific issues in ALI/ARDS not cited in this article can be found in: Acute respiratory distress syndrome. Matthay MM, ed. Lung biology in health and disease, vol. 179. Lenfant C, series ed. New York and Basel: Marcel Dekker; 2003.
Work cited in this article was supported by individual grants to the authors from the NIH, a SCOR in ARDS (HL 50153), and a SCCOR in ALI/ARDS (HL74005).
Conflict of Interest Statement:Neither author has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet 1967;2:319–323. [DOI] [PubMed] [Google Scholar]
- 2.Flori HR, Glidden DV, Rutherford GW, Matthay MA. Pediatric acute lung injury: prospective evaluation of risk factors associated with mortality. Am J Respir Crit Care Med 2005;171:995–1001. [DOI] [PubMed] [Google Scholar]
- 3.Falke KJ, Pontoppidan H, Kumar A, Leith DE, Geffin B, Laver MB. Ventilation with end-expiratory pressure in acute lung disease. J Clin Invest 1972;51:2315–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nuckton TJ, Alonso JA, Kallet RH, Daniel BM, Pittet J-F, Eisner MD, Matthay MA. Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med 2002;346:1281–1286. [DOI] [PubMed] [Google Scholar]
- 5.Matthay MA, Zimmerman GA, Esmon C, Bhattacharya J, Coller B, Doerschuk CM, Floros J, Gimbrone MA Jr, Joffman E, Hubmayr RD, et al. Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med 2003;167:1027–1035. [DOI] [PubMed] [Google Scholar]
- 6.The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000;342:1301–1308. [DOI] [PubMed] [Google Scholar]
- 7.The Acute Respiratory Distress Syndrome Network. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med 2004;351:327–336. [DOI] [PubMed] [Google Scholar]
- 8.Brigham KL, Woolverton WC, Blake LH, Staub NC. Increased sheep lung vascular permeability caused by pseudomonas bacteremia. J Clin Invest 1974;54:792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohkuda K, Nakahara K, Weidner WJ, Binder A, Staub NC. Lung fluid exchange after uneven pulmonary artery obstruction in sheep. Circ Res 1978;43:152–161. [DOI] [PubMed] [Google Scholar]
- 10.Brigham KL, Bowers R, Haynes J. Increased lung vascular permeability by Escherichia coli endotoxin. Circ Res 1979;45:292–297. [DOI] [PubMed] [Google Scholar]
- 11.Fein A, Grossman RF, Jones JG, Overland E, Pitts L, Murray JF, Staub NC. The value of edema fluid protein measurement in patients with pulmonary edema. Am J Med 1979;67:32–38. [DOI] [PubMed] [Google Scholar]
- 12.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000;342:1334–1349. [DOI] [PubMed] [Google Scholar]
- 13.Albert RK, Lakshminarayan S, Hildebrandt J, Kirk W, Butler J. Increased surface tension favors pulmonary edema formation in anesthetized dogs' lungs. J Clin Invest 1979;63:1015–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol 2005;5:58–68. [DOI] [PubMed] [Google Scholar]
- 15.Gregory TJ, Longmore WJ, Moxley MA, Whitsett JA, Reed CR, Fowler III AA, Hudson LD, Maunder RJ, Crim C, Hyers TM. Surfactant chemical composition and biophysical activity in acute respiratory distress syndrome. J Clin Invest 1991;88:1976–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spragg RG, Lewis JF, Walmrath HD, Johannigman J, Bellingan G, Laterre PF, Witte MC, Richards GA, Rippin G, Rathgeb F, et al. Effect of recombinant surfactant protein C-based surfactant on the acute respiratory distress syndrome. N Engl J Med 2004;351:884–892. [DOI] [PubMed] [Google Scholar]
- 17.Anzueto A, Baughman RP, Guntupalli KK, Weg JG, Wiedemann HP, Raventos AA, Lemaire F, Long W, Zaccardelli DS, Pattishall EN. Aerosolized surfactant in adults with sepsis-induced acute respiratory distress syndrome. Exosurf Acute Respiratory Distress Syndrome Sepsis Study Group. N Engl J Med 1996;334:1417–1421. [DOI] [PubMed] [Google Scholar]
- 18.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev 2002;82:569–600. [DOI] [PubMed] [Google Scholar]
- 19.Matthay MA, Wiener-Kronish JP. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis 1990;142:1250–1257. [DOI] [PubMed] [Google Scholar]
- 20.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;163:1376–1383. [DOI] [PubMed] [Google Scholar]
- 21.Prewitt RM, McCarthy J, Wood LDH. Treatment of acute low pressure pulmonary edema in dogs: relative effects of hydrostatic and oncotic pressure, nitroprusside, and positive end-expiratory pressure. J Clin Invest 1981;67:409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitchell JP, Schuller D, Calandrino FS, Schuster DP. Improved outcome based on fluid management in critically ill patients requiring pulmonary artery catheterization. Am Rev Respir Dis 1992;145:990–998. [DOI] [PubMed] [Google Scholar]
- 23.Lamy M, Fallat RJ, Koeniger E, Dietrich HP, Ratliff JL, Eberhart RC, Tucker HJ, Hill JD. Pathologic features and mechanisms of hypoexemia in adult respiratory distress syndrome. Am Rev Respir Dis 1976;114:267–284. [DOI] [PubMed] [Google Scholar]
- 24.Bachofen A, Weibel ER. Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am Rev Respir Dis 1977;116:589–615. [DOI] [PubMed] [Google Scholar]
- 25.Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med 1982;3:35–56. [PubMed] [Google Scholar]
- 26.Albertine KH. Ultrastructural abnormalities in increased-permeability pulmonary edema. Clin Chest Med 1985;6:345–369. [PubMed] [Google Scholar]
- 27.Pittet JF, MacKersie RC, Martin TR, Matthay MA. Biological markers of acute lung injury: prognostic and pathogenetic significance. Am J Respir Crit Care Med 1997;155:1187–1205. [DOI] [PubMed] [Google Scholar]
- 28.Brigham KL. Mechanisms of lung injury. Clin Chest Med 1982;3:9–24. [PubMed] [Google Scholar]
- 29.Cochrane CG, Spragg R, Revak SD. Pathogenesis of the adult respiratory distress syndrome. Evidence of oxidant activity in bronchoalveolar lavage fluid. J Clin Invest 1893;71:754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Folkesson HG, Matthay MA, Hébert C, Broaddus VC. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanism. J Clin Invest 1995;96:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiser MR, Pechet TT, Williams JP, Ma M, Frenette PS, Moore FD, Kobzik L, Hines RO, Wagner DD, Carroll MC, et al. Experimental murine acid aspiration injury is mediated by neutrophils and the alternative complement pathway. J Appl Physiol 1997;83:1090–1095. [DOI] [PubMed] [Google Scholar]
- 32.Hammerschmidt DE, Weaver LJ, Hudson LD, Craddock PR, Jacob HS. Association of complement activation and elevated plasma-C5a with adult respiratory distress syndrome. Pathophysiological relevance and possible prognostic value. Lancet 1980;1:947–949. [DOI] [PubMed] [Google Scholar]
- 33.Jacob HS, Craddock PR, Hammerschmidt DE, Moldow CF. Complement-induced granulocyte aggregation: an unsuspected mechanism of disease. N Engl J Med 1980;302:789–794. [DOI] [PubMed] [Google Scholar]
- 34.Zimmerman GA, Albertine KH, McIntyre TM. Pathogenesis of sepsis and septic-induced lung inury. In: Acute respiratory distress syndrome. Lenfant C, Matthay MA, eds. New York: Marcel Dekker; 2003. pp. 245–287.
- 35.Zimmerman GA, Renzetti AD, Hill HR. Functional and metabolic activity of granulocytes from patients with adult respiratory distress syndrome: evidence for activated neutrophils in the pulmonary circulation. Am Rev Respir Dis 1983;127:290–300. [DOI] [PubMed] [Google Scholar]
- 36.Zimmerman GA, Renzetti AD, Hill HR. Granulocyte adherence in pulmonary and systemic arterial blood samples from patients with adult respiratory distress syndrome. Am Rev Respir Dis 1984;129:798–804. [DOI] [PubMed] [Google Scholar]
- 37.Fowler AA, Fowler AA, Fisher BJ, Centor RM, Carchman RA. Development of the adult respiratory distress syndrome: progressive alteration of neutrophil chemotactic and secretory processes. Am J Pathol 1984;116:427–435. [PMC free article] [PubMed] [Google Scholar]
- 38.Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA Jr. Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J Clin Invest 1985;76:2003–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zimmerman GA, McIntyre TM, Prescott SM. Thrombin stimulates the adherence of neutrophils to human endothelial cells in vitro. J Clin Invest 1985;76:2235–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zimmerman GA, Albertine KH, Carveth HJ, Gill EA, Grissom CK, Hoidal JR, Imaizumi T, Maloney CG, McIntyre TM, Michael JR, et al. Endothelial activation in ARDS. Chest 1999;116:18S–24S. [DOI] [PubMed] [Google Scholar]
- 41.Ward PA, Hunninghake GW. Lung inflammation and fibrosis. Am J Respir Crit Care Med 1998;157:S123–S129. [DOI] [PubMed] [Google Scholar]
- 42.Doerschuk CM, Mizgerd JP, Kubo H, Qin L, Kumasaka T. Adhesion molecules and cellular biomechanical changes in acute lung injury: Giles F. Filley Lecture. Chest 1999;116:37S–43S. [DOI] [PubMed] [Google Scholar]
- 43.Kuebler WM, Ying X, Singh B, Issekutz AC, Bhattacharya J. Pressure is proinflammatory in lung venular capillaries. J Clin Invest 1999;104:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rinaldo JE, Rogers RM. Adult respiratory-distress syndrome: changing concepts of lung injury and repair. N Engl J Med 1982;306:900–909. [DOI] [PubMed] [Google Scholar]
- 45.Tate RM, Repine JE. Neutrophils and the adult respiratory distress syndrome. Am Rev Respir Dis 1983;128:552–559. [DOI] [PubMed] [Google Scholar]
- 46.Repine JE, Beehler CJ. Neutrophils and adult respiratory distress syndrome: two interlocking perspectives in 1991. Am Rev Respir Dis 1991;144:251–252. [DOI] [PubMed] [Google Scholar]
- 47.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R, et al. The American-European Consensus Conference on ARDS: definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 1994;149:818–824. [DOI] [PubMed]
- 48.Kollef MH, Schuster DP. The acute respiratory distress syndrome. N Engl J Med 1995;332:27–37. [DOI] [PubMed] [Google Scholar]
- 49.Downey GP, Dong Q, Kruger J, Dedhar S, Cherapanov V. Regulation of neutrophil activation in acute lung injury. Chest 1999;116:46S–54S. [PubMed] [Google Scholar]
- 50.Matthay MA. Conference summary: acute lung injury. Chest 1999;116:119S–126S. [DOI] [PubMed]
- 51.Abraham E. Neutrophils and acute lung injury. Crit Care Med 2003;31:S195–S199. [DOI] [PubMed] [Google Scholar]
- 52.Riedemann NC, Guo RF, Neff TA, Laudes IJ, Keller KA, Sarma VJ, Markiewski MM, Mastellos D, Strey CW, Pierson CL, et al. Increased C5a receptor expression in sepsis. J Clin Invest 2002;110:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerard C. Complement C5a in the sepsis syndrome–too much of a good thing? N Engl J Med 2003;348:167–169. [DOI] [PubMed] [Google Scholar]
- 54.Strieter RM, Kunkel SL, Keane MP, Standiford TJ. Chemokines in lung injury: Thomas A. Neff Lecture. Chest 1999;116:103S–110S. [DOI] [PubMed] [Google Scholar]
- 55.Martin TR. Lung cytokines and ARDS: Roger S. Mitchell Lecture. Chest 1999;116:2S–8S. [DOI] [PubMed] [Google Scholar]
- 56.Park WY, Goodman RB, Steinberg KP, Ruzinski JT, Radella F II, Park DR, Pugin J, Skerrett SJ, Hudson LD, Martin TR. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;164:1896–1903. [DOI] [PubMed] [Google Scholar]
- 57.Keane MP, Donnelly SC, Belperio JA, Goodman RB, Dy M, Burdick MD, Fishbein MC, Strieter RM. Imbalance in the expression of CXC chemokines correlates with bronchoalveolar lavage fluid angiogenic activity and procollagen levels in acute respiratory distress syndrome. J Immunol 2002;169:6515–6521. [DOI] [PubMed] [Google Scholar]
- 58.Zimmerman GA, McIntyre TM, Prescott SM, Stafforini DM. The platelet-activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit Care Med 2002;30:S294–S301. [DOI] [PubMed] [Google Scholar]
- 59.Dull RO, Garcia JG. Leukocyte-induced microvascular permeability: how contractile tweaks lead to leaks. Circ Res 2002;90:1143–1144. [DOI] [PubMed] [Google Scholar]
- 60.Matute-Bello G, Liles WC, Radella F II, Steinberg KP, Ruzinski JT, Jonas M, Chi EY, Hudson LD, Martin TR. Neutrophil apoptosis in the acute respiratory distress syndrome. Am J Respir Crit Care Med 1997;156:1969–1977. [DOI] [PubMed] [Google Scholar]
- 61.Martin TR, Pistorese BP, Hudson LD, Maunder RJ. The function of lung and blood neutrophils in patients with the adult respiratory distress syndrome: implications for the pathogenesis of lung infections. Am Rev Respir Dis 1991;144:254–262. [DOI] [PubMed] [Google Scholar]
- 62.Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med 1994;150:113–122. [DOI] [PubMed] [Google Scholar]
- 63.Baughman RP, Gunther KL, Rashkin MC, Keeton DA, Pattishall EN. Changes in the inflammatory response of the lung during acute respiratory distress syndrome: prognostic indicators. Am J Respir Crit Care Med 1996;154:76–81. [DOI] [PubMed] [Google Scholar]
- 64.Thickett DR, Armstrong L, Christie SJ, Millar AB. Vascular endothelial growth factor may contribute to increased vascular permeability in acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;164:1601–1605. [DOI] [PubMed] [Google Scholar]
- 65.Rosseau S, Hammerl P, Maus U, Walmrath HD, Schutte H, Grimminger F, Seeger W, Lohmeyer J. Phenotypic characterization of alveolar monocyte recruitment in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol 2000;279:L25–L35. [DOI] [PubMed] [Google Scholar]
- 66.Bone RC, Francis PB, Pierce AK. Intravascular coagulation associated with the adult respiratory distress syndrome. Am J Med 1976;61:585–589. [DOI] [PubMed] [Google Scholar]
- 67.Idell S. Anticoagulants for acute respiratory distress syndrome: can they work? Am J Respir Crit Care Med 2001;164:517–520. [DOI] [PubMed] [Google Scholar]
- 68.Esmon CT. Protein C anticoagulant pathway and its role in controlling microvascular thrombosis and inflammation. Crit Care Med 2001;29:S48–S51. [DOI] [PubMed] [Google Scholar]
- 69.Hoffmann JN, Vollmar B, Laschke MW, Inthorn D, Fertmann J, Schildberg FW, Menger MD. Microhemodynamic and cellular mechanisms of activated protein C action during endotoxemia. Crit Care Med 2004;32:1011–1017. [DOI] [PubMed] [Google Scholar]
- 70.Nick JA, Coldren CD, Geraci MW, Poch KR, Fouty BW, O'Brien J, Gruber M, Zarini S, Murphy RC, Kuhn K, et al. Recombinant human activated protein C reduces human endotoxin-induced pulmonary inflammation via inhibition of neutrophil chemotaxis. Blood 2004;104:3878–3885. [DOI] [PubMed] [Google Scholar]
- 71.Heffner JE, Sahn SA, Repine JE. The role of platelets in the adult respiratory distress syndrome. Culprits or bystanders? Am Rev Respir Dis 1987;135:482–492. [DOI] [PubMed] [Google Scholar]
- 72.Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha'afi RI, Hla T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 1999;99:301–312. [DOI] [PubMed] [Google Scholar]
- 73.McVerry BJ, Peng X, Hassoun PM, Sammani S, Simon BA, Garcia JG. Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am J Respir Crit Care Med 2004;170:987–993. [DOI] [PubMed] [Google Scholar]
- 74.Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med 2004;169:1245–1251. [DOI] [PubMed] [Google Scholar]
- 75.Frank JA, Gutierrez JA, Jones K, Allen L, Dobbs L, Matthay MA. Low tidal volume reduces epithelial and endothelial injury in acid-induced rat lungs. Am J Respir Crit Care Med 2002;165:2492–2499. [DOI] [PubMed] [Google Scholar]
- 76.Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, Bruno F, Slutsky AS. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 1999;282:54–61. [DOI] [PubMed] [Google Scholar]
- 77.Uhlig S, Ranieri M, Slutsky AS. Biotrauma hypothesis of ventilator-induced lung injury. Am J Respir Crit Care Med 2004;69:314–315. [DOI] [PubMed] [Google Scholar]
- 78.Baleeiro CE, Wilcoxen SE, Morris SB, Standiford TJ, Paine R III. Sublethal hyperoxia impairs pulmonary innate immunity. J Immunol 2003;171:955–963. [DOI] [PubMed] [Google Scholar]
- 79.He CH, Waxman AB, Lee CG, Link H, Rabach ME, Ma B, Chen Q, Zhu Z, Zhong M, Nakayama K, et al. Bcl-2-related protein A1 is an endogenous and cytokine-stimulated mediator of cytoprotection in hyperoxic acute lung injury. J Clin Invest 2005;115:1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wiener-Kronish JP, Broaddus VC, Albertine K, Gropper MA, Matthay MA, Staub NC. The relationship of pleural effusions to increased permeability pulmonary edema in anesthetized sheep. J Clin Invest 1988;82:1422–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sakuma T, Okaniwa G, Nakada T, Nishimura T, Fujimura S, Matthay MA. Alveolar fluid clearance in the resected human lung. Am J Respir Crit Care Med 1994;150:305–310. [DOI] [PubMed] [Google Scholar]
- 82.Fang X, Fukuda N, Barbry P, Sartori C, Verkman AS, Matthay MA. Novel role of CFTR in fluid absorption from the distal airspaces of the lung. J Gen Physiol 2002;119:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Johnson MD, Widdicombe JH, Allen L, Barbry P, Dobbs LG. Alveolar epithelial type I cells contain transport proteins and transport sodium, supporting an active role for type I cells in regulation of lung liquid homeostasis. Proc Natl Acad Sci USA 2002;99:1966–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Borok Z, Liebler JM, Lubman RL, Foster MJ, Zhou B, Li X, Zabski SM, Kim K-J, Crandall ED. Sodium transport proteins are expressed by rat alveolar epithelial type I cells. Am J Physiol Lung Cell Mol Physiol 2002;282:L599–L608. [DOI] [PubMed] [Google Scholar]
- 85.Ridge KM, Olivera WG, Saldias F, Azzam Z, Horowitz S, Rutschman DH, Dumasius V, Factor P, Sznajder JI. Alveolar type 1 cells express the alpha2 Na,K-ATPase, which contributes to lung liquid clearance. Circ Res 2003;92:453–460. [DOI] [PubMed] [Google Scholar]
- 86.Lecuona E, Saldias F, Comellas A, Ridge K, Guerrero C, Sznajder JI. Ventilator-associated lung injury decreases lung ability to clear edema in rats. Am J Respir Crit Care Med 1999;159:603–609. [DOI] [PubMed] [Google Scholar]
- 87.Wiener-Kronish JP, Albertine KH, Matthay MA. Differential responses of the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. J Clin Invest 1991;88:864–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Eisner MD, Thompson T, Hudson LD, Luce JM, Hayden D, Schoenfeld D, Matthay MA and the ARDS Network. Efficacy of low tidal volume ventilation in patients with different clinical risk factors for acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;164:231–236. [DOI] [PubMed] [Google Scholar]
- 89.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest 2002;109:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Henson PM. Possible roles for apoptosis and apoptotic cell recognition in inflammation and fibrosis. Am J Respir Cell Mol Biol 2003;29:S70–S76. [PubMed] [Google Scholar]
- 91.Pittet JF, Griffiths MJ, Geiser T, Matthay MA, Sheppard D. TGF-beta is a critical mediator of acute lung injury. J Clin Invest 2001;107:1537–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hastings RH, Wright JR, Albertine KH, Ciriales R, Matthay MA. Effect of endocytosis inhibitors on alveolar clearance of albumin, immunoglobulin G, and SP-A in rabbits. Am J Physiol 1994;266:L544–L552. [DOI] [PubMed] [Google Scholar]
- 93.Frank J, Roux J, Kawakatsu H, Su G, Dagenais A, Berthiaume Y, Howard M, Canessa CM, Fang X, Sheppard D, et al. Transforming growth factor-β1 decreases expression of the epithelial sodium channel αENaC and alveolar epithelial vectorial sodium and fluid transport via an ERK1/2-dependent mechanism. J Biol Chem 2003;278:43939–43950. [DOI] [PubMed] [Google Scholar]
- 94.Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, Zimmerman GA, Matthay MA, Ware LB. Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol 2002;161:1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tomashefski JF Jr. Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med 2003;21:435–466. [DOI] [PubMed] [Google Scholar]
- 96.Goggel R, Winoto-Morbach S, Vielhaber G, Imai Y, Lindner K, Brade L, Brade H, Ehlers S, Slutsky AS, Schutze S, et al. PAF-mediated pulmonary edema: a new role for acid sphingomyelinase and ceramide. Nat Med 2004;10:155–160. [DOI] [PubMed] [Google Scholar]
- 97.Tasaka S, Koh H, Yamada W, Shimizu M, Ogawa Y, Hasegawa N, Yamaguchi K, Ishii Y, Richer SE, Doerschuk CM, et al. Attenuation of endotoxin-induced acute lung injury by the Rho-associated kinase inhibitor, Y-27632. Am J Respir Cell Mol Biol 2005;32:504–510. [DOI] [PubMed] [Google Scholar]
- 98.Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005;436:112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures: protection by positive end-expiratory pressure. Am Rev Respir Dis 1974;10:556–565. [DOI] [PubMed] [Google Scholar]
- 100.Dreyfuss D, Soler P, Basset G, Saumon G. High inflation pressure pulmonary edema: respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis 1998;137:1159–1164. [DOI] [PubMed] [Google Scholar]
- 101.Parker JC, Townsley MI, Rippe B, Taylor AE, Thigpen J. Increased microvascular permeability in dog lungs due to high peak airway pressures. J Appl Physiol 1984;57:1809–1816. [DOI] [PubMed] [Google Scholar]
- 102.Parsons PE, Eisner MD, Thompson BT, Matthay MA, Ancukiewicz M, Bernard GR, Wheeler AP; NHLBI Acute Respiratory Distress Syndrome Clinical Trials Network. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit Care Med 2005;33:1–6. [DOI] [PubMed] [Google Scholar]
- 103.Eisner MD, Parsons P, Matthay MA, Ware L, Greene K; Acute Respiratory Distress Syndrome Network. Plasma surfactant protein levels and clinical outcomes in patients with acute lung injury. Thorax 2003;58:983–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Parsons PE, Matthay MA, Ware LB, Eisner MD; National Heart, Lung, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials Network. Elevated plasma levels of soluble TNF receptors are associated with morbidity and mortality in patients with acute lung injury. Am J Physiol Lung Cell Mol Physiol 2005;288:L426–L431. [DOI] [PubMed] [Google Scholar]
- 105.Rubenfeld GD, Caldwell EC, Martin DM, Steinberg KP, Hudson LD. The incidence of acute lung injury in adults in the United States: Results of the King County Lung Injury Project. Am J Respir Crit Care Med 2002;165:A219. [Google Scholar]
- 106.Varki A, Rosenberg LE. Emerging opportunities and career paths for the young physician-scientist. Nat Med 2002;8:437–439. [DOI] [PubMed] [Google Scholar]