

Abstract
L-arginine is metabolized to nitric oxide (NO) by NO synthase (NOS), or to urea and L-ornithine by arginase. L-ornithine contributes to vascular remodeling in pulmonary hypertension via metabolism to polyamines and proline. Previously we found that cytokines upregulate both NOS and arginase in pulmonary arterial endothelial cells. We hypothesized that cytokine-induced arginase I and II expression depend on epidermal growth factor (EGF) receptor (EGFR) activity. Bovine pulmonary arterial endothelial cells were treated with lipopolysaccharide and tumor necrosis factor-α (L/T). L/T treatment resulted in a substantial increase in urea production, and this increase in urea production was potently inhibited by both genistein and AG1478, inhibitors of EGFR. Levels of arginase I protein and arginase II mRNA were increased in response to L/T treatment, and genistein prevented the L/T-induced elevations in both arginase I protein and arginase II mRNA levels. L/T treatment increased production of nitrites and inducible NOS mRNA accumulation, and genistein and AG1478 had little effect on these changes. EGF (50 ng/ml) treatment resulted in enhanced urea production. Finally, a 170-kD protein was phosphorylated upon treatment with either EGF or L/T. Our results indicate that arginase induction by L/T depends in part on EGFR activity. We speculate that EGFR inhibitors may attenuate vascular remodeling without affecting NO release, and thus may represent novel therapeutic modalities for pulmonary hypertensive disorders.
Keywords: nitric oxide, pulmonary hypertension, inducible nitric oxide synthase, cell signalling
Endothelial cells respond to inflammatory stimuli by releasing various substances, including nitric oxide (NO). NO and L-citrulline are produced by the NO synthases (NOS) from L-arginine (L-arg). At least two isoforms of NOS are expressed in endothelial cells: inducible NOS (iNOS or NOS2) and endothelial NOS (eNOS or NOS3) (1, 2). The NO produced by NOS causes vasodilation and can act as an antimitogenic agent to prevent vascular remodeling (3), thus preventing pulmonary hypertension. Furthermore, NO has a critical role in the antimicrobial action of the innate immune response (4, 5). Endothelial cells can also metabolize L-arg to urea and L-ornithine (L-orn) via arginase (6–8). At least two isoforms of arginase are expressed in endothelial cells: arginase I and arginase II (2, 9, 10). L-orn is required for proline and polyamine synthesis, which are necessary for cellular proliferation (8, 11). An increase in arginase activity is critical for the resolution of inflammatory injury and repair of tissue damage (12, 13). Thus, it has been postulated that in inflammatory diseases, such as acute respiratory distress syndrome (ARDS), NO production from L-arg is involved in the initial host response, whereas L-orn production from L-arg is involved in healing (8, 14, 15). However, polyamines and proline produced by L-orn metabolism may also be involved in lung fibrosis and vascular remodeling in chronic inflammatory lung diseases such as chronic obstructive pulmonary disease (COPD) and bronchopulmonary dysplasia (BPD).
Because arginase I and/or arginase II have important roles in tissue repair and remodeling, and NO production by NOS may decrease vascular smooth muscle proliferation, then arginase and NOS may have opposing effects on tissue repair and vascular remodeling. This led us to the question: Is arginase upregulated by a signal transduction module that does not affect NOS? Growth factor receptors have been shown to play a critical role in cell proliferation and tissue repair (16, 17). Because epidermal growth factor (EGF) receptor (EGFR) is expressed in endothelial cells (18) and involved in many cellular functions, including angiogenesis (19, 20), we hypothesized that treatment of pulmonary arterial endothelial cells (PAEC) with lipopolysaccharide and tumor necrosis factor-α (L/T) would result in EGFR-dependent arginase upregulation without affecting NOS. In the present article, we studied the role of EGFR in the regulation of L/T-mediated induction of both arginase and NOS in bovine PAEC (bPAEC) using assays of urea and nitrites, as well as immunoblotting and RT-PCR techniques. Our studies indicate that arginase I and II induction by inflammatory stimuli depends on EGFR activity, whereas NOS induction by inflammatory stimuli does not depend on EGFR.
MATERIALS AND METHODS
PAEC Culture
bPAEC were obtained from Clonetics (San Diego, CA), and were cultured in endothelial growth media (EGM; Clonetics), which contains ∼ 250 μM L-arg and 10% fetal bovine serum, as previously described (2, 6). bPAEC between passages 3 and 8 were used for these studies. On the day of study, bPAEC were washed three times with 4 ml of HEPES Balanced Salt Solution (HBSS; Clonetics). Then 5 ml of EGM was placed on the bPAEC (control) and the bPAEC were returned to the incubator at 37°C in 5% CO2, balance air for 24 h. In the L/T-treated bPAEC, 1.5 μg/ml LPS (from Escherichia coli serotype 0127:B8; Sigma Chemical, St. Louis, MO) and 1.5 ng/ml TNF-α (Sigma) was included in the EGM as previously described (2, 6). To examine the role of EGFR L/T bPAEC were treated with 10 μM genistein (Sigma), or 30 nM AG1478 (Calbiochem, San Diego, CA); the genistein or AG1478 were added to the bPAEC at the same time as the L/T was added. The dose of AG1478 was based on previous studies from our laboratory (21). After 24 h, the media was harvested and stored in 1-ml aliquots, frozen at −70°C.
bPAEC Protein Isolation
Protein was isolated from the bPAEC as previously described (2, 6). Briefly, bPAEC were washed twice with ice-cold HBSS and harvested in 750 μl ml of lysis buffer (0.2M NaOH, 0.2% SDS with the following added to each ml 30 min before use: 2 μg aprotinin, 5 μg leupeptin, 0.7 μg pepstatin A, and 174 μg phenylmethylsulfonyl fluoride). The bPAEC were scraped and 100-μl aliquots were stored at −70°C for subsequent Western blot analysis. Total protein concentration was determined by the Bradford method (BioRad, Hercules, CA).
bPAEC RNA Isolation
Total RNA was isolated from the bPAEC using Trizol as previously described (2, 6).
Urea Assay
The samples of medium were assayed in duplicate for urea concentration colorimeterically as previously described (2, 6).
Nitrite Assay
The samples of medium were assayed in duplicate for NO−2 using a chemiluminescence NO analyzer (Model 270B; Sievers Instruments, Boulder, CO) as previously described (2, 6).
Immunoblotting
The lysed bPAEC were assayed for arginase I protein using Western blot analysis as previously described (2, 6, 22). Aliquots of cell lysate were diluted 1:1 with SDS sample buffer, heated to 80°C for 15 min, and then centrifuged at 10,000 × g at room temperature for 2 min. Aliquots of the supernatant were used for SDS-PAGE. The proteins were transferred to PVDF membranes, and blocked overnight in PBS with 0.1% Tween (PBS-T) containing 5% skim milk and 3% bovine serum albumin. The membranes were incubated with a primary antibody against arginase I (1:1,000; Transduction Laboratories, San Diego, CA), or phosphotyrosine (4G10; Upstate Biotechnology, Lake Placid, NY) for 4 h and subsequently washed three times with PBS-T with 1% skim milk. The membranes were then incubated with the biotinylated IgG secondary antibody (1:5,000; Vector Laboratories, Burlingame, CA) for 1 h, washed, and then incubated with streptavidin–horseradish peroxidase conjugate (1:1,500; BioRad) for 30 min. The bands for arginase I, phospho-eNOS, or phosphotyrosine were visualized using chemiluminescence (Amersham ECL, Piscataway, NJ) and quantified using densitometry (Sigma Gel; Jandel Scientific, San Rafael, CA). To control for protein loading, the blots were stripped and reprobed for β-actin using a monoclonal antibody (1:10,000; Abcam, Cambridge, MA).
RT-PCR
RT-PCR for iNOS and arginase II was performed as previously described (2, 6). Briefly, 2 μg of total RNA were reverse-transcribed in a 40-μl reaction containing 2.5 μM dT16 (Applied Biosystems, Foster City, CA), 20 U AMV-RT, 1 mM dNTP, 1× Buffer (Promega, Madison, WI) and balance RNase-free water. The samples were incubated in a PCR-iCycler (BioRad) at 42°C for 60 min, 95°C for 5 min, and stored at −20°C. PCR reactions (total volume 50 μl) contained 5 μl of RT product, 1 mM MgCl2, 1.25 U AmpliTaqGold (Applied Biosystems), 0.2 mM dNTP (Promega), 0.3 μM forward (5′-TTGGTGTGATCTGGGTTGATGC-3′) and reverse (5′-TGCCTTCTCGATAGGTCAGTCC-3′) primers for arginase II, or 0.3 μM of forward (5′-TGGACTTGGCTACGGAACTGG-3′) and reverse (5′-TTCTGGTGAAGCGTGTCTTGG-3′) primers for iNOS were added to each sample. The mixed samples were heated to 94°C for 4 min, and then cycled as follows: 94°C for 1 min, 53°C for 1 min, and 72°C for 2 min for 35 cycles for arginase II and 38 cycles for iNOS. The PCR products were visualized and sized by 2.0% agarose gel electrophoresis and post-stained with Syber Gold (Molecular Probes, Eugene, OR) for 30 min. The gels were scanned and densitized using a MultiGenius Bio Imaging System (Syngene, Frederick, MD), and band density analysis was performed on a PC computer with SigmaGel (Jandel Scientific) software. The PCR product sizes were the expected 422 bp and 356 bp for arginase II and iNOS, respectively. Preliminary PCR reactions run at various total cycle numbers between 20 and 45 demonstrated that 35 and 38 total cycles were well within the linear range for each reaction product.
Statistical Analysis
Values are mean ± SE. One-way ANOVA was used to compare the effect of treatment on either production of nitrites or urea, and to compare the densitometry data between groups. Significant differences were identified using a Neuman-Keuls post hoc test. Differences were considered significant when P < 0.05.
RESULTS
Attenuation of L/T-Induced Urea Production by EGFR Inhibitors
We have previously shown that L/T treatment of bPAEC increased both NO and urea production (2, 6). To decipher the signaling pathways involved in this regulation, we examined the effects of EGFR inhibition on L/T-induced NO or urea production in bPAEC. bPAEC were washed three times and then 5 ml of either EGM (n = 10), EGM containing L/T (n = 11), or EGM containing L/T and 10 μM genistein (n = 11) was placed on the bPAEC. After 24 h the medium was removed and protein was isolated. The samples of medium were assayed for urea and NO−2 production. Consistent with our previous results (2, 6), L/T treatment resulted in greater production of urea than in control bPAEC, and genistein (10 μM) completely prevented the L/T-induced increase in urea production in bPAEC (Figure 1A). L/T treatment also resulted in greater NO−2 production than in control bPAEC, and interestingly genistein (10 μM) had little effect on the L/T-induced increase in NO−2 production in bPAEC (Figure 1B). In a second set of similar experiments, 30 nM AG1478 (n = 6 in each of the three groups), a highly selective inhibitor of EGFR, was substituted for genistein. AG1478 completely inhibited L/T-induced urea production (Figure 1C), but had little effect on L/T-induced production of nitrites (Figure 1D). These results suggest that EGFR kinase is involved in L/T-induced arginase upregulation but does not play a substantial role in L/T-induced NOS upregulation.
Figure 1.

Both genistein and AG1478 prevented the L/T-induced increase in urea production without affecting L/T-induced NO production. (A) production of urea and (B) production of nitrites NO−2 by control bPAEC, L/T-treated bPAEC, and L/T + 10 μM genistein–treated bPAEC after a 24-h incubation. *Different from control bPAEC, P < 0.05. (C) Production of urea and and (D) production of nitrites NO−2 by control bPAEC, L/T-treated bPAEC, and L/T + 30 nM AG1478–treated bPAEC after a 24-h incubation. *Different from control bPAEC, P < 0.05.
EGFR Inhibitors Attenuate L/T-Induced Arginase Expression
We have previously shown that L/T treatment increases arginase I protein levels and arginase II mRNA levels in bPAEC (6). To determine the role of EGFR in arginase I protein induction by L/T, bPAEC were grown in either EGM (n = 6), EGM containing L/T (n = 6), or EGM containing L/T and 10 μM genistein (n = 6) for 24 h. The protein lysates were analyzed for arginase I protein levels by immunoblotting as described above. Treatment of bPAEC with L/T increased arginase I protein levels by ∼ 2-fold (Figure 2). However, the addition of 10 μM genistein almost completely prevented the L/T-induced increase in arginase I protein levels in bPAEC (Figure 2).
Figure 2.

EGFR inhibition prevented the L/T-induced upregulation of arginase I protein levels. (A) Representative immunoblot. The top blot is probed for arginase I protein, and the bottom blot is the same blot reprobed for β-actin protein to demonstrate equal protein loading. The first two lanes of the blots are from control bPAEC, the next two lanes are from L/T-treated bPAEC, and the final two lanes are from L/T + 10 μM genistein–treated bPAEC. (B) The relative densitometry (density of arginase I band/density of β-actin band) for arginase I (n = 6 in each group). *Different from control, P < 0.05.
Currently there are no commercially available antibodies directed against bovine arginase II; therefore, we determined changes in arginase II mRNA expression. To examine the time course of L/T-induced changes in arginase II mRNA expression levels, total RNA was harvested from bPAEC 0.5, 1, 2, 4, 6, and 24 h after treatment with vehicle or L/T. Vehicle-treated bPAEC had very low levels of arginase II mRNA at all time-points tested (Figure 3). Conversely, L/T-treatment resulted in a rapid and sustained elevation in arginase II mRNA levels, which reached peak levels at 4 h, and these peak levels were maintained throughout the 24-h experimental period (Figure 3). To examine the effect of EGFR inhibitors on arginase II mRNA levels, total RNA was harvested from bPAEC after 24 h and arginase II mRNA levels were determined by RT-PCR. Control bPAEC had detectable arginase II mRNA, and treatment with L/T resulted in an increase in arginase II mRNA levels (Figure 4). Similar to the effect of genistein on arginase I protein levels, treatment with 10 μM genistein essentially prevented the L/T-induced increase in arginase II mRNA expression at 6 and 24 h after treatment (Figure 4). Treatment with AG1478 also prevented the L/T-induced increase in arginase II mRNA levels (Figure 4C).
Figure 3.

L/T treatment resulted in a time-dependent and sustained upregulation of arginase II mRNA expression in bPAEC. (A) A representative RT-PCR for arginase II from control bPAEC and L/T-treated bPAEC, there is the appearance of an easily determined band by 4 h in the L/T-treated bPAEC. (B) Normalized density for AII bands from L/T-treated bPAEC demonstrate a time-dependent increase in expression starting at ∼ 2 h, with a sustained peak between 6 and 24 h. The data were fit with an exponential of the form y = a(1 − e−bt), which demonstrated a significant relationship with AII density and time in the L/T-treated bPAEC, r = 0.98, P < 0.001, with a time constant of ∼ 2.5 h. On the other hand, there was no significant relationship with AII density and time in the control bPAEC, r = 0.63, P = 0.17. A straight line was also fit to the control data, which also demonstrated no correlation between AII density in control bPAEC and time (y = 0.002x + 0.12, r = 0.62, P = 0.19). n = 3 for each time-point, *different from 0.5 h time-point in same group, P < 0.01. # L/T different from control at same time-point, P < 0.05.
Figure 4.
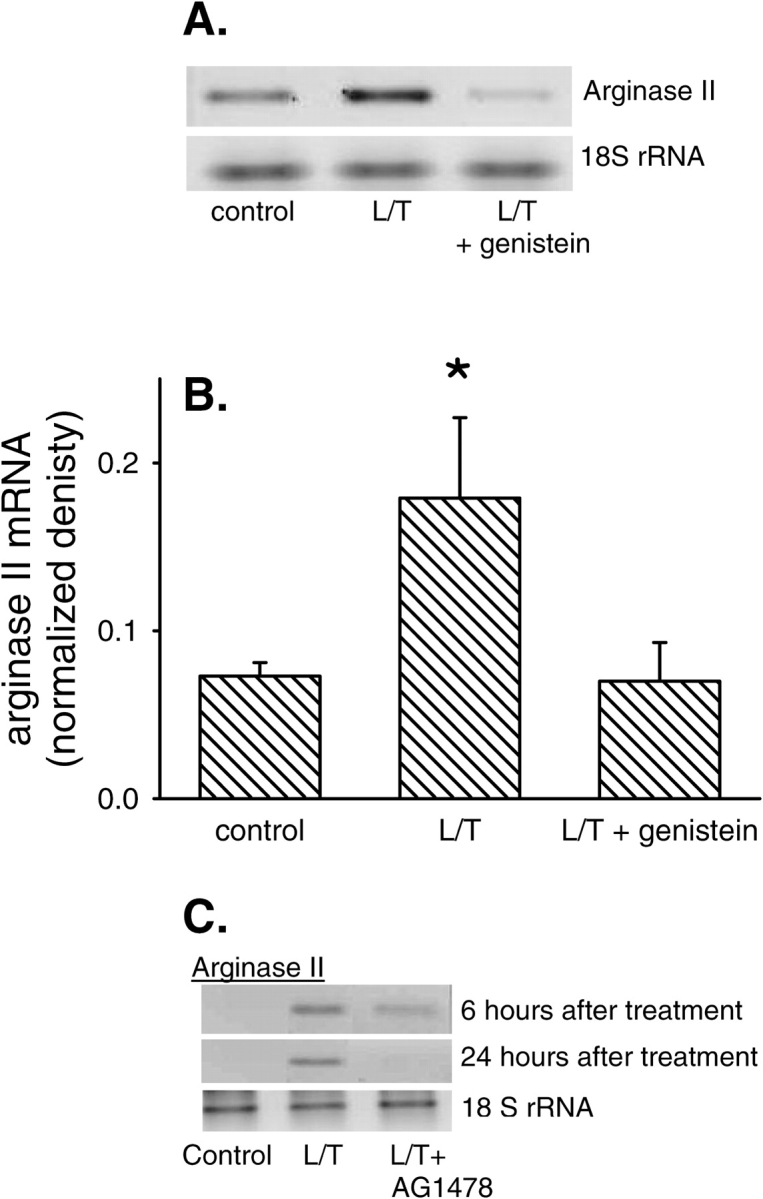
EGFR inhibition prevented the L/T-induced upregulation of arginase II mRNA levels. (A) A representative RT-PCR gel for arginase II after L/T treatment from control, L/T and L/T + AG1478 treated bPAEC 24 h after treatment. (B) The normalized densitometry (density of arginase II mRNA band/density of 18S rRNA band) for arginase II mRNA bands from RT-PCR at 24 h after L/T treatment from control, L/T-, and L/T + genestein–treated bPAEC (n = 6 in each group). *Different from control, P < 0.05. (C) A representative RT-PCR gel for arginase II from control, L/T-, and L/T + AG1478–treated bPAEC 6 and 24 h after treatment. AG 1478 prevented L/T-induced arginase II expression in these bPAEC.
EGFR Inhibitors Have Little Effect on L/T-Induced NOS Expression
We have previously shown that L/T treatment increases eNOS and iNOS expression in bPAEC (2). To examine the time course of L/T-induced changes in iNOS mRNA expression levels, total RNA was harvested from bPAEC 0.5, 1, 2, 4, 6, and 24 h after treatment with vehicle or L/T. Vehicle-treated bPAEC had very low levels of iNOS mRNA at all time-points tested (Figure 5). Conversely, L/T-treatment resulted in a rapid elevation in iNOS mRNA levels, which reached peak levels at 4 and 6 h, and the iNOS mRNA levels decreased by 24 h (Figure 5). In a similar series of experiments after 24 h bPAEC were harvested for total RNA, and iNOS mRNA levels were determined by RT-PCR (n = 3 in each group). Basal levels of iNOS mRNA were very low. However, L/T treatment resulted in a substantial increase in iNOS mRNA levels. Treatment with genistein moderately inhibited the L/T-induced upregulation of iNOS mRNA (Figure 6A), suggesting that EGFR activity may contribute to iNOS induction in bPAEC. However, the levels of iNOS mRNA in the L/T + genistein–treated bPAEC were significantly greater than in controls and did not differ statistically from the iNOS mRNA levels in the L/T-treated bPAEC (Figure 6B). Treatment with AG1478 also resulted in a moderate attenuation of the L/T-induced upregulation of iNOS mRNA levels (Figure 6C).
Figure 5.

L/T treatment resulted in a time-dependent upregulation of iNOS mRNA expression in bPAEC. (A) A representative RT-PCR for iNOS from control bPAEC and L/T-treated bPAEC. In the L/T-treated bPAEC there is the appearance of an easily detected band which peaks at 4–6 h, and band intensity decreases but remains significantly greater than controls at 24 h. (B) Normalized density for iNOS bands from L/T-treated bPAEC demonstrate a time-dependent increase in expression starting at ∼ 2 h, with a peak between 4 and 6 h. The band intensity decreases by 24 h but remains well above levels seen in the control bPAEC (n = 3–5 for each time-point). *Different from 0.5 h time-point in same group, P < 0.01. + different from previous time in same group, P < 0.05.
Figure 6.

EGFR inhibition did not prevent the L/T-induced upregulation of iNOS mRNA expression levels. (A) Representative RT-PCR gel for iNOS. L/T-treatment resulted in readily discernable bands, and 10 μM genistein did not prevent the appearance of discernable iNOS bands. The 18S rRNA bands demonstrate equal RNA loading. (B) The normalized densitometry (density of iNOS bands/density of 18S rRNA bands) data for the iNOS bands in the three conditions. *Different from control, P < 0.01. (C) Representative RT-PCR gel for iNOS. L/T treatment resulted in readily discernable bands, and 10 μM AG1478 did not prevent the appearance of discernable iNOS bands. The 18S rRNA bands demonstrate equal loading of lanes in gel.
Direct Effects of EGFR Activation on Urea and NO2− Production
To determine if EGFR activation by EGF would result in increased urea production, the following experiment was done. bPAEC were grown in either EGM (n = 9), EGM containing L/T (n = 8) as a positive control, or EGM containing 50 ng/ml EGF (n = 8). After 24 h the medium was harvested for determination of nitrites and urea, and the protein was harvested as described above. Treating bPAEC with EGF (50 ng/ml) resulted in a substantial increase in urea production. In fact, the urea production due to EGF treatment was similar to that after L/T treatment (Figure 7A). Although we found that L/T treatment increased production of nitrites, the production of nitrites by bPAEC treated with EGF was not different from the production of nitrites by control bPAEC (Figure 7B).
Figure 7.

Treatment of bPAEC with EGF increased urea production. (A) demonstrates urea production and (B) demonstrates production of nitrites NO−2 after a 24-h incubation of bPAEC in either normal medium (control), L/T added to medium as a positive control, or 50 ng/ml EGF added to medium. *Different from control, P < 0.05.
Stimulation of EGFR Tyrosine Phosphorylation by L/T
We tested a panel of antibodies (EGFR sc-03, p-EGFR sc-23420-R, sc-23421-R, and sc21429-R [Santa Cruz Biotechnology; EGFR catalog #610016, BD Biosciences; and EGFR catalog #05–104, Upstate]) recognizing either total or phosphorylated human EGFR in HeLa cells (data not shown), but none of them reacted with bPAEC EGFR. Thus, to examine whether L/T treatment enhances EGFR activation, bPAEC were serum starved overnight by reducing the fetal bovine serum to 0.5%, then treated with EGF (50 ng/ml) for 5, 15, 30, 60, or 1,440 min. The protein lysates from these bPAEC were subjected to immunoblotting for protein tyrosine phosphorylation using an antibody against phosphotyrosine (4G10; Upstate Laboratories, Charlottesville, VA). In control bPAEC, tyrosine phosphorylation of a protein in the 170-kD range was almost undetectable. However, upon EGF-stimulation a protein of ∼ 170 kD became tyrosine-phosphorylated within 15 min, and the tyrosine phosphorylation was sustained (Figure 8A). This finding is consistent with phosphorylation of EGFR in these bPAEC after EGF treatment. In a similar set of experiments, bPAEC were serum starved overnight and then treated with L/T for 5, 15, 30, 60, or 1,440 min. The protein was harvested and immunoblotting with 4G10 was performed. After 15 min there was phosphorylation of a 170-kD band, and by 60 min the phosphorylation of this 170-kD band began to wane (Figure 8B). Importantly, tyrosine phosphorylation of this 170-kD protein was blocked by genistein and AG1478. bPAEC were treated as above with 50 ng/ml EGF after serum starvation; some bPAEC had 10 μM genistein added to the medium and some bPAEC had 30 nM AG1478 added to the medium. After 60 min, protein was harvested for immunoblotting with 4G10. Figure 8C demonstrates that EGF-induced tyrosine phosphorylation of the 170-kD protein was prevented by both genistein and AG1478. Taken together, these experiments strongly suggest that EGFR plays an important role in mediating L/T-induced upregulation of urea production in bPAEC.
Figure 8.

Treatment of bPAEC with either EGF or L/T resulted in tyrosine phosphorylation of a 170-kD protein. (A) Treatment of bPAEC with EGF resulted in tyrosine phosphorylation of a 170-kD protein. Immunoblot for phosphotyrosine (4G10; Upstate Biotechnology) of bPAEC protein isolated 0, 5, 15, 30, 60, and 1,440 min after adding EGF (50 ng/ml) to medium. The bPAEC were serum starved overnight by reducing the fetal bovine serum in the medium to 0.5% from 10%. (B) Treatment of bPAEC with L/T resulted in tyrosine phosphorylation of a 170-kD protein. Immunoblot for phosphotyrosine is shown in the top panel and the mean densities are shown in the bottom panel. The bPAEC were serum starved (0.5%) overnight and protein harvested 5, 15, 30, 60, and 1,440 min after treatment with L/T. (C) Treatment with either genistein (10 μM) or AG1478 (30 nM) prevented the EGF-induced increase in phosphorylation of a 170-kD protein. All bPAEC were treated with 50 ng/ml EGF after serum starvation overnight; some bPAEC had 10 μM genistein added to the medium, and some had 30 nM AG1478 added to the medium. After 60 min, protein was harvested for immunoblotting for phosphotyrosine. The first two lanes are from EGF-treated bPAEC, the next two lanes are from EGF + genistein–treated bPAEC, and the final two lanes are from EGF + AG1478–treated bPAEC.
DISCUSSION
The main findings of this study in bPAEC were that: (1) pharmacologic inhibition of EGFR prevented the L/T-induced increase in urea, but not NO, production; (2) pharmacologic inhibition of EGFR prevented the L/T-induced increase in both arginase I protein and arginase II mRNA levels; (3) pharmacologic inhibition of EGFR did not prevent the L/T-induced increase in iNOS mRNA levels; (4) addition of EGF to the culture medium increased production of urea, but not of NO; and (5) addition of either EGF or L/T resulted in the phosphorylation of a 170-kD protein, which was prevented by pharmacologic inhibition of EGFR. Taken together, these results demonstrate that EGFR activation by L/T in bPAEC results in upregulation of arginase without having a major affect on iNOS. These findings support our hypothesis that EGFR represents a proximal signal transduction module that differentially affects arginase and iNOS expression and/or activation in response to cytokines in endothelial cells.
We utilized genistein and AG1478 to pharmacologically inhibit EGFR activity. Genistein, an isoflavone, was first described in 1987 as an EGFR tyrosine kinase inhibitor, which prevented EGF-stimulated phosphotyrosine levels in A431 cells (23). In a bronchial epithelial cell line, genistein inhibited EGFR phosphorylation caused by either EGF or H2O2 stimulation (24). AG1478 is a tyrophostin, which was described in 1989 as a potent and selective EGFR protein kinase inhibitor (25). AG1478 has been shown to prevent EGF-induced EGFR phosphorylation and growth arrest in vascular smooth muscle cells (26). We found that treatment of cultured endothelial cells with the EGFR inhibitors genistein and AG1478, led to alterations in L/T-induced protein expression, strongly suggesting that EGFR is involved in L/T-triggered signal transduction in bPAEC. We attempted to directly examine EGFR protein expression and alterations in EGFR phosphorylation in bPAEC. However, currently there is no commercially available antibody against bovine EGFR. Although both genistein and AG1478 may have nonspecific effects, the fact that both genistein and AG1478 inhibited EGF-induced phosphorylation of a 170-kD protein and blocked L/T-induced urea production and arginase II mRNA expression strongly support an inhibitory effect on EGFR in these bPAEC.
There is a great deal of information on the role of EGFR in tumor growth and vascularization associated with tumor growth. Nonetheless, there is relatively little information on the function of EGFR in endothelial cells. In human umbilical vein endothelial cells (HUVEC), it has been shown that H2O2 or EGF treatment results in EGFR phosphorylation and subsequent c-Jun N-terminal kinase (JNK) activation (18). Similarly, it has been reported that in human dermal microvascular endothelial cells EGF treatment results in EGFR phosphorylation (20), which is associated with cytoskeletal alterations and cell migration. Furthermore, Fujiyama and coworkers (19) have demonstrated that angiotensin II stimulation of rat cardiac microvascular endothelial cells results in EGFR phosphorylation and increases expression of proteins involved in angiogenesis. Thus, although we were unable to directly demonstrate EGFR phosphorylation in our bovine cell culture system, our results with genistein, AG1478, and EGF, taken in context with data from the literature, support a role for EGFR in the upregulation of urea production in bPAEC. Our findings suggest that EGFR does not appear to play a major role in mediating NOS regulation in bPAEC. First, pharmacologic inhibition of EGFR had relatively little effect on L/T-induced NO production or iNOS expression. Second, the addition of EGF to the culture medium did not result in alterations in NO production.
The biological significance of increased urea production in the L/T-treated bPAEC remains unclear. We speculate that arginase could represent a molecular mechanism that cells use to attenuate NO production and dampen inflammatory responses (27, 28). That is, by upregulating arginase, cells would limit the availability of L-arg to NOS and thereby decrease NO production. Consistent with this concept are previous studies in which inhibiting arginase activity led to enhanced NO production (4, 6). Furthermore, increased arginase activity may favor the formation of polyamines and/or L-proline from L-orn, which are important in tissue repair after injury (7, 11). For example, in murine macrophages and mice it has been found that Th2 cytokines are potent inducers of arginase, whereas Th1 cytokines are potent inducers of iNOS (5, 12, 29). Given the key role of arginase in polyamine and proline synthesis, the induction of arginase independent of the induction of NOS may be vital to allow for cellular proliferation (8). Supporting this concept, Ignarro and colleagues (13) recently reported that treatment of vascular smooth muscle cells with NO donors decreased cellular proliferation, whereas overexpression of arginase I in these cells enhanced cellular proliferation. Furthermore, it has been shown recently in human endothelial cells that EGFR activation was necessary for cellular proliferation (20), and inhibiting arginase decreased cellular proliferation (30). Thus, the upregulation of arginase may be an important mechanism to prevent damage associated with excessive NO production and augment tissue repair in cytokine-associated lung diseases such as ARDS.
On the other hand, cellular proliferation may lead to vascular remodeling and/or lung fibrosis. There is evidence that EGFR has a central role in the signaling cascade involved in lung fibrosis. For example, treatment with AG1478 prevented both vanadium pentoxide-induced lung fibrosis in rats (31) and lung remodeling in an ovalbumin model of asthma in mice (32). Similarly, an increase in arginase activity may have negative consequences in diseases characterized by pulmonary hypertension. Arginase may limit endothelial NO production and increase vascular cell proliferation, thus contributing to vascular constriction and vascular remodeling. This is supported by the findings that vascular smooth muscle cells transfected with arginase I exhibit enhanced proliferation (13). Moreover, endothelial cells from humans with primary pulmonary hypertension were found to express higher levels of arginase protein and to produce less NO than in cells from normal control subjects, despite similar levels of eNOS protein (33). In this regard, inhibiting signal transduction pathways leading to arginase without affecting NOS levels may be beneficial for patients with pulmonary hypertension. We postulate that pharmacologic inhibition of EGFR may represent a viable therapeutic strategy in diseases characterized by fibrosis or pulmonary hypertension.
This study was supported by grants from the National Heart Lung and Blood Institute HL-04050 (L.G.C.), and from the National Institute of Allergy and Infectious Diseases AI-57798 (Y.L.).
Conflict of Interest Statement: None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Buga GM, Singh R, Pervin S, Rogers NE, Schmitz DA, Jenkinson CP, Cederbaum SD, Ignarro LJ. Arginase activity in endothelial cell: Inhibition by NG-hydroxy-L-arginine during high-output NO production. Am J Physiol Heart Circ Physiol 1996;271:H1988–H1998. [DOI] [PubMed] [Google Scholar]
- 2.Nelin LD, Nash HE, Chicoine LG. Cytokine treatment increases arginine metabolism and uptake in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol 2001;281:L1232–L1239. [DOI] [PubMed] [Google Scholar]
- 3.Michelakis ED. The role of the NO axis and its therapeutic implications in pulmonary arterial hypertension. Heart Fail Rev 2003;8:5–21. [DOI] [PubMed] [Google Scholar]
- 4.Chang IC, Liao JC, Kuo L. Arginase modulated nitric oxide production in activated macrophages. Am J Physiol Heart Circ Physiol 1998;274:H342–H348. [DOI] [PubMed] [Google Scholar]
- 5.Chang IC, Zoghi B, Liao JC, Kuo L. The involvement of tyrosine kinases, cyclic AMP/protein kinase A, and p38 mitogen-activated protein kinase in IL-13-mediated arginase I induction in macrophages: Its implications in IL-13-inhibited nitric oxide production. J Immunol 2000;165:2134–2141. [DOI] [PubMed] [Google Scholar]
- 6.Chicoine LG, Paffett ML, Young TL, Nelin LD. Arginase inhibition increases nitric oxide production in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol 2004;287:L60–L68. [DOI] [PubMed] [Google Scholar]
- 7.Li H, Meininger CJ, Kelly KA, Hawker JR, Morris SM, Wu G. Activities of arginase I and II are limiting for endothelial cell proliferation. Am J Physiol Regul Integr Comp Physiol 2002;282:R64–R69. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Meininger CJ, Hawker JR, Haynes TE, Kepka-Lenhart D, Mistry SK, Morris SM, Wu G. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Endocrinol Metab 2001;280:E75–E80. [DOI] [PubMed] [Google Scholar]
- 9.Louis CA, Reichner JS, Henry WL, Mastrofrancesco B, Gotoh T, Mori M, Albina JE. Distinct arginase isoforms expressed in primary and transformed macrophages: regulation by oxygen tension. Am J Physiol Regul Integr Comp Physiol 1998;274:R775–R782. [DOI] [PubMed] [Google Scholar]
- 10.Que LG, Kantrow SP, Jenkinson CP, Piantadosi CA, Huang YCT. Induction of arginase isoforms in the lung during hyperoxia. Am J Physiol Lung Cell Mol Physiol 1998;275:L96–L102. [DOI] [PubMed] [Google Scholar]
- 11.Janne J, Alhonen L, Leinonen P. Polyamines: from molecular biology to clinical applications. Ann Med 1991;23:241–259. [DOI] [PubMed] [Google Scholar]
- 12.Morris SM, Kepka-Lenhart D, Chen LC. Differential regulation of arginases and inducible nitric oxide synthase in murine macrophage cells. Am J Physiol Endocrinol Metab 1998;275:E740–E747. [DOI] [PubMed] [Google Scholar]
- 13.Ignarro LJ, Buga GM, Wei LH, Bauer PM, Wu G, del Soldato P. Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc Natl Acad Sci USA 2001;98:4202–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salimuddin, A Nagasaki, Gotoh T, Isobe H, Mori M. Regulation of the genes for arginase isoforms and related enzymes in mouse macrophages by lipopolysaccharide. Am J Physiol Endocrinol Metab 1999;277:E110–E117. [DOI] [PubMed] [Google Scholar]
- 15.Schearer JD, Richards JR, Mills JD, Caldwell MD. Differential regulation of macrophage arginine metabolism: a proposed role in wound healing. Am J Physiol Endocrinol Metab 1997;272:E181–E190. [DOI] [PubMed] [Google Scholar]
- 16.Geiser T, Jarreau PH, Atabai K, Matthay MA. Interleukin-1β augments in vitro alveolar epithelial repair. Am J Physiol Lung Cell Mol Physiol 2000;279:L1184–L1190. [DOI] [PubMed] [Google Scholar]
- 17.Madtes DK, Busby HK, Strandjord TP, Clark JG. Expression of transforming growth factor-alpha and epidermal growth factor receptor is increased following bleomycin-induced lung injury in rats. Am J Respir Cell Mol Biol 1994;11:540–551. [DOI] [PubMed] [Google Scholar]
- 18.Chen K, Vita JA, Berk BC, Keaney JF Jr. c-Jun N-terminal kinase activation by hydrogen peroxide in endothelial cells involves src-dependent epidermal growth factor receptor transactivation. J Biol Chem 2001;276:16045–16050. [DOI] [PubMed] [Google Scholar]
- 19.Fujiyama S, Matsubara H, Nozawa Y, Maruyama K, Mori Y, Tsutsumi Y, Masaki H, Uchiyama Y, Koyama Y, Nose A, et al. Angiotensin AT1 and AT2 receptors differentially regulate angiopoietin-2 and vascular endothelial growth factor expression and angiogenesis by modulating heparin binding-epidermal growth factor (EGF)-mediated EGF receptor transactivation. Circ Res 2001;88:22–29. [DOI] [PubMed] [Google Scholar]
- 20.Schraufstatter IU, Trieu K, Sikora L, Sriramarao P, DiScipio R. Complement C3a and C5a induce different signal transduction cascades in endothelial cells. J Immunol 2002;169:2102–2110. [DOI] [PubMed] [Google Scholar]
- 21.Chen W, Martindale JL, Holbrook NJ, Liu Y. Tumor promoter arsenite activates extracellular signal-regulated kinase through a signaling pathway mediated by epidermal growth factor receptor and Shc. Mol Cell Biol 1998;18:5178–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y. Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J Immunol 2002;169:6408–6416. [DOI] [PubMed] [Google Scholar]
- 23.Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem 1987;262:5592–5595. [PubMed] [Google Scholar]
- 24.Goldkorn T, Balaban N, Matsukuma K, Chea V, Gould R, Last J, Chan C, Chavez C. EGF-receptor phosphorylation and signaling are targeted by H2O2 redox stress. Am J Respir Cell Mol Biol 1998;19:786–798. [DOI] [PubMed] [Google Scholar]
- 25.Gazit A, Yaish P, Gilon C, Levitzki A. Tyrphostins I: synthesis and biological activity of protein tyrosine k inase inhibitors. J Med Chem 1989;32:2344–2352. [DOI] [PubMed] [Google Scholar]
- 26.Gui Y, Zheng XL. Epidermal growth factor induction of phenotype-dependent cell cycle arrest in vascular smooth muscle cells is through the mitogen-activated protein kinase pathway. J Biol Chem 2003;278:53017–53025. [DOI] [PubMed] [Google Scholar]
- 27.Koga T, Koshiyama Y, Gotoh T, Yonemura N, Hirata A, Tanihara H, Negi A, Mori M. Coinduction of nitric oxide synthase and arginine metabolic enzymes in endotoxin-induced uveitis rats. Exp Eye Res 2002;75:659–667. [DOI] [PubMed] [Google Scholar]
- 28.Sonoki T, Nagasaki A, Gotoh T, Takiguchi M, Takeya M, Matsuzaki H, Mori M. Coinduction of nitric oxide synthase and arginase I in cultured rat peritoneal macrophages and rat tissues in vivo by lipopolysaccharide. J Biol Chem 1997;272:3689–3693. [DOI] [PubMed] [Google Scholar]
- 29.Modolell M, Corraliza IM, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur J Immunol 1995;25:1101–1104. [DOI] [PubMed] [Google Scholar]
- 30.Bachetti T, Comini L, Francolini G, Bastianon D, Valetti B, Cadei M, Grigolato P, Suzuki H, Finazzi D, Albertini A, et al. Arginase pathway in human endothelial cells in pathophysiological conditions. J Mol Cell Cardiol 2004;37:515–523. [DOI] [PubMed] [Google Scholar]
- 31.Rice AB, Moomaw CR, Morgan DL, Bonner JC. Specific inhibitors of platelet-derived growth factor or epidermal growth factor receptor tyrosine kinase reduce pulmonary fibrosis in rats. Am J Pathol 1999;155:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vargaftig BB, Singer M. Leukotrienes mediate part of Ova-induced lung effects in mice via EGFR. Am J Physiol Lung Cell Mol Physiol 2003;285:L808–L815. [DOI] [PubMed] [Google Scholar]
- 33.Xu W, Kaneko FT, Zheng S, Comhair SAA, Janocha AJ, Goggans T, Thunnissen FBJM, Farver C, Hazen SL, Jennings C, et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J 2004;18:1746–1748. [DOI] [PubMed] [Google Scholar]