

Abstract
Lung injury is associated with increased concentrations of hyaluronan (hyaluronic acid, HA). HA modifies cell behavior through interaction with cell-associated receptors such as receptor for HA-mediated motility (RHAMM, CD168). Using a function blocking anti-RHAMM antibody (R36), we investigated the expression and role of RHAMM in the inflammatory response to intratracheal bleomycin in rats. Immunostaining showed increased expression of RHAMM in macrophages 4–7 d after injury. Surface biotin labeling of cells isolated by lavage confirmed increased surface expression of a 70-kD RHAMM after lung injury, and in situ hybridization demonstrated increased RHAMM mRNA in macrophages responding to injury. Time-lapse cinemicrography demonstrated a 5-fold increase in motility of alveolar macrophages from bleomycin-treated animals that was completely blocked by R36 in vitro. Further, HA-stimulated macrophage chemotaxis was also inhibited by R36. Daily administration of R36 to injured animals resulted in a 40% decrease in macrophage accumulation 7 d after injury. Further, H&E staining of tissue sections showed that bleomycin-mediated changes in lung architecture were improved with R36 treatment. Taken together with previous results showing the inhibitory effects of HA-binding peptide on inflammation and fibrosis, we conclude that the interaction of RHAMM with HA is a critical component of the recruitment of inflammatory cells to the lung after injury.
Keywords: lung injury, bleomycin, inflammation, hyaluronan, RHAMM
Acute lung injury is characterized by an initial inflammatory response that precedes pulmonary fibrosis (1–3). Pulmonary fibrotic disorders such as acute respiratory distress syndrome (ARDS) in adults and bronchopulmonary dysplasia in infants are characterized by a chronic inflammatory state consisting largely of macrophages (4, 5). The prevailing model of acute lung injury asserts that injury to the epithelium results in the release of numerous inflammatory mediators, promoting the influx initially of neutrophils and subsequently macrophages into sites of injury, with further increases in cytokine production and modulation of the extracellular matrix, including fibronectin, elastin, hyaluronan, and collagen (6–10).
Hyaluronan (hyaluronic acid, HA) is a nonsulfated glycosaminoglycan that consists of a polymer of repeating disaccharide units of N-acetyl glucosamine and glucuronic acid (11). Increased HA concentrations are found coincident with periods of rapid cell proliferation and migration such as developing, regenerating, and remodeling tissues, as well as in tumorigenesis (reviewed in Ref. 10). Specifically, increased recovery of HA has been shown in bronchoalveolar lavage (BAL) from fibrotic lung diseases such as sarcoidosis (12), occupational lung disorders (13), and ARDS (14). Interestingly, HA-binding peptides administered to rodents have been shown to inhibit macrophage cell motility and reduce inflammation and fibrosis during skin wound repair (15), in the carageenen model of cutaneous inflammation (16), and after acute lung injury (17). These data suggest that HA is an integral part of the cause rather than a consequence of the inflammatory response to tissue injury.
HA modifies cell behavior through interactions with at least two molecularly distinct cell-associated receptors. CD44, a widely distributed cell surface glycoprotein (18, 19), exists in several molecular forms derived by alternate splicing of a single mRNA and exhibits different HA-binding characteristics depending upon the cell type and conditions involved (20). It has been implicated in lymphocyte homing to tissues (21), tumorigenesis (22), and is clearly expressed in tissues undergoing repair after wounding (23). In acute lung injury, CD44 appears to mediate fibroblast migration (24) and HA-induced gene expression in mouse alveolar macrophages (25). CD44-deficient mice challenged with noninfectious lung injury exhibit sustained infiltration of inflammatory cells within the alveolar interstitium, increased mortality, and LMW HA accumulation at 14 d, as well as impaired clearance of neutrophils in association with decreased TGF-β activation (26). This phenotype was significantly reversed with bone marrow reconstitution from CD44+/+ mice, suggesting an important role for CD44 in HA clearance after lung injury and resolution of the inflammatory response after tissue injury (26).
Since recruitment of inflammatory cells to the lung after injury is not affected in the absence of CD44, and blockade of HA inhibits macrophage accumulation, an HA receptor other than CD44 must mediate inflammatory cell accumulation. Receptor for HA-mediated motility (RHAMM) is an HA receptor expressed at the cell surface that regulates cell locomotion and proliferation (15, 27–31). In several injury models, RHAMM and HA are overexpressed in macrophages (15, 31), fibroblasts (32), epithelial cells (15), and smooth muscle cells (29) responding to injury. We therefore focused on the expression and role of RHAMM in inflammation using the function-blocking anti-RHAMM antibody, R36. We hypothesized that RHAMM plays an important role in the recruitment of inflammatory cells to the lung after bleomycin injury.
MATERIALS AND METHODS
Reagents and Macrophage Cell Line
A rabbit polyclonal anti-RHAMM antibody, R36, was raised against amino acids 585–605 encoded in the full-length RHAMM cDNA (33, 34) and purified to IgG fraction. Its specificity for RHAMM has been demonstrated previously (28, 29).
A six-mer hyaluronan oligosaccharide (HA-6) was the kind gift of Dr. Akira Asari (Seikagaku Corporation, Tokyo, Japan). This oligosaccharide is produced by enzymatic digestion of high-molecular-weight HA extracted from rooster comb and purified using high-performance liquid chromatography. HA-6 was demonstrated to be free of endotoxin, protein, and DNA, and the molecular size and purity were confirmed by fluorescence-assisted carbohydrate electrophoresis analysis and gas chromatography/mass spectroscopy (data not shown).
RAW 264.7 murine macrophages were obtained from ATCC (Cat# TIB-71; Rockville, MD). Cells were maintained in DMEM supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 100 μg/ml streptomycin, and 100 U/ml penicillin at 37°C under 5% CO2.
Animals
The Institutional Animal Care and Utilization Committees at both The Children's Hospital of Philadelphia and The University of Pennsylvania School of Medicine approved all animal care procedures. Six-week-old (200–250 g) male Sprague-Dawley rats (Charles River Breeding Laboratories, N. Wilmington, MA) were housed in the Animal Care Facility of The Children's Hospital of Philadelphia under standard conditions with free access to food and water. Animals were anesthetized with ketamine:xylazine:atropine (16:8:0.01 mg/kg), and the trachea was visualized through a vertical incision in the neck. Using an insulin syringe, 250 μl of either LPS-free saline or 8 U/kg bleomycin sulfate (Bristol Myers Squibb, Princeton, NJ) in 250 μl of saline was injected into the trachea. The incision was closed with surgical clips, and animals were immediately injected intraperitoneally with either 0.5 mg of anti-RHAMM antibody (R36) or nonimmune IgG. Antibody treatment was continued with daily injections of 0.5 mg of either R36 or IgG for either 3 or 6 d. At least three animals were examined for each condition, including one set that remained unmanipulated throughout.
Animals were killed on Days 4 or 7 after injury. BAL was obtained (36 ml/kg, total lung capacity) and immediately centrifuged at 500 × g for 10 min to remove all cells and cellular debris. Approximately 80–90% of lavage was routinely recovered. The cellular pellet and the supernatant were separated and frozen at −70°C until further analysis. The pulmonary artery was perfused with PBS to remove all blood from the lungs. The left lung was either inflated to 25 cm H2O with 1% paraformaldehyde and placed in 10% neutral formalin for full fixation before processing for paraffin sectioning, or inflated with OCT/PBS solution to 25 cm H2O and immediately blocked in OCT and stored at −70°C for frozen sectioning. The three lobes of the right lung were immediately frozen in liquid nitrogen and stored at −70°C.
Immunostaining
RHAMM expression after bleomycin injury was determined using the Rabbit IgG Vectastain ABC kit (Vector Laboratories, Inc., Burlingame, CA) as previously described (17). Briefly, after removal of paraffin and stepwise hydration, slides were blocked with goat serum per manufacturer instructions. Primary antibody (R36, 25 μg/ml) was applied overnight at 4°C in a humid slide box. After washes in 0.01 M TBS and 0.01 M TBS/0.1% Tween, endogenous peroxidase activity was blocked with 0.6% hydrogen peroxide/methanol for 30 min at room temperature. Secondary antibody, biotinylated goat anti-rabbit IgG, was applied for 1 h at room temperature, and specific staining was obtained with avidin–biotin complex (ABC) and diaminobenzadine (DAB). Staining was enhanced with 0.5% CuSO4 in 0.9% NaCl and slides were counterstained with 0.25% methyl green in ddH2O. Slides were dipped quickly in n-Butanol, then xylene, and finally mounted with Permount (Fisher Scientific, Fair Lawn, NJ). Frozen sections were processed similarly except that they did not require removal of paraffin and rehydration, and were studied using immunofluorescence. Experiments to determine the changes in expression of RHAMM over time after intratracheal treatments were repeated three times.
Macrophage accumulation was quantified by immunofluorescent staining with the rat macrophage–specific marker ED1 (Serotec, Raleigh, NC). Endogenous fluorescence was blocked with separate exposure to sodium borohydrate (1 g/10 ml PBS, 3 min × 2) and 1 M glycine in PBS for 30 min. Nonspecific sites were blocked with PBS/100% goat serum/0.02% azide for 30 min at room temperature. Sections were incubated with fluorescein isothiocyanate (FITC)-conjugated ED1 overnight at 4°C. Slides were dried and mounted with Fluoromount-G (Southern Biotechnology Associates, Birmingham, AL). Three blinded, independent observers counted ED1-positive cells per high-power field to quantify macrophage accumulation in at least three lung sections from each animal. Each observer selected three random fields from different portions of the lung for counting and used sections obtained from different areas of lung tissue to ensure adequate sampling. In addition, all sections were observed under low-power magnification to ensure that all areas of the lung section had been included in the determinations. Good concordance of results between the observers provided reassurance of the accuracy of the counting.
Immunoblot Analysis
Western blots were performed for RHAMM in lysates of macrophages obtained by BAL. Isolated cells were incubated for 10 min with lysis buffer (25 mM Tris-HCl, 0.15 M NaCl, 1.0 mM EDTA, 1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, and Sigma protease inhibitor at 1:20 dilution). Cells were then scraped, transferred to microcentrifuge tubes, centrifuged at 14,000 rpm for 10 min, and the resulting supernatant collected and stored at −80°C. The protein content of each sample was determined using the Bradford assay (35), and 10 μg of each sample was loaded and subjected to electrophoresis at 150 mV in Novex NuPAGE 12% Bis-Tris gels (Invitrogen, Carlsbad, CA) in MES-SDS buffer (from 40× stock from Invitrogen), transferred to nitrocellulose membranes, blocked with 5% nonfat dry milk (NFDM) reconstituted in Tris-buffered saline with 0.1% Tween 20 (TTBS) for 1 h at room temperature, and probed with R36 (5 μg/ml) diluted in 2% NFDM-TTBS overnight at 4°C. After probing with the secondary antibody conjugated to horseradish peroxidase, protein bands were detected by enhanced chemiluminescence (Amersham, Piscataway, NJ). Semiquantitative densitometry was performed on the resulting films with MacBASE version 2.4 (FUJIFilm, Elmsford, NY) as described previously (36).
Surface Biotin Labeling
Lavage cells were isolated from BAL by centrifugation at 5,000 × g for 5 min. Pelleted cells were resuspended in DMEM with no FBS. Cells were plated equally and macrophages adhered after 10 min at 37°C/5%CO2. Surface biotin labeling (Pierce, Rockford, IL) was performed 2 h after plating per manufacturer's instructions. Whole cell lysate was collected and 25 μg of each sample was immunoprecipitated with streptavidin-agarose as per manufacturer's instructions. The precipitate was resuspended in NuPAGE LDS sample buffer with DTT (Invitrogen) and separated by gel electrophoresis. Proteins in the gel were transferred to nitrocellulose membranes, and immunoblots were performed for RHAMM as described above.
Probe Generation for In Situ Hybridization
A partial cDNA for rat RHAMM was amplified from a rat smooth muscle cell cDNA library (the kind gift of Dr. C. Giachelli, The University of Washington, Seattle) by PCR using primers (5′: GTT GGT TGG TTG GAA AAA TCT ; 3′: GCA GCA GTT CGG GTT GCC TTC TTT CAA) specific for positions 13–1,645 of the cDNA (GenBank Accession No. U87983). The amplification was performed using a hot start Mg++ Bead (Invitrogen) with a final concentration of 200 μM dNTPs, 1.5 mM Mg, 0.2 mM primers, 50 mM KCl, 10 mM Tris HCl, pH 8.3, and 0.25 Taq polymerase (Gibco BRL, Gaithersburg, MD). Amplification was for 30 cycles on a programmable thermal cycler PTC-100 with a denaturation temperature of 94°C for 1 min, annealing temperature of 60°C for 1 min, and extension temperature of 72°C for 2 min, and a terminal extension at 72°C for 5 min. An aliquot was run on a 1% low-melting-point agarose gel, and the single band obtained was purified using the Wizard Mini Prep system (Promega, Madison, WI). To generate sense and antisense single-stranded digoxigenin-labeled probes, 20 ng of the initial PCR product was reamplified with a single primer using 50 μM of dATP, dCTP, dGTP, 37 μM dTTP, and 12.5 μM digoxigenin-labeled dUTP using the same amplification parameters and 50 cycles. A single band was generated on agarose gel and was purified again by Wizard Mini Prep. The single-stranded probe generated by amplification with the 5′ primer (5′ GAA ATA GAA GAT CTT AAA CTG GAG AAT TTG 3′) generated a sense strand, whereas the 3′ primer (5′ CAA ATT CTC CAG TTT AAG ATC TTC TAT TTC 3′) generated an antisense probe (both from position 1,067 of the rat RHAMM cDNA). This protocol is a modification of the technique described by Finckh and coworkers (37).
In Situ Hybridization
Immediately after harvest, small portions of lung tissue were fixed in 4% paraformaldehyde in PBS with freshly added 0.1% DEPC. After 2 h, the tissue was cryoprotected in 30% sucrose overnight and then blocked in OCT (Miles, Elkhart, IN). Four-micron sections were cut and placed on silanated ProbeOn Slides (Fisher). In situ hybridization was performed as previously reported (38) with the exception of the use of the Microprobe apparatus (Fisher) and 0.1% Brij 35 detergent in all prehybridization and posthybridization steps. High stringency posthybridization washes were with 0.1× SSC at room temperature 2 min per wash for seven washes. The overnight hybridization was performed at 42°C under coverslips to reduce the volume of reagents. Detection was with NBT-BCIP performed for 4 h. At least two animals were studied for each treatment group.
Time-Lapse Cinemicrography
Freshly isolated cells from BAL of saline- or bleomycin-treated animals were monitored for their motility using a Nikon TE-300 inverted microscope (Nikon Corp., Tokyo, Japan) to which a video camera (Hamamatsu CCD, Inc., Hamamatsu City, Japan) was attached. Cell locomotion of at least 20 cells per animal was determined using Metamorph (Universal Imaging Corp., Downingtown, PA). Cells were plated and maintained at 37°C and followed for 2 h with mean velocities calculated every 10 min. The effect of anti-RHAMM antibody was examined by adding 5 μg/ml of antibody per plate per treatment 15 min before measurement of motility. Nonimmune IgG was used as a control. Each experiment was repeated at least three times.
Chemotaxis Assay
Chemotaxis of macrophages was performed using a modified Boyden chemotaxis chamber containing a 96-well microchemotaxis plate (MBA-96; Neuro Probe, Cabin John, MD) as described previously (39) with minor modifications. Briefly, the bottom wells of the chamber contained 40 μl of HA-6 (4 mM) dissolved in defined medium (DM), without FCS. Positive and negative controls included 10% FCS or DM, respectively. The upper wells were filled with 1 × 106 cells/ml suspended in 100 μl DM in the presence or absence of R36 or normal rabbit IgG. A 5-μm-pore polycarbonate membrane filter was placed between the bottom and top chamber. The chamber was incubated for 6 h at 37°C. Nonmigratory cells on the upper surface of the membrane were treated with 200 μl of 1 mM EDTA for 15–20 min and wiped off. Cells that had migrated into the membrane were stained with Diff-Quick (Baxter Healthcare, McGaw Park, IL) and counted in five randomly selected high-power fields in each well. Each chemoattractant solution was tested in six wells and each experiment was repeated at least three times. Data were expressed as the number of macrophages that migrated into the membrane for each condition and converted to a percentage of control (DM) and combined for three separate experiments.
Statistical Analysis
Statistical comparisons between uninjured animals and saline- or bleomycin-treated groups were performed using ANOVA with Bonferroni correction for individual comparisons. All P values < 0.05 were considered significant.
RESULTS
RHAMM Expression Increases in Macrophages after Bleomycin Lung Injury
To determine the changes in RHAMM expression in the lungs of animals after intratracheal bleomycin, we first examined lung tissue sections by immunocytochemistry at various time points after injury (Figure 1). Constitutive expression of RHAMM was observed in the bronchiolar epithelium, smooth muscle, and resident alveolar macrophages (Figure 1b). Four days after injury, staining for RHAMM was observed in macrophages accumulating both in the alveolar walls and in the alveolar spaces (Figure 1c). In contrast, saline-instilled rat lungs were identical to uninjured, control lungs (data not shown). To ensure specificity of immunostaining, sections were incubated with antibody that had been preincubated with 3-fold excess RHAMM fusion protein (Figure 1a), or by the use of antibody that had been passed over a GST-RHAMM fusion protein column to remove RHAMM-specific antibody (data not shown). In both cases, staining was almost completely abrogated.
Figure 1.

Localization of RHAMM after intratracheal treatments. RHAMM distribution was determined using diaminobenzadine-based immunohistochemistry using R36. (a) Uninjured control lung section stained with R36 preincubated with recombinant RHAMM protein shows no staining, confirming antibody specificity. (b) Normal RHAMM expression (brown staining) is restricted to the apex of airway epithelial cells, resident alveolar macrophages, and in bronchiolar smooth muscle. (c) At 4 d after injury, accumulating macrophages stain strongly for RHAMM. Macrophages were identified using the cell-specific marker, ED1 (data not shown; see Ref. 36). Photomicrographs shown are representative of at least five animals examined at each time and for each condition.
We next examined the expression of RHAMM in inflammatory cells obtained by lavage as a function of time after intratracheal treatments. Immunoblot analysis using R36 demonstrated 70-kD and 80-kD bands for RHAMM (Figure 2a). Since equal protein was loaded for each lane, the data represent the changes in RHAMM independent of the changes in numbers of cells accumulating after intratracheal treatments. Densitometric analysis of each band showed that bleomycin, but not saline treatment, resulted in an increase in both isoforms to a maximum 7 d after bleomycin injury (Figure 2a).
Figure 2.


Immunoblot analysis of RHAMM after intratracheal treatments. (a) Expression of RHAMM was assessed in macrophages obtained by BAL. Equal protein was loaded from samples obtained from uninjured animals and compared with those from rats given either intratracheal saline or bleomycin. Constitutive expression of 70-kD and 80-kD forms of RHAMM were noted. Densitometric analysis was performed as described in MATERIALS AND METHODS. The expression of both isoforms increased after intratracheal bleomycin and both forms were maximally expressed at 7 d. Animals given intratracheal saline did not increase expression of either form of RHAMM. Blot shown is representative of three separate experiments showing similar results. (b) To determine surface expression of RHAMM, macrophages obtained by lavage were cultured for 2 h, then surface labeled with biotin. Cell lysates were then obtained and immunoprecipitated with streptavidin-sepharose. Subsequent immunoblotting for RHAMM showed that bleomycin injury was associated with a 2-fold increase in surface expression of the 70-kD form and that the 80-kD form was not expressed on the cell surface. Blot shown is representative of three separate experiments.
Since cell surface expression of RHAMM has been correlated with increased cell motility (29, 40), and we had previously shown increased surface expression of RHAMM by flow cytometry of lavage cells from bleomycin-injured animals (31), we also examined the expression of cell surface RHAMM by surface biotin labeling 7 d after intratracheal treatments. Compared with saline-treated rats, a 2-fold increase in the 70-kD band was observed in lavage macrophages at 7 d after bleomycin, indicating the upregulation of the 70-kD RHAMM isoform at the cell surface after bleomycin injury (Figure 2b, P = 0.02, n = 3).
To determine the cells responsible for synthesizing RHAMM, in situ hybridization was employed. Single-stranded sense and antisense DNA probes were used in a nonradioactive digoxigenin-labeled in situ hybridization. Consistent with the immunohistochemical data, 4 d after intratracheal saline administration RHAMM mRNA was localized to resident alveolar macrophages (Figure 3b). This distribution did not differ from that of uninjured normal rat lungs (data not shown). Intense positive staining with the antisense probe was obtained in macrophages accumulating in areas of lung injury 4 d after bleomycin injury, confirming that these cells were the sites of RHAMM synthesis (Figure 3c). No staining was observed with the sense probe, thereby confirming the specificity of the antisense probe (Figure 3a).
Figure 3.

In situ hybridization for RHAMM at 4 d. Single-strand digoxigenin-labeled DNA probes were used for nonradioactive in situ hybridization. a and b are both sections from saline-treated animals. (a) Lack of hybridization using sense DNA on tissue sections from an uninjured control animal confirmed probe specificity. (b) Antisense RHAMM probe hybridized to resident alveolar macrophages (arrows). (c) Four days after injury, antisense probe hybridized intensely to accumulating macrophages (arrowheads), suggesting that these cells are the site of RHAMM synthesis.
RHAMM Is Necessary for Increased Macrophage Motility after Injury
To determine whether the increased expression of RHAMM observed in macrophages 4 d after injury corresponded to changes in motility, we used time-lapse cinemicrography to measure the locomotion of macrophages in the first 2 h after isolation by BAL from control and bleomycin-treated animals 4 d after intratracheal treatments (Figure 4). Macrophages after bleomycin treatment showed significantly higher velocities than saline-treated controls (Figure 4, **P < 0.001, n = 4). Macrophages isolated from saline-treated animals also showed a small increase in cell locomotion as compared with untreated controls (Figure 4, *P < 0.05, n = 4). This may represent injury as a result of instillation of saline into the lungs. In both saline- and bleomycin-treated animals, treatment with R36 inhibited increased motility to baseline (Figure 4, *P < 0.05, n = 4). Nonimmune IgG, used as a control, had no effect on the motility of cells from bleomycin-treated animals. These data suggest that, as described for HA (17), RHAMM fully accounts for increased macrophage motility observed after bleomycin injury.
Figure 4.


Effect of anti-RHAMM antibody on macrophage motility 4 d after treatments. (a) Time-lapse cinemicrography was used to determine the effect of R36 on macrophage motility. Cells isolated from lavage of injured animals were plated and adherent macrophages were followed for 2 h after the addition of 5 μg/ml of R36. Nonimmune IgG was used as a control. Saline treatment alone caused some increase in macrophage motility. Bleomycin treatment resulted in a 5-fold increase in motility of macrophages. In both cases, motility was inhibited to baseline with R36 treatment, but not by nonimmune IgG. These data suggest that RHAMM fully accounts for increased macrophage motility after intratracheal treatment. (b) HA oligosaccharide-stimulated chemotaxis of RAW 264.7 cells was assessed using a modified Boyden chamber assay. HA-6 stimulated a 2.5-fold increase in macrophage chemotaxis that was completely blocked in the presence of R36, but not with normal rabbit IgG (P < 0.05, n = 6/condition, representative experiment of three repeats).
RHAMM Is Necessary for HA-Mediated Macrophage Chemotaxis
To determine whether RHAMM contributes to HA-stimulated chemotaxis, a modified Boyden chamber assay was performed using a six-mer HA oligosaccharide (HA-6) as the chemoattractant for RAW264.7 murine macrophages as described in Materials and Methods. HA-6 stimulated a 2.5-fold increase in macrophage chemotaxis over defined medium as a control (Figure 4b, *P < 0.01, n = 6 per condition, experiment repeated three times). Addition of R36 to the top chamber with the cells inhibited HA-stimulated chemotaxis to baseline (Figure 4b, **P < 0.01 versus HA-6 and HA-6 + Rabbit IgG, n = 6 per condition, repeated three times), whereas normal rabbit IgG had no effect. These data suggest that in addition to the random migration of macrophages, RHAMM:HA interactions mediate macrophage chemotaxis.
In Vivo Effects of RHAMM Antibody
We next evaluated the effects of R36 on the inflammatory response to bleomycin. To quantify the effect of R36 on macrophage accumulation, tissue sections were immunostained with ED1 and positive cells per high-power field (hpf) were counted (Figure 5). Saline treatment resulted in a trend to higher macrophage number as compared with controls (saline 14.9 ± 3.3 versus control 10.9 ± 0.21 cells per hpf, n = 4, P = 0.07). However, bleomycin treatment resulted in a 3-fold increase in macrophage number (28.4 ± 4.7 cells per hpf, P < 0.05, n = 4). Daily R36 treatment resulted in a 40% reduction in macrophage number after injury as compared with nonimmune IgG-treated animals (Figure 5, 17.7 ± 4.9 cells per hpf, P < 0.05, n = 4).
Figure 5.

In vivo effect of anti-RHAMM antibody on lavage HA content and macrophage accumulation The effect of R36 on macrophage accumulation was determined by staining tissue sections with the rat macrophage–specific marker ED1. Three blinded observers independently counted positive cells per hpf. Saline treatment resulted in a small trend toward increased macrophage number as compared with controls (P = 0.07). Bleomycin treatment resulted in a 3-fold increase in macrophage number (P < 0.05 versus saline). While normal IgG demonstrated nonspecific effects, daily R36 treatment resulted in a further significant reduction in macrophage number after injury (P < 0.05 versus bleomycin + IgG).
Finally, we determined the effect of antibody treatment on lung architecture at 7 d. Saline-treated animals showed normal lung architecture (Figure 6a). Bleomycin treatment resulted in an influx of inflammatory cells with thickening of alveolar septae and early fibrosis (Figure 6b). Nonimmune IgG also resulted in accumulation of inflammatory cells with evidence of fibrosis (Figure 6c). However, treatment with R36 resulted in decreased inflammatory cell accumulation and a return to more normal architecture (Figure 6d).
Figure 6.
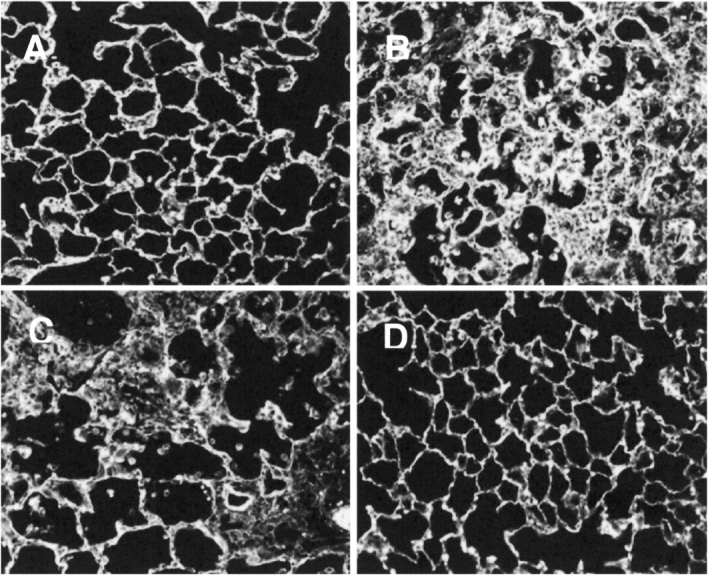
Effect of R36 on lung architecture at 7 d. Fluorescent images of paraffin tissue sections illustrate that (A) saline-treated animals had adequate alveoli and normal architecture. (B) Bleomycin treatment resulted in an influx of inflammatory cells with thickening of alveolar septae and early fibrosis. (C) Nonimmune IgG also resulted in accumulation of inflammatory cells with evidence of fibrosis. (D) However, treatment with R36 resulted in decreased inflammatory cell accumulation and a return to more normal architecture. Data shown are representative of at least three animals examined for each condition.
DISCUSSION
In the present study, we demonstrate increased synthesis and expression of the promigratory molecule RHAMM in macrophages accumulating in areas of the lung injured by intratracheal instillation of bleomycin. Elevated expression of RHAMM occurred in association with increased macrophage motility observed after injury. In addition, using antibody inhibition, we show that cell surface RHAMM is not only necessary for this increased motility, but also contributes to lung macrophage accumulation after injury. Taken together with our previous studies demonstrating the ability of HA-binding peptide to decrease inflammation and fibrosis after bleomycin injury (17), the results of the current study suggest that RHAMM:HA interactions play a critical role in the recruitment of inflammatory cells after lung injury by bleomycin.
HA exerts direct effects on cells and on the extracellular matrix that may be relevant to its role in wound repair. Thus, the proinflammatory cytokines TNF-α and IL-1β have been shown to induce cell surface expression of HA (41), which in turn promotes adhesion of leukocytes to the endothelium (41, 42). Further, HA has been shown to stimulate the migration and proliferation of smooth muscle cells (29), fibroblasts (27), immune cells (15), and endothelial cells (43, 44). HA also activates monocytes into macrophages (45), and increases cytokine gene expression by macrophages (25) and fibroblasts (46). Increased accumulation of HA is associated with inflammation after acute injury to several organ systems including the lung (reviewed in Refs. 10 and 31). However, recent studies have suggested that HA promotes inflammation. Thus, HA-binding peptide specifically blocks macrophage motility in vitro and, when systemically administered to rats during bleomycin injury, results in decreased alveolar macrophage motility and accumulation with subsequently reduced collagen content in the lung (17). In rats, subcutaneous injection of HA-binding peptide before mechanical ventilation blocks neutrophil influx in the lung (47). Importance of HA to inflammation and the likely universal nature of this response was confirmed by Mummert and colleagues (16). In a model of contact hypersensitivity, this group demonstrated that systemic, local, or topical administration of a different HA-binding peptide to reactive hapten-sensitized mice blocked skin-directed homing of inflammatory leukocytes. Together, these studies suggest that the interaction of HA with its receptors precedes and promotes the inflammatory response to injury.
Several other authors have also demonstrated increased cell surface RHAMM expression in leukocytes (48–50). Further, we have previously shown that inflammatory cell chemotaxis and random migration is dependent on RHAMM (15). Interestingly, RHAMM expression is unaltered in CD44 null mice that exhibit increased inflammation after collagen-induced arthritis and anti-RHAMM antibody blocks the inflammatory cell influx in the absence of CD44 (51). Thus, RHAMM:HA interactions may be responsible for leukocyte transmigration through the endothelium and for chemotaxis within injured areas of the lung. However, other reports have noted an absence of cell surface RHAMM after injury. Teder and colleagues (52, 53) proposed two mechanisms for the increased accumulation of HA after bleomycin injury in rats. First, they showed that fibroblasts exposed to BAL from injured animals increase their production of HA, a response that was largely abrogated by blocking antibodies to TGF-β1 (52). Further, alveolar macrophages obtained 5 d after bleomycin injury bound less [3H]-hyaluronan than those from saline-treated controls, suggesting lower HA receptor expression in macrophages after injury (53). This lower expression of HA receptors was also thought to contribute to elevated HA, since less HA would be internalized by these cells. Indeed, in their study, no RHAMM could be detected on the surface of these cells. Further, Weiss and coworkers (9) examined the HA-binding properties of activated monocytes. Immunostaining of skin samples from allergic contact dermatitis found little HA on vascular endothelial cells or activated lymphocytes and no RHAMM expression in monocytes infiltrating sites of cutaneous inflammation. In vitro activation of peripheral blood monocytes by tissue culture, plastic adhesion, and treatment with LPS and IFN-γ did increase HA binding, but did not upregulate RHAMM expression. Instead, FACS analysis showed that activated monocytes expressed increased cell surface CD44, coinciding with increased CD44 mRNA and protein (9).
The lack of increased HA receptor expression in macrophages after bleomycin injury as reported by Teder and colleagues (53) is contradictory to other reports and the data presented in the current study. For instance, the expression of the standard form of CD44 (CD44 s) is increased in alveolar macrophages and in the interstitium of the lung in areas of thickened alveolar wall, while the expression of CD44v6, thought to be the epithelial form, is decreased in type II pneumocytes (54). Further, CD44, presumably acting thorough its ability to bind HA, has been implicated in novel rolling and adhesion pathways leading to leukocyte adhesion to activated endothelium (41, 42). Further, the recruitment of lymphocytes to the lung after antigenic challenge to sensitized mice could be blocked using an anti-CD44 antibody (55–58).
Thus, there are conflicting data with respect to increased expression of CD44 and RHAMM after lung injury. It is unclear why these differences in observations exist. However, it is relevant to this discrepancy that we have noticed that the increased motility observed in macrophages isolated from bleomycin-injured animals is rapidly lost after 2 h following culture (data not shown). Since Teder and colleagues (53) examined macrophages after 24 h in culture, the decreased surface expression of HA receptors observed could be the result of downregulated cell surface expression of HA receptors.
Upregulation of growth factors such as TNF and TGF-β after bleomycin-induced lung injury coincides with increased HA production (59, 60). In lungs injured by bleomycin, the increased expression of HA receptors in macrophages coincides with the temporal increase in TGF-β1 expression observed in the same cells after similar injury (8, 54). This similarity in expression suggests in vivo regulation of HA receptors in macrophages by TGF-β1. In fibroblasts, the expression of RHAMM has been shown to be regulated by TGF-β1 by a mechanism that involves increased message stability (61), and the RHAMM:HA interaction is required for TGF-β1–stimulated increases in cell locomotion (62). TGF-β1 may thus be responsible for an autocrine system in which production of this growth factor results in upregulation of HA and its receptors in macrophages and a paracrine effect on fibroblasts to produce HA and increased collagen deposition, thereby influencing both inflammation and fibrosis after injury. Our data, however, showing a decrease in macrophage accumulation with HA-binding peptide treatment (17), and with anti-RHAMM antibody treatment (this study) suggests that RHAMM: HA interactions are, at least in part, responsible for the inflammatory response to acute lung injury.
In summary, the results of this study suggest that HA and RHAMM are critical components of the inflammatory and fibrotic processes resulting from acute lung injury and that these molecules represent novel targets to limit the adverse consequences of lung injury in humans.
Acknowledgments
The authors thank Dr. Cecila Giachelli for the rat smooth muscle cell cDNA library, as well as Dasha Pechersky and Melane Ferenbach for technical help. They also thank Drs. Akira Asari, Masaki Kosemura, and Takahiro Masa (Seikagaku Corporation, Tokyo, Japan) for preparation of the HA-6 oligosaccharide.
This study was funded by grants from the American Lung Association (SE Pennsylvania), the March of Dimes, and the National Institutes of Health (HL42672 and HL073896) (all to R.C.S.).
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Selman M, King TE Jr, Pardo A. Idiopathic Pulmonary Fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001;134:136–151. [DOI] [PubMed] [Google Scholar]
- 2.Downey GP, Dong Q, Kruger J, Dedhar S, Cherapanov V. Regulation of neutrophil activation in acute lung injury. Chest 1999;116:46S–54S. [PubMed] [Google Scholar]
- 3.Hasleton PS, Roberts TE. Adult Respiratory Distress Syndrome: an update. Histopathology 1999;34:285–294. [DOI] [PubMed] [Google Scholar]
- 4.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000;342:1334–1349. [DOI] [PubMed] [Google Scholar]
- 5.Groneck P, Speer CP. Pulmonary inflammation in the pathogenesis of bronchopulmonary dysplasia. Pediatr. Pulmonol. 1997;16:29–30. [DOI] [PubMed] [Google Scholar]
- 6.Raghow R, Lurie S, Seyer JM, Kang AH. Profiles of steady state levels of messenger RNAs coding for Type I procollagen, elastin and fibronectin in hamster lungs undergoing bleomycin-induced interstitial pulmonary fibrosis. J Clin Invest 1985;76:1733–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernnas J, Nettelbladt O, Bjermer L, Sarnstrand B, Malmstrom A, Hällgren R. Alveolar accumulation of fibronectin precedes fibrosis in bleomycin-induced pulmonary injury. Eur Respir J 1992;5:404–410. [PubMed] [Google Scholar]
- 8.Khalil N, Whitman C, Zuo L, Danielpour D, Greenberg A. Regulation of alveolar macrophage transforming growth factor-beta secretion by corticosteroids in bleomycin-induced pulmonary inflammation. J Clin Invest 1993;92:1812–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss JM, Renkl AC, Ahrens T, Moll J, Mai BH, Denfeld RW, Schopf E, Ponta H, Herrlich P, Simon JC. Activation-dependent modulation of hyaluronate-receptor expression and of hyaluronate avidity in human monocytes. J Invest Dermatol 1998;111:227–232. [DOI] [PubMed] [Google Scholar]
- 10.Savani RC, DeLisser HM. Hyaluronan and its receptors in lung health and disease. In: Garg HG, Roughley PJ, Hales CA, editors. Proteoglycans and lung disease. Lung biology in health and disease. New York: Marcel Dekker; 2003. pp. 73–106.
- 11.Laurent TC, Fraser JRE. Hyaluronan. FASEB J 1992;6:2397–2404. [PubMed] [Google Scholar]
- 12.Hällgren R, Eklund A, Engstrom-Laurent A, Schmekel B. Hyaluronate in bronchoalveolar lavage fluid: a new marker in sarcoidosis reflecting pulmonary disease. BMJ 1985;290:1778–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bjermer L, Engstrom-Laurent A, Lundgren R, Rosenhall L, Hallgren R. Hyaluronate and type III procollagen peptide concentrations in bronchoalveolar lavage fluid as markers of disease activity in farmer's lung. BMJ 1987;295:803–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hällgren R, Samuelsson T, Laurent TC, Modig J. Accumulation of hyaluronan (hyaluronic acid) in the lung in Adult Respiratory Distress Syndrome. Am Rev Respir Dis 1989;139:682–687. [DOI] [PubMed] [Google Scholar]
- 15.Savani RC, Khalil N, Turley EA. Hyaluronan receptor antagonists alter skin inflammation and fibrosis following injury. Proc West Pharmacol Soc 1995;38:131–136. [PubMed] [Google Scholar]
- 16.Mummert ME, Mohamadzadeh M, Mummert DI, Mizumoto N, Takashima A. Development of a peptide inhibitor of hyaluronan-mediated leukocyte trafficking. J Exp Med 2000;192:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savani RC, Hou G, Liu P, Wang C, Simons E, Grimm PC, Stern R, Greenberg AH, DeLisser H, Khalil N. A role for hyaluronan (HA) in macrophage accumulation and collagen deposition after bleomycin-induced lung injury. Am J Respir Cell Mol Biol 2000;23:475–484. [DOI] [PubMed] [Google Scholar]
- 18.Entwistle J, Hall CL, Turley EA. HA receptors: regulators of cell signaling to the cytoskeleton. J Cell Biochem 1996;61:569–577. [DOI] [PubMed] [Google Scholar]
- 19.Naor D, Sionov RV, Ish-Shalom D. CD44: structure, function and association with the malignant process. Adv Cancer Res 1997;71:241–319. [DOI] [PubMed] [Google Scholar]
- 20.Day AJ. The structure and regulation of hyaluronan-binding proteins. Biochem Soc Trans 1999;27:115–121. [DOI] [PubMed] [Google Scholar]
- 21.Miyake K, Medina KL, Hayashi S-I, Ono S, Hamaoka T, Kincade PW. Monoclonal antibodies to Pgp-1/CD44 block lympho-hemopoiesis in long-term bone marrow cultures. J Exp Med 1990;171:477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salmi M, Gron-Virta K, Sointu P, Grenman R, Kalimo H, Jalkanen S. Regulated expression of exon v6 containing isoforms of CD44 in man: downregulation during malignant transformation of tumors of squamocellular origin. J Cell Biol 1993;122:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Penneys NS. CD44 expression in normal and inflamed skin. J Cutan Pathol 1993;20:250–253. [DOI] [PubMed] [Google Scholar]
- 24.Svee K, White J, Vaillant P, Jessurun J, Roongta U, Krumwide M, Johnson D, Henke C. Acute lung injury fibroblast migration and invasion of a fibrin matrix is mediated by CD44. J Clin Invest 1996;98:1713–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKee CM, Penno MB, Cowman M, Burdick MD, Strieter RM, Bao C, Noble PW. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages: The role of HA size and CD44. J Clin Invest 1996;98:2403–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, Henson PM, Noble PW. Resolution of lung inflammation by CD44. Science 2002;296:155–158. [DOI] [PubMed] [Google Scholar]
- 27.Hardwick C, Hoare K, Owens R, Hohn HP, Hook M, Moore D, Cripps V, Austen L, Nance DM, Turley EA. Molecular cloning of a novel Hyaluronan receptor that mediates tumor cell motility. J Cell Biol 1992;117:1343–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hall CL, Yang B, Yang X, Zhang S, Turley M, Samuel S, Lange LA, Wang C, Curpen GD, Savani RC, et al. Overexpression of the hyaluronan receptor RHAMM is transforming and is required for transformation by H-ras. Cell 1995;82:19–28. [DOI] [PubMed] [Google Scholar]
- 29.Savani RC, Wang C, Yang B, Zhang S, Kinsella MG, Wight TN, Stern R, Nance DM, Turley EA. Migration of bovine aortic smooth muscle cells following wounding injury: the role of Hyaluronan and RHAMM. J Clin Invest 1995;95:1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S, Chang MC, Zylka D, Turley S, Harrison R, Turley EA. The hyaluronan receptor RHAMM regulates extracellular-regulated kinase. J Biol Chem 1998;273:11342–11348. [DOI] [PubMed] [Google Scholar]
- 31.Savani RC, Bagli DJ, Harrison RE, Turley EA. The role of hyaluronan-receptor interactions in wound repair. In: Garg HG, Longaker MT, editors. Scarless wound healing. Basic and clinical dermatology, 19th ed. New York-Basel: Marcel Dekker; 2000. pp. 115–142.
- 32.Lovvorn HN, Cass DL, Sylvester KG, Yang EY, Crombleholme TM, Adzick NS, Savani RC. Hyaluronan receptor expression increases in fetal excisional skin wounds and correlates with fibroplasia. J Pediatr Surg 1998;33:1062–1070. [DOI] [PubMed] [Google Scholar]
- 33.Entwistle J, Zhang S, Yang B, Wong C, Qun L, Hall CL, Jingbo A, Mowat M, Greenberg AH, Turley EA. Characterization of the murine gene encoding the hyaluronan receptor RHAMM. Gene 1995;163:233–238. [DOI] [PubMed] [Google Scholar]
- 34.Fieber C, Plug R, Sleeman J, Dall P, Ponta H, Hofmann M. Characterization of the murine gene encoding the intracellular hyaluronan receptor IHABP (RHAMM). Gene 1999;226:41–50. [DOI] [PubMed] [Google Scholar]
- 35.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 36.Savani RC, Godinez RI, Godinez MH, Wentz E, Zaman A, Cui Z, Pooler PM, Guttentag SH, Beers MF, Gonzales LW, et al. Respiratory distress after intratracheal bleomycin: selective deficiency of surfactant proteins B and C. Am J Physiol Lung Cell Mol Physiol 2001;281:L685–L696. [DOI] [PubMed] [Google Scholar]
- 37.Finckh U, Lingenfelter P, Myerson D. Producing single-stranded DNA probes with the Taq DNA polymerase: A high yield protocol. Biotechniques 1991;10:35–39. [PubMed] [Google Scholar]
- 38.Meltzer JC, Sanders V, Grimm PC, Stern E, River C, Lee S, Rennie SL, Gietz RD, Hole AK, Watson PH, et al. Production of digoxigenin-labelled RNA probes and the detection of cytokine mRNA in rat spleen and brain by in situ hybridization. Brain Res Brain Res Protoc 1998;2:339–351. [DOI] [PubMed] [Google Scholar]
- 39.Shi Y, Kornovski BS, Savani RC, Turley EA. A rapid, multiwell colorometric assay for chemotaxis. J Immunol Methods 1993;164:149–154. [DOI] [PubMed] [Google Scholar]
- 40.Hall CL, Wang C, Lange LA, Turley EA. Hyaluronan and the hyaluronan receptor RHAMM promote focal adhesion turnover and transient tyrosine kinase turnover. J Cell Biol 1994;126:575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mohamadzadeh M, DeGrendele H, Arizpe H, Estess P, Siegelman M. Proinflammatory stimuli regulate endothelial hyaluronan expression and CD44/HA-dependent primary adhesion. J Clin Invest 1998;101:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeGrendele HC, Estess P, Picker LJ, Siegelman MH. CD44 and its ligand hyaluronate mediate rolling under physiologic flow: a novel lymphocyte-endothelial cell primary adhesive pathway. J Exp Med 1996;183:1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trochon V, Mabilat C, Bertrand P, Legrand Y, Smadja-Joffe F, Soria C, Delpech B, Lu H. Evidence of involvement of CD44 in endothelial cell proliferation, migration and angiogenesis in vitro. Int J Cancer 1996;66:664–668. [DOI] [PubMed] [Google Scholar]
- 44.Savani RC, Cao G, Pooler PM, Zaman A, Zhou Z, DeLisser HM. Differential involvement of the hyaluronan (HA) receptors CD44 and Receptor for HA-Mediated Motility in endothelial cell function and angiogenesis. J Biol Chem 2001;276:36770–36778. [DOI] [PubMed] [Google Scholar]
- 45.Noble PW, Lake FR, Henson PM, Riches DW. Hyaluronate activation of CD44 induces insulin-like growth factor-1 expression by a tumor necrosis factor-alpha-dependent mechanism in murine macrophages. J Clin Invest 1993;91:2368–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kobayashi H, Terao T. Hyaluronic acid-specific regulation of cytokines by human uterine fibroblasts. Am J Physiol 1997;273:C1151–C1159. [DOI] [PubMed] [Google Scholar]
- 47.Syrkina OL, Mascarenhas MM, Garg HG, Hales CA, Quinn DA. Neutrophil influx with ventilator-induced lung injury (VILI) is dependent on low molecular weight hyaluronic acid. Am J Respir Crit Care Med 2003;167:A563. [Google Scholar]
- 48.Doherty DE, Hirose N, Zagarella L, Cherniack RM. Prolonged monocyte accumulation in the lung during bleomycin-induced pulmonary fibrosis: a non-invasive assesment of monocyte kinetics by scintigraphy. Lab Invest 1992;66:231–242. [PubMed] [Google Scholar]
- 49.Bazil V, Horejsi V. Shedding of the CD44 adhesion molecule from leukocytes induced by anti-CD44 monoclonal antibody stimulating the effect of a natural receptor ligand. J Immunol 1992;149:747–753. [PubMed] [Google Scholar]
- 50.Pilarski LM, Pruski E, Wizniak J, Paine D, Seeberger K, Mant MJ, Brown CB, Belch AR. Potential role for hyaluronan and the hyaluronan receptor RHAMM in mobilization and trafficking of hematopoietic progenitor cells. Blood 1999;93:2918–2927. [PubMed] [Google Scholar]
- 51.Nedvetzki S, Gonen E, Assayag N, Reich R, Williams RO, Thurmond RL, Huang JF, Neudecker BA, Wang FS, Turley EA, et al. RHAMM, a receptor for hyaluronan-mediated motility, compensates for CD44 in inflamed CD44-knockout mice: a different interpretation of redundancy. Proc Natl Acad Sci USA 2004;101:18081–18086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teder P, Nettelbladt O, Heldin P. Characterization of the mechanism involved in bleomycin-induced increased hyaluronan production in rat lung. Am J Respir Cell Mol Biol 1995;12:181–189. [DOI] [PubMed] [Google Scholar]
- 53.Teder P, Heldin P. Mechanism of impaired local hyaluronan turnover in bleomycin-induced lung injury in rat. Am J Respir Cell Mol Biol 1997;17:376–385. [DOI] [PubMed] [Google Scholar]
- 54.Kasper M, Bierhaus A, Whyte A, Binns RM, Schuh D, Muller M. Expression of CD44 isoforms during bleomycin- or radiation-induced pulmonary fibrosis in rats and mini-pigs. Histochem Cell Biol 1996;105:221–230. [DOI] [PubMed] [Google Scholar]
- 55.Curtis JL, Kim S, Scott PJ, Buechner-Maxwell VA. Adhesion receptor phenotypes of the murine lung CD4+ T cells during the pulmonary immune response to sheep erythrocytes. Am J Respir Cell Mol Biol 1995;12:520–530. [DOI] [PubMed] [Google Scholar]
- 56.Wolber FM, Curtis JL, Milik AM, Fields T, Seitzman GD, Kim K, Kim S, Sonstein J, Stoolman LM. Lymphocyte recruitment and kinetics of adhesion receptor expression during the pulmonary response to particulate antigen. Am J Pathol 1997;151:1715–1727. [PMC free article] [PubMed] [Google Scholar]
- 57.Wolber FM, Curtis JL, Maly P, Kelly J, Smith P, Yednock TA, Lowe JB, Stoolman LM. Endothelial selectins and alpha-integrins regulate independent pathways of T lymphocyte recruitment in the pulmonary immune response. J Immunol 1998;161:4396–4403. [PubMed] [Google Scholar]
- 58.Curtis JL, Wolber FM, Sonstein J, Craig RA, Polak T, Knibbs RN, Todt J, Seitzman GD, Stoolman LM. Lymphocyte-endothelia cell adhesive interactions in lung immunity: lessons from the murine reponse to particulate antigen. Immunopharmacology 2000;48:223–229. [DOI] [PubMed] [Google Scholar]
- 59.Heldin P, Laurent TC, Heldin CH. Effect of growth factors on hyaluronan synthesis in cultured human fibroblasts. Biochem J 1989;258:919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heldin P, Asplund T, Ytterberg D, Thelin S, Laurent TC. Characterization of the molecular mechanism involved in the activation of hyaluronan synthetase by platelet-derived growth factor in human mesothelial cells. Biochem J 1992;283:165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amara FM, Entwistle J, Kuschak TI, Turley EA, Wright JA. Transforming growth factor-beta 1 stimulates multiple protein interactions at a unique cis-element in the 3′-untranslated region of the hyaluronan receptor RHAMM mRNA. J Biol Chem 1996;271:15279–15284. [DOI] [PubMed] [Google Scholar]
- 62.Samuel SK, Hurta RAR, Spearman MA, Wright JA, Turley EA, Greenberg AH. TGF-β1 stimulation of cell locomotion utilizes the hyaluronan receptor RHAMM and hyaluronan. J Cell Biol 1993;123:749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]