

Abstract
Pseudomonas aeruginosa is a gram-negative bacterium that is an opportunistic pathogen in patients with cystic fibrosis and in immunocompromised hosts. This bacterium produces a variety of proteolytic enzymes, including alkaline protease (AP), which has multiple biological effects. This study investigated the effects of AP on the A549 pulmonary epithelial cell line. Results demonstrate that AP inhibited tumor necrosis factor (TNF)-α–induced RANTES gene expression and secretion in a concentration-dependent manner. The TNF-α–induced RANTES gene expression and secretion was attenuated with a neutralizing monoclonal antibody directed against the TNF receptor type 1 (TNFR1). Conversely, a neutralizing monoclonal antibody directed against TNF receptor type II had no effect, suggesting that these events were regulated through the TNFR1 receptor. In addition, we observed that soluble TNF receptor type 1 (sTNFR1) levels were significantly increased in culture supernatants of AP-treated cells in a concentration-dependent manner. Finally, membrane-associated TNFR1 was decreased after AP exposures. In these studies, the enzymatically inactive form of AP had no effect on TNF-α–induced RANTES secretion, shedding of sTNFR1, or membrane-associated TNFR1. These results demonstrate that AP stimulates shedding of cell-surface TNFR1, resulting in an increase in sTNFR1. Consequently, these events decrease the cells' ability to stimulate RANTES gene expression and secretion through TNFR1.
Keywords: Pseudomonas aeruginosa, tumor necrosis factor alpha, cytokine, airway epithelium
Pseudomonas aeruginosa is an opportunistic gram-negative bacterium, which causes severe pneumonia in immunocompromised hosts and in patients with cystic fibrosis (1, 2). P. aeruginosa causes airway tissue damage by producing several virulence factors, including exotoxins, proteases, lipopolysaccharide (LPS), and pigments, many of which play roles in the toxicity and aggressivity of the bacteria (3–5). Virulence factors induce numerous biological effects in host cells. They are known to modulate inflammatory and immune responses by altering the bioavailability of cytokines. For example, metalloproteases from P. aeruginosa are known to degrade tumor necrosis factor (TNF)-α (6), IFN-γ (7), and “regulated on activation, normal T cells expressed and secreted” (RANTES) (8), thereby decreasing the bioavailability of these cytokines. RANTES plays an important role in the immune responses in the airway by functioning as chemoattractant for both T cells and eosinophils. In addition, Pseudomonas pyocyanin has been shown to increase the release of IL-8 and decrease cytokine-dependent RANTES expression from human airway epithelial cell lines (9).
TNF-α is a cytokine that is involved in regulating airway inflammation. TNF-α exerts its biological effect through two high-affinity TNF receptors, TNF receptor type 1 (TNFR1, p55) and TNF receptor type 2 (TNFR2, p75). Both receptors have been identified and located on the surface of airway epithelial cells and A549 cells. TNFR1 has been implicated as an important signaling receptor, resulting in activation of pathways that regulate inflammatory and immune responses (10). For example, TNF-α can increase cytokines and adhesion molecules in airway epithelial cells via a pathway involving TNFR1 (11, 12).
Both types of TNF receptors can be released from the cell surface by the proteolytic activity of the metalloprotease TNF-α–cleaving enzyme (TACE) (13). Receptor shedding reduces the surface expression of TNF receptors and may desensitize cells to TNF receptor–mediated events. In addition, shed receptors are present as soluble forms and can bind and neutralize TNF-α, which may allow them to function as physiologic neutralizing agents for TNF-α (14).
The purpose of this study was to determine whether alkaline protease isolated from P. aeruginosa altered TNF-α–induced RANTES gene expression and secretion. We report that the regulation of TNF-α–induced RANTES gene expression in A549 cells appears to be mediated through TNFR1. We demonstrated that AP inhibits TNF-α–induced RANTES gene expression and secretion in the A549 pulmonary epithelial cell line. In addition, we observed that AP stimulated shedding of membrane-associated TNFR1, which resulted in an increase in soluble TNF receptor 1 (sTNFR1) in cell culture supernatants. This response correlated with the transiently diminished TNF-induced RANTES expression and secretion. These findings suggest that AP treatment results in shedding of cell-associated surface TNFR1, leading to an increase in sTNFR1 and a decrease in the cells' responsiveness to the cytokine TNF-α.
MATERIALS AND METHODS
Airway Epithelial Cell Culture
A549 cells (ATCC CCL-185) were cultured in Ham's F-12K medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml). Cells were grown at 37°C in a humidified 5% CO2 atmosphere on plastic culture plates. Cultures were used when confluent.
Purification of Alkaline Protease
P. aeruginosa (ATCC 22248) was grown in a semisynthetic medium, as described by Maeda and Morihara (15), for 5 d. Bacterial cells were removed from the medium by centrifugation (3,000 × g, 20 min) and alkaline protease was purified from the supernatant (15). Briefly, proteins in the supernatant were precipitated with ammonium sulfate. The precipitate was dissolved in 0.2 M Tris-HCl buffer, pH 8.0, dialyzed, and loaded onto a DEAE-Sephacel anion-exchange column equilibrated with the same buffer. Proteins were eluted with a NaCl gradient. Fractions containing proteolytic activity were pooled and applied onto a Mono Q FPLC column equilibrated in 0.2 M Tris-HCl, pH 8.0. The proteolytically active fraction was eluted with a NaCl gradient. Elutions containing pure alkaline protease were pooled and concentrated by ultrafiltration. The homogeneity of the enzyme was confirmed by SDS-PAGE, and the enzyme concentration was determined using the BCA method with bovine serum albumin as a standard. The purified alkaline protease was tested for endotoxin levels with the Limulus Amebocyte Lysate assay (BioWhittaker, Walkersville, MD). The purified protease contained the following endotoxin units (EU): 25 μg/ml protease samples contained 500 EU, 10 μg/ml protease samples contained 200 EU, 1 μg/ml protease samples contained 20 EU, and 0.1 μg/ml protease samples contained 2 EU. The equivalent or a greater concentration of endotoxin detected in the alkaline protease was used in each experiment to verify that the results were not due to endotoxin contamination.
Measurement of RANTES Secretion and Soluble TNFR1
The secreted RANTES and soluble TNFR1 in the culture supernatant were measured with an enzyme-linked immunosorbant assay (ELISA) using a commercially available sandwich-type ELISA (R&D Systems Inc., Minneapolis, MN).
Measurement of AP Degradation of Soluble TNFR1
To determine whether or not active AP could degrade soluble TNFR1, we used a commercially available ELISA that incorporated a cell-free assay. In this assay, recombinant human sTNFR1 (200 pg/ml) was incubated at 37°C in the presence or absence of 0.1, 1.0, and 10 μg/ml AP in A549 cell culture medium void of cells for 15 min. At the end of the incubation period, sTNFR1 was measured using a commercially available Quantikine human TNFR1 ELISA kit to determine if active protease would degrade sTNFR1 (R&D Systems).
Measurement of RANTES mRNA by Real-Time PCR
Total RNA was isolated from the cells by using the Absolutely RNA RT-PCR Miniprep Kit (Stratagene, La Jolla, CA). Briefly, medium was completely removed from the 12-well plates and 350 μl of lysis buffer was added to each well. The cell lysate was transferred into microcentrifuge tubes and the RNA was isolated according to the manufacturer's protocol. After isolation, RNA was stored at −80°C. The quantity and purity of the RNA was determined by measuring the absorbance at 260 nm and the A260/A280 ratio on a spectrophotometer. RANTES gene expression was quantified in a two-step RT-PCR. Complimentary DNA was reverse-transcribed from total RNA samples (0.625 μg/50 μl) using random hexamers from TaqMan RT reagents (Applied Biosystems, Foster City, CA). PCR products were synthesized from cDNA (22.5 ng/20 μl) using the TaqMan universal PCR master mix and Assays on Demand gene expression reagents for human RANTES (Assay ID:Hs00174575_m1; Applied Biosystems). Measurements were done using the ABI Prism 7900 HT sequence detection system according to the manufacturer's protocols. As an endogenous control for these PCR quantification studies, 18 S ribosomal RNA gene expression was measured using TaqMan ribosomal RNA control reagents (Applied Biosystems). Results represent normalized RANTES mRNA amounts relative to control cultures using the 2-ΔΔCT method (16). Each experiment was repeated in triplicate.
Sample Preparation and Cell-Surface Labeling for Membrane-Associated TNFR1
Confluent cultures of 6-well plates were exposed to purified AP for 15 min. After exposure, cultures were washed three times with ice-cold PBS, pH 8.0, containing 0.1 mM CaCl2 and 1.0 mM MgCl2. Monolayers were then treated twice with PBS containing 0.5 mg/ml N-hydroxysuccinimidyl (NHS)-biotin for 15 min at room temperature. After incubations, samples were washed twice with 50 mM Tris-HCl buffer, pH 8.0, and once with PBS-CaCl2-MgCl2, pH 8.0. Cell monolayers were treated with lysis buffer (50 mM HEPES, 150 mM NaCl, 1% Triton X-100, 1 mM AEBSF, 20 μg/ml aprotinin, and 20 μg/ml leupeptin) and the cell lysates were clarified by centrifugation at 13,000 rpm for 10 min. Sample volumes were adjusted to a constant protein concentration (1.25 mg/ml) using the Bradford dye-binding procedure (Bio-Rad Protein Assay; Bio-Rad, Richmond, CA). Samples were transferred to tubes containing ImmunoPure Immobilized avidin beads (Pierce, Rockford, IL) and incubated overnight at 4°C with end-over-end rotation. After incubation, the beads (containing the biotinylated membrane proteins) were washed with lysis buffer, boiled in sample buffer (125 mM Tris/HCl, 15% sucrose, 4% SDS, 10 mM EDTA, 0.1 mg/ml bromphenol blue, 4% mercaptoethanol) for 5 min, and analyzed by immunoblotting as described below.
Immunoblotting of Biotinylated Proteins
Biotinylated protein samples were resolved by 12% SDS-PAGE and transferred onto nitrocellulose membranes. Blots were blocked in buffer containing 5% nonfat dried milk in 20 mMTris, 500 mM NaCl, 0.1% Tween-20 (TBST), pH 7.5. After blocking, the membrane was immunoblotted with a rabbit polyclonal anti-TNFR1 antibody (1:500) (Santa Cruz Biotechnology, Santa Cruz, CA) in blocking buffer overnight at 4°C. After incubation, blots were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (1:25,000) (Pierce). Blots were then treated with SuperSignal West Dura extended duration chemiluminescence substrate solution (Pierce) and analyzed using a Fluor-S Max2 (Bio-Rad) MultiImager system and associated software.
Cytotoxicity Assessment
Cytotoxicity was assessed by lactate dehydrogenase (LDH) release using the commercially available colorimetric assay CytoTox 96 (Promega, Madison, WI) according to the manufacturer's instructions. Cells were exposed to various concentrations of AP (0.1, 1, 10, 25, or 50 μg/ml) with or without the protease inhibitor (o-phenanthroline). After incubation for 6 and 24 h, both supernatants and cell lysates were collected and assessed for LDH content. Percentage of LDH release was calculated by taking the ratio of LDH released into the supernatant to the total LDH in the supernatant and the cell lysate. Cultures showed > 96% cell viability and no cytotoxicity at all concentrations and incubation periods tested.
Cell Treatments
A549 cells were cultured in 96-well plates for ELISA experiments, 12-well plates for RT-PCR, and 6-well plates for Western analysis.
RANTES secretion.
To study the effects of AP on TNF-α–induced RANTES secretion, confluent monolayers were washed with serum-free medium and exposed to AP for various incubation periods or concentrations in serum-free medium. After AP exposure, cultures were washed with medium containing 10% FBS and subsequently stimulated with TNF-α (10 ng/ml) for 1 h. After stimulation, cultures were washed with medium containing 10% FBS to remove any unbound TNF-α and were incubated in medium. Supernatants were collected at 24 h. Protein concentrations from each well were determined and used to normalize for well-to-well variation.
Membrane-associated TNFR1 and sTNFR1.
To investigate the effects of AP on membrane-bound TNFR1 and sTNFR1, monolayers were washed with serum-free medium and exposed to various AP concentrations in serum-free medium for 15 min. After AP exposure, supernatants were mixed with EDTA (final concentration 10 mM), stored at −70°C, and assessed for sTNFR1 by ELISA. Cells were subsequently washed, biotinylated, and assayed for membrane-associated TNFR1 by Western analysis.
RANTES mRNA expression.
To study the effects of AP on TNF-α–induced RANTES mRNA expression, cultures were washed with serum-free medium and exposed to various AP concentrations in serum-free medium for 15 min. After AP exposure, cells were washed and exposed to 10 ng/ml TNF-α for 15 min. After stimulation, cultures were washed with medium to remove any unbound TNF-α and incubated for 1 h in complete medium. RNA was then subsequently isolated for RT-PCR.
Inactivation of alkaline protease.
To inactivate AP proteolytic activity, the enzyme was incubated with 5 mM 1,10-phenanthroline overnight at 37°C. After incubation, inactivated AP was dialyzed for 2 h and adjusted to the appropriate protein concentration. After treatment, the enzyme activity was determined to be reduced by 95%.
Statistical Analysis
Data were analyzed for significance using a one-way ANOVA with Bonferroni post-test correction for multiple comparisons (17). Data were considered significant at P < 0.05.
RESULTS
Inhibition of TNF-α–Induced RANTES Secretion in A549 Cells by AP
Utilizing an ELISA to detect RANTES secretion, an initial TNF-α dose–response curve to RANTES secretion was established as shown in Figure 1A. In addition, we investigated the effects of exposure time and concentration of AP on TNF-α–induced RANTES secretion. As shown in Figure 1B, AP treatment inhibited TNF-α–mediated RANTES secretion in a dose-dependent manner. Treatment of the cultures with the inactive form of AP had no effect on TNF-α–mediated RANTES secretion from A549 cells (Figure 1B). As illustrated in Figure 1C, TNF-α–mediated RANTES secretion was inhibited with relatively short exposures of 10 μg/ml AP, with significant decreases in RANTES secretion exhibited after 5 and 15 min of exposure. Interestingly, after longer preincubation times, RANTES secretion returned to the level in the control. To address possible cytotoxic events, cultures were exposed to both active and inactive AP for 6 and 24 h. Cultures showed > 96% cell viability and no cytotoxicity at all concentrations tested (data not shown).
Figure 1.
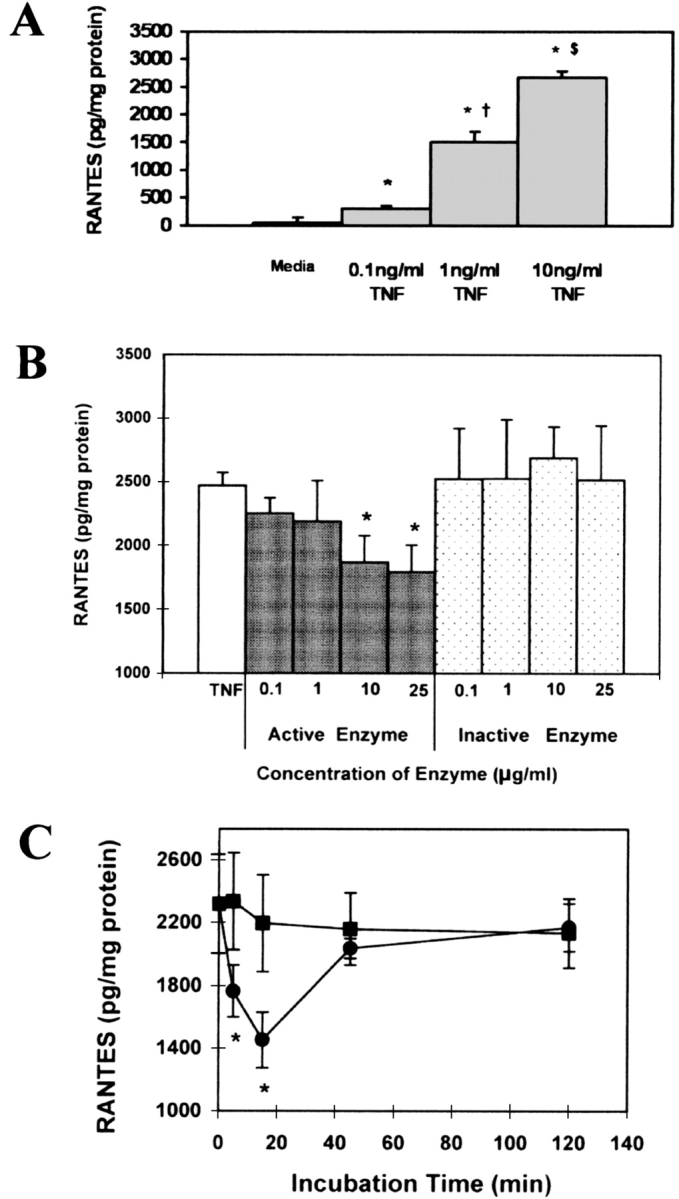
AP inhibits TNF-α–induced RANTES secretion from A549 cells. (A) A549 cells were exposed to increasing concentrations of TNF-α for 1 h. After TNF-α stimulation, cells were washed and incubated in media alone for 24 h and supernatants were assayed for secreted RANTES (n = 6; * significantly different from control; † significantly different from 0.1 ng/ml TNF; $ significantly different from 1 ng/ml TNF; P < 0.05). (B) A549 cells were exposed for 15 min with increasing concentrations of active (gray columns) and inactive (dotted columns) protease. Cells were washed to inactivate any enzymatic activity and stimulated with 10 ng/ml TNF-α for 1 h. After TNF-α stimulation, cells were incubated in media alone for 24 h and supernatants were analyzed for secreted RANTES by ELISA. (Values shown are mean ± SD, n = 6 at each point; * significantly different from TNF-α; P < 0.05.) (C) A549 cells were exposed to 10 μg/ml of active (circles) and inactive (squares) protease for indicated periods. Treatments were washed to inactivate any enzymatic activity and stimulated with 10 ng/ml TNF-α for 1 h. After TNF-α stimulation, all treatments were incubated in media alone for 24 h and supernatants were assayed for secreted RANTES by ELISA. The graph summarize the results from three separate experiments (n = 6; * significantly different from inactive protease; P < 0.05).
Inhibition of TNF-α–Induced RANTES mRNA Expression in A549 Cells by AP
To investigate the effects of AP on TNF-α–mediated RANTES gene expression, we employed real-time PCR. As shown in Figure 2, 10 μg/ml of AP treatment for 15 min significantly suppressed the expression of the TNF-α–induced RANTES mRNA in A549 cells. Interestingly, the inactivated AP (IAP) also had an effect on the RANTES gene expression in A549 cells. These data suggest that the decreased levels of secreted RANTES may be, at least in part, due to the effects of both the active and inactive protease on steady-state levels of RANTES mRNA.
Figure 2.

AP inhibits TNF-α–induced RANTES mRNA expression in A549 cells. A549 cells were exposed to 10 μg/ml of active AP (AP) and inactive AP (IAP) for 15 min, washed to inactivate any enzymatic activity, then stimulated with 10 ng/ml TNF-α for 15 min. After stimulation, cell cultures were washed again and after 1 h incubation in media alone RNA was isolated. The expression levels reported are relative to untreated A549 cells. These experiments were performed in triplicate (n = 6; * significantly different from TNF; † significantly different from TNF + IAP; P < 0.05).
Inhibition of TNF-α–Induced RANTES Gene Expression and RANTES Secretion with TNFR1-Neutralizing Antibodies
In these studies we assessed which TNF receptor was mediating the TNF-α–induced RANTES secretion and gene expression in the airway cultures. As illustrated in Figure 3A, preincubation and coincubation of the epithelial cell cultures with a neutralizing monoclonal antibody against the TNFR1 (neutralizing dose of 50%) suppressed TNF-α–dependent stimulation of RANTES gene expression. This resulted in substantially decreased secretion of the cytokine in the antibody-treated cultures compared with control cell cultures (Figure 3B). Significantly, this response was not observed when treating the cultures with the neutralizing antibody for TNFR2 (Figures 3A and 3B). Collectively, these results suggest that TNF-α functions through TNFR1 to induce RANTES gene expression and secretion.
Figure 3.

Inhibition of TNF-α–induced RANTES gene expression and RANTES secretion with TNFR1 neutralizing antibodies. (A) TNF-α (10 ng/ml) was incubated with cultures for 1 h in media alone (open column), or TNF-α in the presence of a neutralizing monoclonal antibody against the human TNF receptor 1 (TNFR1, neutralizing dose [ND of 50%]) (gray column) or TNF-α in the presence of a neutralizing monoclonal antibody against the human TNF receptor 2 (TNFR2, neutralizing dose [ND50 of 50%]) (dotted column). RT-PCR was performed on RNA isolated from each group of cells. The expression levels reported are relative to control, untreated A549 cells. This experiment was performed in duplicate with triplicate samples (n = 6; * significantly different from TNF; P < 0.05). (B) TNF-α (10 ng/ml) was incubated with cultures for 1 h in media alone (open column), or TNF-α in the presence of a neutralizing monoclonal antibody against the human TNF receptor 1 (TNFR1, neutralizing dose [ND of 50%]) (gray column) or TNF-α in the presence of a neutralizing monoclonal antibody against the human TNF receptor 2 (TNFR2, neutralizing dose [ND of 50%]) (dotted column). After TNF-α stimulation cell cultures were incubated in media alone for 24 h and the level of secreted RANTES in the conditioned media was determined using an ELISA. This experiment was performed in duplicate with triplicate samples. (Values shown are mean ± SD, n = 6 at each point. * Significantly different from TNF-α alone; P < 0.05.)
AP Stimulates Shedding of sTNFR1 in A549 Cells
To determine whether AP could stimulate shedding of sTNFR1, we used a commercially available ELISA to detect sTNFR1 in supernatants of the airway cultures. As shown in Figure 4, there was a significant increase in sTNFR1 levels in the supernatants of AP-treated cells in a concentration-dependent manner. Exposure to the inactive form of AP had no effect on sTNFR1 levels in the supernatant. In addition, we tested whether LPS, applied at concentrations detected in the purified protease, would have an effect on shedding of sTNFR1. Cultures exposed to 500 EU of LPS, which were concentrations present in 25 μg/ml purified protease were not significantly different from control or inactivated protease exposures (Figure 4). In addition, 500 EU of LPS in combination with the inactive form of AP had no effect on sTNFR1 levels in the supernatant suggesting that LPS in combination with a 1,10-phenanthroline sensitive protease was not involved in the TNFR1-induced shedding (data not shown).
Figure 4.

AP stimulates TNFR1 shedding (sTNFR1) from A549 cells. A549 cells were exposed to increasing concentrations of active (gray columns) and inactive (dotted columns) protease and 500 EU/ml LPS (striped columns) for 15 min. After treatments, sTNFR1 in the media was measured by ELISA. Results are expressed as percentage of medium alone. (All values are mean ± SEM, n = 6 at each point. * Significantly different from control; P < 0.05.)
We also tested by ELISA whether active AP degraded sTNFR1 in culture medium alone in a cell-free assay during the 15 min when culture supernatants were exposed to the protease. Experiments demonstrated that exposure of 0.1, 1.0, and 10 μg/ml active AP to purified sTNFR1 for 15 min resulted 99.4 ± 3.7%, 93.2 ± 3.6%, and 93.1 ± 5.7% detection of sTNFR1, respectively, when comparing sTNFR1 incubated in media alone as a control. This suggests that a 15-min AP exposure had a minimal effect on sTNFR1 degradation. Longer exposures, however, of 1.5 and 2 h, resulted in an 87.9 ± 8.1% and 83.2 ± 7.0% detection of sTNFR1, respectively. Collectively, these results suggest that exposure of A549 cells to active AP results in shedding of sTNFR1 into the supernatant, subsequently inhibiting TNF-α–induced RANTES regulation via proteolytic cleavage of TNFR1. In addition, longer exposures (hours) of sTNFR1 to active protease may lead to degradation of sTNFR1.
AP Exposure to Airway Epithelial Cells Decreases Membrane-Associated TNFR1
To investigate the effect of AP on membrane-associated TNFR1, cultures were exposed for 15 min or 2 h to 10 μg/ml AP. After exposures, cells were surface biotinylated and subsequently examined by Western blot analysis to detect membrane-associated TNFR1 in the airway cell cultures. As detected with immunoblots, cultures exposed for 15 min resulted in a decrease in the membrane-associated TNFR1 (Figure 5). Exposure to the inactive form of AP (Figure 5) or to LPS did not alter the membrane-associated TNFR (data not shown). Interestingly, cultures exposed for 2 h did not result in a decrease in the membrane-associated TNFR1 (Figure 5). In addition, we investigated whether or not TNF-α cleaving enzyme (TACE) was involved in the AP-induced TNFR1 shedding. In these studies, cultures were pretreated in the presence or absence of the TACE inhibitor, TAPI (Peptides International, Louisville, KY), a specific hydroxamate inhibitor of metalloprotease disintegrins. Exposure to AP for 15 min in the presence of 1 uM TAPI in vitro in culture medium had no effect on altering AP activity (data not shown). In addition, preincubation and coincubation with TAPI, followed by exposure of cultures to AP, did not prevent the shedding of sTNFR1 (Figure 6) nor alter the effect of AP on TNF-α–induced RANTES secretion (Figure 7). Preincubation with TAPI did not alter the AP-induced reduction in membrane-associated TNFR1 in the cultures (data not shown). Finally, to confirm that TNFR1 can be shed in A549 cells through a TACE-dependent pathway, cultures were exposed to PMA in the presence or absence of TAPI. As demonstrated in Figure 6, PMA stimulated shedding of TNFR1 through a TACE-dependent pathway. The results presented here suggest that AP stimulates shedding of sTNFR1 and may reduce cell surface TNFR1 directly by proteolytic cleavage independent of TACE.
Figure 5.
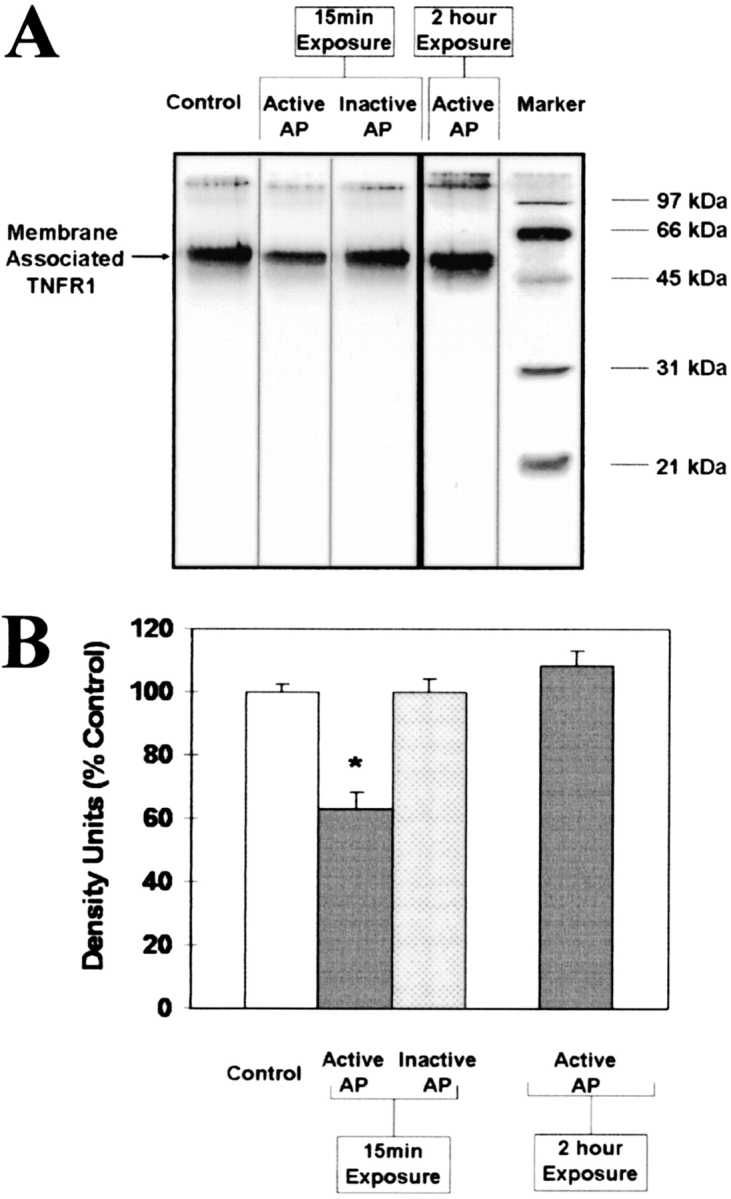
AP reduces membrane-associated TNFR1 on A549 cells. (A) A549 cells were exposed to 10 μg/ml active AP (AP) and inactive AP (IAP) for 15 min and 10 μg/ml active AP (AP) for 2 h. After exposures, cells were collected for surface biotinylation and immunoblotting for membrane-associated TNFR1. (B) Graph summary of data from four separate experiments. Data are expressed as mean percentage of control densitometry units ± SEM (n = 4; *P < 0.05).
Figure 6.

AP-induced shedding of sTNFR1 from A549 cells is not dependent on TACE. Cultures exposed to 10 μg/ml of active AP for 15 min were preincubated (30 min) and coincubated (15 min) in the presence or absence of increasing concentrations of the TACE inhibitor TAPI. In addition, cultures exposed to 0.1 μM PMA for 15 min were preincubated (30 min) and coincubated (15 min) in the presence or absence of TAPI. After incubations, sTNFR1 in the media was measured by ELISA. Results are expressed as percentage of medium alone. (All values are mean ± SD, n = 8 at each point. * Significantly different from medium alone as control; P < 0.05).
Figure 7.

Inhibition of TNF-α–induced RANTES secretion in A549 cells by AP is not dependent on TACE. A549 cells were preincubated for 30 min in the presence or absence of 1 μM TAPI. After preincubations, cell cultures were exposed to AP for 15 min, washed, stimulated with 10 ng/ml TNF-α for 1 h. After TNF-α stimulation, cell supernatants were measured for RANTES by ELISA after 24 h incubation in medium alone. Open column, medium alone; gray column, 10 μg/ml AP; dotted column, 1 μM TAPI and 10 μg/ml AP; black column, 1 μM TAPI alone. All values are mean ± SD (n = 8; * significantly different from control; P < 0.05).
DISCUSSION
In this study, we report that the metalloprotease from P. aeruginosa, referred to as alkaline protease (AP), can inhibit cytokine-induced signaling in A549 cells. Specifically, we have shown that exposure of alkaline protease to the A549 pulmonary epithelial cell line inhibits TNF-α–induced RANTES gene expression and secretion in vitro. Airway exposure to environmental organisms such as P. aeruginosa and their secretory factors activates a cascade of events resulting in airway inflammation. This inflammatory response is critical to the subsequent clearance of organisms within the airway. The airway initiates a response against these organisms with proinflammatory cytokines, such as TNF-α and RANTES, which originate in part from epithelial cells within the respiratory tract (18–21). TNF-α is a particularly potent stimulus of RANTES release in the airway (22); therefore, inhibiting this response would benefit the invading organism. TNF-α can also stimulate inflammation by upregulating the expression of adhesion molecules such as the intercellular adhesion molecule 1 (ICAM-1), which are essential for the recruitment of leukocytes (10, 23, 24).
The role of TNF-α in mediating lung inflammation in the course of gram-negative bacterial pneumonia is incompletely defined. One mechanism by which TNF-α amplifies lung inflammation is by augmenting chemokine production (25, 26). Chemokines are a major component of chemotactic activity, and the chemokine RANTES plays an important role in the recruitment of monocytes and T cells to the site of bacterial infection (27). Recruited monocytes contribute to bacterial killing and tissue repair. Furthermore, macrophages play an important role in resolving the inflammatory responses (28). Similarly, T lymphocytes play major roles in cell-mediated immune responses. The finding that the P. aeruginosa AP inhibits TNF-α–induced RANTES secretion suggests that RANTES levels may be reduced at sites of bacterial protease release even in the early stage of bacterial infection.
The biological effects of TNF-α that result in activation of pathways which regulate inflammatory mediators such as chemokines have been reported to be mediated primarily through TNFR1, and not TNFR2 (11, 29–32). For example, human airway smooth muscle cells require TNFR1 and not TNFR2 for induction of TNF-α–induced RANTES secretion (33). While TNF-α–induced RANTES secretion has been reported in human airway smooth muscle cells to be dependent on TNFR1, it has not been reported in A549 cells. In the present study we demonstrate that TNF-α–induced RANTES secretion is regulated through TNFR1 in A549 cells. Using a neutralizing antibody directed against TNFR1 resulted in a diminished cellular response to TNF-α with respect to RANTES gene expression and secretion. A neutralizing antibody directed against TNFR2 had no effect on these events. Therefore, we conclude that TNFR1 appears to be the main receptor mediating TNF-α–induced RANTES secretion and gene expression in A549 cells.
Our findings demonstrate that AP inhibits TNF-α–induced RANTES gene expression and secretion, stimulates shedding of membrane-associated TNFR1, and increases soluble TNFR1 (sTNFR1) in culture supernatants in A549 cultures. Interestingly, both active and inactive protease altered TNF-α–induced RANTES gene expression. One explanation of these data is that there may be multiple pathways by which TNF-α–induced RANTES gene expression may be inhibited. One of these pathways may be dependent on the active enzyme to decrease the availability of TNFR1, and an additional pathway that is independent of proteolytic activity. Data presented here suggests that while the active alkaline protease was more effective inhibiting TNF-α–induced RANTES gene expression, both active and inactive protease were capable of inhibiting TNF-α–induced RANTES gene expression.
Inhibition of TNF-α–induced RANTES gene expression, however, did not appear to be a result of LPS contamination because cultures exposed to 500 EU of LPS, which are concentrations greater than what was identified in our purified protease (200 EU/10 μg/ml AP), did not alter TNF-α–induced RANTES gene expression (data not shown). Data presented here also demonstrate that neither the inactive form of AP at all concentrations, nor 500 EU of LPS, had an effect on TNF-α–induced RANTES secretion, shedding of membrane associated TNFR1, or sTNFR1 levels; these results suggest that the active protease is required for these inhibitory events to occur. Data presented here, however, do not exclude the possibility that the effect of purified active AP may require some level of LPS. Since all of the LPS could not be completely removed in the purification process of the active AP, we could not determine whether or not LPS, in combination with the active protease, was involved in the biological responses.
AP-induced TNFR1 shedding may occur by direct cleavage of TNFR1 on the cell membrane or through the activation of TNF-α–cleaving enzyme (TACE, ADAM-17). TACE is a member of the disintegrin and metalloproteinase (ADAM) family of zinc-metalloproteases located within the membrane of cells in its inactive form (13). TACE has a pro-domain, and pro-domain removal is thought to be a perquisite for TACE protease activity (33). Once activated, TACE cleaves the membrane-associated TNFR1, resulting in an increase in soluble TNFR1 (sTNFR1) and a subsequent decrease in membrane-bound TNFR1.
To explore the potential involvement of TACE, we used a specific inhibitor of TACE, TAPI, and investigated its effect on AP-induced shedding of sTNFR1 and its effect on inhibiting TNF-α–induced RANTES secretion. We report that AP-induced TNFR1 shedding could not be blocked with up to 1 μM concentrations of the TACE inhibitor TAPI. In addition, both preincubation and coincubation of cells with TAPI did not alter the inhibitory effect that AP exerted on TNF-α–induced RANTES secretion. These results suggest that AP exerts its effects directly on TNFR1 at the cell surface rather than mediating receptor release through a TACE-dependent mechanism as seen with PMA-induced TNFR1 shedding in the A549 cells. Further, our in vitro studies investigating the interactions of AP and TAPI demonstrated that AP activity was not altered in the presence of 1 μM TAPI (data not shown). The effect of AP on soluble TNF-α release was not surprising, since it is in agreement with recent studies demonstrating that matrix metalloproteinases (MMPs), such as MMP1, MMP7, MMP9, and MMP17, can stimulate shedding of soluble TNF (34–36). In addition, a study reported in monocytes suggested that MMP-7 may stimulate TNFR1 shedding from the cell surface through a direct mechanism independent of TACE (34). Further studies investigating the direct effect of AP on TNF receptors in vitro are currently under investigation in our laboratory.
Although TNFR1 signaling, including TNF-α–induced RANTES gene expression and secretion, is mediated through cell-surface receptors, the majority of TNFR1 molecules are stored in the Golgi apparatus (37, 38). A recent study investigating TNFR1 molecules in endothelial cells suggests that the TNFR1 stored in the Golgi pool serves to replenish the cell's surface receptors and may also provide a source for shed receptors (39). A substance such as histamine, which decreases cell surface TNFR1, results in mobilization of TNFR1 from the Golgi to the cell surface in cultured endothelial cells (39). Our data support a similar effect, in that exposure to AP causes a transient decrease of both cell surface TNFR1 and TNF-α–induced RANTES signaling. Data presented here demonstrate that after 15 min of AP exposure, both cell surface TNFR1 and TNF-α–induced RANTES signaling were inhibited. Each of these events appeared to be transient in that both cell surface TNFR1 and TNF-α–induced RANTES signaling recovered after 2 h. These findings may be a result of mobilization of the TNFR1 from the Golgi pool to the cell surface after AP-mediated shedding of receptors on the A549 cells. Future experiments are being considered to investigate the effect of both short and long exposures of AP on transcriptional regulation of TNFR1, cytosolic TNFR1, and membrane-associated TNFR1 in both A549 cells and primary cultures of fully differentiated normal human bronchial epithelial cells in our laboratory.
In summary, we report that alkaline protease (alias aeruginolysin), isolated from P. aeruginosa, induces a transient inhibition of TNF-α–stimulated RANTES gene expression and secretion. The TNF-α–induced RANTES regulation in the A549 pulmonary epithelial cell line is mediated through TNFR1. In addition, AP exposure resulted in a decrease in membrane-associated TNFR1 and a subsequent increase in sTNFR1 in the culture supernantants of A549 cells. The orchestrated release of cytokines and other immunomodulatory factors from airway cells play an important role in the immune response to bacterial infections. The imbalance of cytokines as a result of proteolysis may interfere with critical responses regulated by these proteins. We speculate that AP may contribute to the pathophysiology of P. aeruginosa–associated lung disease through alterations in the cytokine–chemokine response of the airway epithelium. Further understanding of the mechanisms involved in AP-induced shedding of TNFR1 in the airway epithelium will enhance our understanding of the role of P. aeruginosa in lung disease.
This work was funded by grants HL 37019 from the NIH and from the University of Georgia Biomedical Health Science Institute.
Conflict of Interest Statement: T.M.K. does not have a financial relationship with an entity that has an interest in the subject of this manuscript. K.M. does not have a financial relationship with an entity that has an interest in the subject of this manuscript. Except for patents that belong to the University of Georgia, J.P has no declared conflicts of interest. C.L.J. does not have a financial relationship with an entity that has an interest in the subject of this manuscript. J.T. does not have a financial relationship with an entity that has an interest in the subject of this manuscript.
References
- 1.Garau J, Gomez L. Pseudomonas aeruginosa pneumonia. Curr Opin Infect Dis 2003;16:135–143. [DOI] [PubMed] [Google Scholar]
- 2.Fick RBJ. Pathogenesis of the Pseudomonas lung lesion in cystic fibrosis. Chest 1989;96:158–164. [DOI] [PubMed] [Google Scholar]
- 3.Leidal KG, Munson KL, Denning GM. Small molecular weight secretory factors from Pseudomonas aeruginosa have opposite effects on IL-8 and RANTES expression by human airway eoithelial cells. Am J Respir Cell Mol Biol 2001;25:186–195. [DOI] [PubMed] [Google Scholar]
- 4.Buret A, Cripps AW. The immuoevasive activities of Pseudomonas aeruginosa: relevance for cystic fibrosis. Am Rev Respir Dis 1993;148:793–805. [DOI] [PubMed] [Google Scholar]
- 5.Suter S. The role of bacterial proteases in the pathogenesis of cystic fibrosis. Am J Respir Crit Care Med 1994;150:S118–S122. [DOI] [PubMed] [Google Scholar]
- 6.Parmely M, Gale A, Clabaugh M, Horvat R, Zhoul WW. Proteolytic inactivation of cytokines by Pseudomonas aeruginosa. Infect Immun 1990;58:3009–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horvat R, Parmely M. Pseudomonas aeruginosa alkaline protease degrades human gamma interferon and inhibits its bioactivity. Infect Immun 1988;56:1673–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leidal KG, Munson KL, Johnson MC, Denning GM. Metalloproteases from Pseudomonas aeruginosa degrade human RANTES, MCP-1, and ENA-78. J Interferon Cytokine Res 2003;23:307–318. [DOI] [PubMed] [Google Scholar]
- 9.Denning GM, Wollenweber LA, Railsback MA, Cox CD, Stoll L, Britigan BE. Pseudomonas pyocyanin increases interleukin-8 expression by human airway epithelial cells. Infect Immun 1998;6:5777–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ermert M, Pantazis C, Duncker HR, Grimminger F, Seeger W, Ermert L. In situ localization of TNFalpha/β, TACE and TNF receptors TNF-R1 and TNF-R2 in control and LPS-treated lung tissue. Cytokine 2003;22:89–100. [DOI] [PubMed] [Google Scholar]
- 11.Krunkosky TM, Fischer BM, Martin LD, Jones N, Akley NJ, Adler KB. Effects of TNF-α on expression of ICAM-1 in human airway epithelial cells in vitro. Am J Respir Cell Mol Biol 2000;22:685–692. [DOI] [PubMed] [Google Scholar]
- 12.Thomas PS. Tumour necrosis factor-alpha: the role of this multifunctional cytokine in asthma. Immunol Cell Biol 2001;79:132–140. [DOI] [PubMed] [Google Scholar]
- 13.Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Showa D, Peschon JJ, Black RA. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J Biol Chem 2000;275:14608–14614. [DOI] [PubMed] [Google Scholar]
- 14.Van Zee KJ, Kohno T, Fischer E, Rock CS, Moldawer LL, Lowry SF. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necrosis factor α in vitro and in vivo. Proc Natl Acad Sci USA 1992;89:4845–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maeda H, Morihara K. Serralysin and related bacterial proteinases. Methods Enzymol 1995;248:395–413. [DOI] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 17.Kleinbaum DG, Kupper LL, Muller KE. Applied regression analysis and multivariate methods, 2nd ed. Boston: PSW-Kent; 1988.
- 18.De Bentzmann S, Plotkowski C, Puchelle E. Receptors in the Pseudomonas aeruginosa adherence to injured and repairing airway epithelium. Am J Respir Crit Care Med 1996;154:S155–S162. [DOI] [PubMed] [Google Scholar]
- 19.Wilson R, Dowling RB, Jackson AD. The effects of bacterial products on airway cells and their function. Am J Respir Crit Care Med 1996;154:S197–S201. [DOI] [PubMed] [Google Scholar]
- 20.Berger M. Inflammatory mediators in cystic fibrosis lung disease. Allergy Asthma Proc 2002;23:19–25. [PubMed] [Google Scholar]
- 21.Chung KF. Cytokines in chronic obstructive pulmonary disease. Eur Respir J Suppl 2002;34:50s–59s. [PubMed] [Google Scholar]
- 22.Terada N, Maesako K, Hamano N, Ikeda T, Sai M, Yamashita T, Fukuda S, Konno A. RANTES production in nasal epithelial cells and endothelial cells. J Allergy Clin Immunol 1996;98:S230–S237. [DOI] [PubMed] [Google Scholar]
- 23.Martin LD, Rochelle LG, Fischer BM, Krunkosky TM, Adler KB. Airway epithelium as an effector of inflammation: molecular regulation of secondary mediators. Eur Respir J 1997;10:2139–2146. [DOI] [PubMed] [Google Scholar]
- 24.Krunkosky TM, Martin LD, Fischer BM, Voynow JA, Adler KB. Effects of TNF alpha on expression of ICAM-1 in human airway epithelial cells in vitro: oxidant-mediated pathways and transcription factors. Free Radic Biol Med 2003;35:1158–1167. [DOI] [PubMed] [Google Scholar]
- 25.Strieter RM, Kunkel SL. Acute lung injury: the role of cytokines in the elicitation of neutrophils. J Investig Med 1994;4:640–651. [PubMed] [Google Scholar]
- 26.Skerrett SJ, Martin TR, Chi EY, Peschon JJ, Mohler KM, Wilson CB. Role of the type 1 TNF receptor in lung inflammation after inhalation of endotoxin or Pseudomonas aerugnosa. Am J Physiol 1999;27:L715–L727. [DOI] [PubMed] [Google Scholar]
- 27.Schall T, Bacon K, Toy K, Goeddel D. Selective attraction of monocytes and T-lymphocytes of the memory phenotype by cytokine RANTES. Nature 1990;347:669–671. [DOI] [PubMed] [Google Scholar]
- 28.Kooguchi K, Hashimoto S, Kobayashi A, Kitamura Y, Kudoh I, Wiener-Kronish J, Sawa T. Role of alveolar macrophages in initiation and regulation of inflammation in Pseudomonas aeruginosa pneumonia. Infect Immun 1998;66:3164–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science 2002;296:1634–1635. [DOI] [PubMed] [Google Scholar]
- 30.Kondo S, Sauder DN. Tumor necrosis factor (TNF) receptor type 1 (p55) is a main mediator for TNF-alpha-induced skin inflammation. Eur J Immunol 1997;27:1713–1718. [DOI] [PubMed] [Google Scholar]
- 31.Kuai J, Wooters J, Hall JP, Rao VR, Nickbarg E, Li B, Kishore MC, Qiu Y, Lin LL. NAK is recruited to the TNFR1 complex in a TNFa-dependent manner and promotes the production of RANTES:Identification of endogenous TNFR-interacting proteins by a proteomic approach. J Biol Chem 2004;279:53266–53271. [DOI] [PubMed] [Google Scholar]
- 32.Amrani Y, Ammit AJ Jr, Panettieri RA. Tumor necrosis factor receptor (TNFR) 1, but not TNFR2, mediates tumor necrosis-α-induced interleukin-6 and RANTES in human airway smooth muscle cells: Role of p38 and p42/44 mitogen-activated protein kinases. Mol Pharmacol 2001;6:646–655. [PubMed] [Google Scholar]
- 33.Black RA. Tumor necrosis factor-α converting enzyme. Int J Biochem Cell Biol 2002;34:1–5. [DOI] [PubMed] [Google Scholar]
- 34.Haro H, Crawford HC, Fingleton B, Shinomiya K, Spengler DM, Matrisian LM. Matrix metalloproteinase-3–dependent generation of a macrophage chemoattractant in a model of herniated disc resorption. J Clin Invest 2000;105:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lunn CA, Fan X, Dalie B, Miller K, Zavodny PJ, Narula SK, Lundell D. Purification of ADAM 10 from bovine spleen as a TNFalpha convertase. FEBS Lett 1997;400:333–335. [DOI] [PubMed] [Google Scholar]
- 36.English WR, Puente XS, Freije JM, Knauper V, Amour A, Merryweather A, Lopez-Otin C, Murphy G. Membrane type 4 matrix metalloproteinase (MMP17) has tumor necrosis factor-alpha convertase activity but does not activate pro-MMP2. J Biol Chem 2000;275:14046–14055. [DOI] [PubMed] [Google Scholar]
- 37.Bladley JR, Thiru S, Pober JS. Disparate localization of 55-kd and 75-kd tumor necrosis factor receptors in human endothelial cells. Am J Pathol 1995;146:27–32. [PMC free article] [PubMed] [Google Scholar]
- 38.Jones SJ, Ledgerwood EC, Prins JB, Galbraith J, Johnson DR, Pober JS, Bradley JR. TNF recruits TRADD to the plasma membrane but not the trans-Golgi network, the principal subcellular location of TNF-R1. J Immunol 1999;162:1042–1048. [PubMed] [Google Scholar]
- 39.Wang J, Al-Lamki RS, Zhang H, Kirkiles-Smith N, Gaeta ML, Thiru S, Pober JS, Bradley JR. Histamine antagonizes tumor necrosis factor (TNF) signaling by stimulating TNF receptor shedding from cell surface and Golgi storage pool. J Biol Chem 2003;278:21751–21760. [DOI] [PubMed] [Google Scholar]