

Abstract
The autism spectrum disorders (ASD) are a heterogeneous set of developmental disorders characterized at their core by deficits in social interaction and communication. Current psychiatric nosology groups this broad set of disorders with strong genetic liability and multiple etiologies into the same diagnostic category. This heterogeneity has challenged genetic analyses. But shared patient resources, genomic technologies, more refined phenotypes, and novel computational approaches have begun to yield dividends in defining the genetic mechanisms at work. Over the last five years, a large number of autism susceptibility loci have emerged, redefining our notion of autism’s etiologies, and reframing how we think about ASD.
Introduction
Autism is a neurodevelopmental disorder defined by three categories of deficits: 1) abnormal development or impairment of social interaction, 2) abnormal development or impairment of communication skills, and 3) stereotypic and repetitive behaviors [1]. Autism is part of a larger family of neurodevelopmental disorders categorized by the Diagnostic and Statistical Manual IV-R under the term Pervasive Developmental Disorders (PDD) [1]. PDD includes Asperger Syndrome (AS), where language appears normal, and pervasive developmental disorder not otherwise specified (PDD-NOS) in which children meet some but not all criteria for autism. Collectively, these disorders are known as the autism spectrum disorders (ASD) [1]. Rett syndrome and Childhood Disintegrative Disorder also are classified as a PDD, but are exclusionary for a diagnosis of autism.
Hypothesized by Kanner to be an innate or inborn disorder in his original description, autism was not formally determined to be a genetic disorder until Folstein and Rutter demonstrated a greater than 50% concordance for monozygotic, versus 0% for dizygotic twins. More recent twin studies have observed as high as 90% trait heritability for monozygotic twins [2], and family studies suggest a 22 fold increased risk over the general population for first-degree relatives [3], although this does not use the current CDC prevalence estimate of 1/152 for ASD [4]. Taken together, these studies indicate a high genetic liability, while leaving some room for environmental factors that may influence the penetrance or expressivity of these disorders with respect to genetic risk factors.
Although ASD is highly heritable, the identification of candidate genes has been hindered by the heterogeneity of the syndrome and insufficient numbers of participants, as compared to whole genome association studies in other complex genetic disorder. The establishment of collaborative groups, such as the International Molecular Genetic Study of Autism Consortium (IMGSAC) and Autism Genome Project Consortium [5,6], and shared resources, such as the Autism Genetic Resource Exchange Consortium (AGRE) [7], were therefore important steps in facilitating the identification of candidate genes. Linkage peaks on chromosomes 7q22–32 [5,8] and chromosome 17q21 [9–11] have been replicated. However most linkage signals have not been replicated, despite large increases in sample size, consistent with significant genetic heterogeneity [6,12]. Recently, a whole genome association study involving over 1500 cases and controls combined from different cohorts has identified and replicated at least one locus at genome-wide significance [13]. This demonstrates the promise of this approach, while at the same time, suggesting that very large sample sizes will be needed to identify additional genetic risk due to common alleles. Currently, there are over 25 different loci that may be considered autism susceptibility candidate genes (ASCG) and many more implicated loci are under investigation [12]. Most of these are rare, Mendelian mutations, including copy number variation (CNV) or syndromic forms of autism, and only a few are due to common genetic variation. In this brief overview, we will try to highlight some of the major advances in the study of autism, as well as discuss what the known ASCG can tell us about the neurodevelopmental mechanisms that may be causative.
A Phenotypic Renaissance
During the 1970s, psychiatric disorders were defined as deviations from the normal spectrum of behavior, but due to practical and economic factors, diagnostic schemes eventually became highly categorical. This categorization, while necessary from a clinical and educational standpoint, resulted in large groups of heterogeneous patients with diverse etiologies being defined by a single terminology. This clinical diagnostic schema based on the DSM was the primary means of phenotypic classification for early genetic work. But for modern genome-wide studies, clinical impression gave way to improved schema for classification using standardized and reliable research tools such as the ADOS and ADI [14,15]. Still the lack of formal replication of most autism linkage peaks [12], despite more than a 10-fold increase in sample size [6], indicates genetic heterogeneity. This, in conjunction with the wide range of phenotypes observed within the categorical classification of autism, suggests that methods reducing these two sources of complexity would increase power to identify ASCG [16].
Newer work has returned to the concept of disease-related spectrums in a more refined manner based on the concept of endophenotypes [17]. In autism, endophenotypes such as presence or lack of language, age at first word, age at first phrase, social cognition, gender, restrictive repetitive behaviors, and best estimate IQ, have been used in an attempt to increase power [9,16,18,19]. Alarcón and coworkers initially utilized this approach to identify a locus related to language delay on chromosome 7q [16,20]. Subsequent linkage directed association identified CNTNAP2 as an ASCG using an age at first word endophenotype, one of the first ASCG identified from a comprehensive evaluation of common variants within a linkage region defined by a whole genome linkage scan [21]. Other support for the more general involvement of this gene in language development were the findings that it is associated with Specific Language Impairment and is related to monogenic disorders affecting language [22,23]. Thus, the further identification and use of endophenotypes related to other autism-related domains will likely increase power to detect ASCG.
The Genomic Revolution
Structural chromosomal variations, including CNVs, have been shown to play an important role in the etiology of ASD [24]. De novo CNVs, hypothesized to be ASD-specific, have been identified in up to 7–10% of sporadic ASD [24,25] De novo CNVs are less frequent in multiplex families, occurring only in about 2% of families screened [24,26], possibly suggesting different genetic liabilities in simplex and multiplex ASD. Recurrent CNVs at 15q11–13 (1–3% of ASD patients), 16p11 (~1% of ASD patients), and 22q11–13 [6,24,25,27,28] have been confirmed in multiple studies. Many of the CNVs identified overlap with previously identified mental retardation (MR) loci or chromosomal syndromes. For example, Phelan-McDermid Syndrome defined by deletions of 22q13 overlaps with deletions and duplications of the gene SHANK3, an ASCG [29]. This is not a surprise as 94% of the original Phelan-McDermid Syndrome patients met ASD criteria via the CARS phenotyping tool. Therefore, analyses of genomic syndromes like Phelan-McDermid Syndrome or other syndromes that are comorbid with ASD diagnoses indicate important genes in the etiology of ASD [12,30].
The syndromes of structural chromosomal abnormalities also may provide insight into the mechanism behind sporadic ASD. For example, advanced paternal age is an established risk factor for ASD [31], and genetic instability through altered recombination efficiency increases as a parent ages. The increased recombination acts as a mechanism for multiple MR-associated syndromes [32]. Therefore, while it is unknown if paternal-age related recombination is a causative agent in CNV formation in autism, it is an attractive hypothesis. Our preliminary analysis of AGRE and other cohorts suggests that this may be the case (Abrahams and Geschwind, unpublished data).
Candidates and Compatriots
Epistasis is a basic and ubiquitous genetic paradigm well known within the developmental biology community. With the advent of large protein-protein interaction maps, full genome expression profiles and large-scale computing resources, network and pathway analyses offer promise of dealing with autism’s complexities. Iossifov and coworkers [33] utilized the idea that interacting proteins involved in linear signaling pathways would have a similar chance of being involved in the etiology of ASD. By overlaying protein interaction datasets with linkage analysis, they were able to identify 24 putative novel ASCG [33], which need further confirmation.
Pathway analysis has also been used to identify novel associations with ASD utilizing a more standard biological approach. In 2006, Campbell and coworkers identified MET as an ASCG [34]. Utilizing knowledge of the MET signaling pathway, they investigated genes involved with this pathway and identified that SERPINE and PLAUR, two components critical for HGF (MET’s ligand) regulation, potentially increase the risk of ASD [35]. Furthermore, MET signals through the PI3K-AKT pathway that contains two known ASCG (PTEN and CYFIP1) and contains three genes that are known to be causative for syndromes that co-occur with ASD [Tuberous Sclerosis (20% diagnosed with ASD) and Fragile X (25% of the males and 6% of females diagnosed with ASD)] [12] (Figure 1). Therefore, it is possible that alleles of SERPINE, PLAUR, MET, PTEN, TSC complex, FMR1, and CYFIP1 might contribute to autism via epistatic interactions. Other than autism, there is not a large overlap of the phenotypes related to each of these genes, but this could be explained by the pleiotropic nature of each of these genes. Therefore, one could hypothesize that the ASD phenotype is common to a large number of epistatic interactions, but due to pleiotropy of the individual loci, a heterogeneous phenotype emerges.
Figure 1. Epistatic and Pleiotropic Relationships of Autism Susceptibility Candidate Genes.
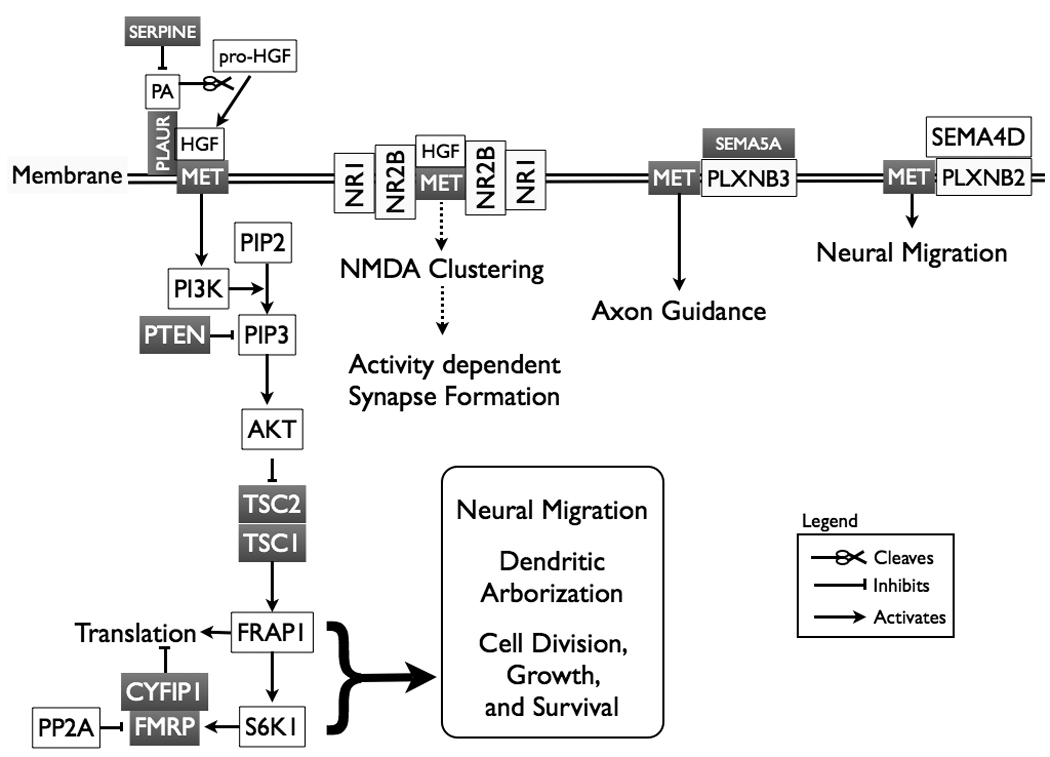
Analyzing the MET-PI3K-AKT pathway can identify epistatic relationships of ASCG. Genes that are in gray have been associated with ASD, Tuberous Sclerosis (TSC1 and 2) or Fragile X (FMRP), syndromes that are comorbid with ASD. In the left most pathway, MET is seen interacting with PLAUR. PLAUR is the receptor for the tissue specific plasminogen activator (PA), a protease that cleaves HGF into its active form. SERPINE inhibits PA from activating HGF. After HGF binds MET, MET signals downstream through the canonical PI3K-AKT pathway that has a potential terminal endpoint of translational regulation by FMR1 and CYFIP. We propose that the core ASD phenotype may be represented by biological pathway convergence, while the pleiotropic nature of each gene provides the overlying phenotypic variability. For example, MET can signal through multiple receptor complexes that define the functional outcome. For example, MET clustering of NMDA receptors can affect long-term potentiation; while MET binding with members of the plexin receptor family dictate neural migration or axon guidance functions.
Following this logic, we utilized the Ingenuity pathway analysis software to identify published direct binding partners, transcriptional regulators, and translational regulators for a set of ASCG [12,26,35–37] to determine if the connections might suggest epistatic relationships. Of 33 candidate genes, direct or indirect interactions (2 degrees of separation at most) can be established, with the obvious caveat that some genes have been recently identified and display a reduced connectivity due to reduced publication volume (Figure 2). The limitations to this analysis lie in validation of the interactions and a realization that interactions are often temporally and spatially specific events. Therefore, while the interaction may be possible, it may not occur in the right tissue or at the right time to affect the pathology of ASD. Despite its limitations, this preliminary network of genes suggests true interconnectedness of several currently known ASCG. The potential for non-linearity and feedback loops suggested by even these simple relationships may obscure phenotypic effects from single polymorphisms or mutations.
Figure 2. A social network for autism susceptibility candidate genes.
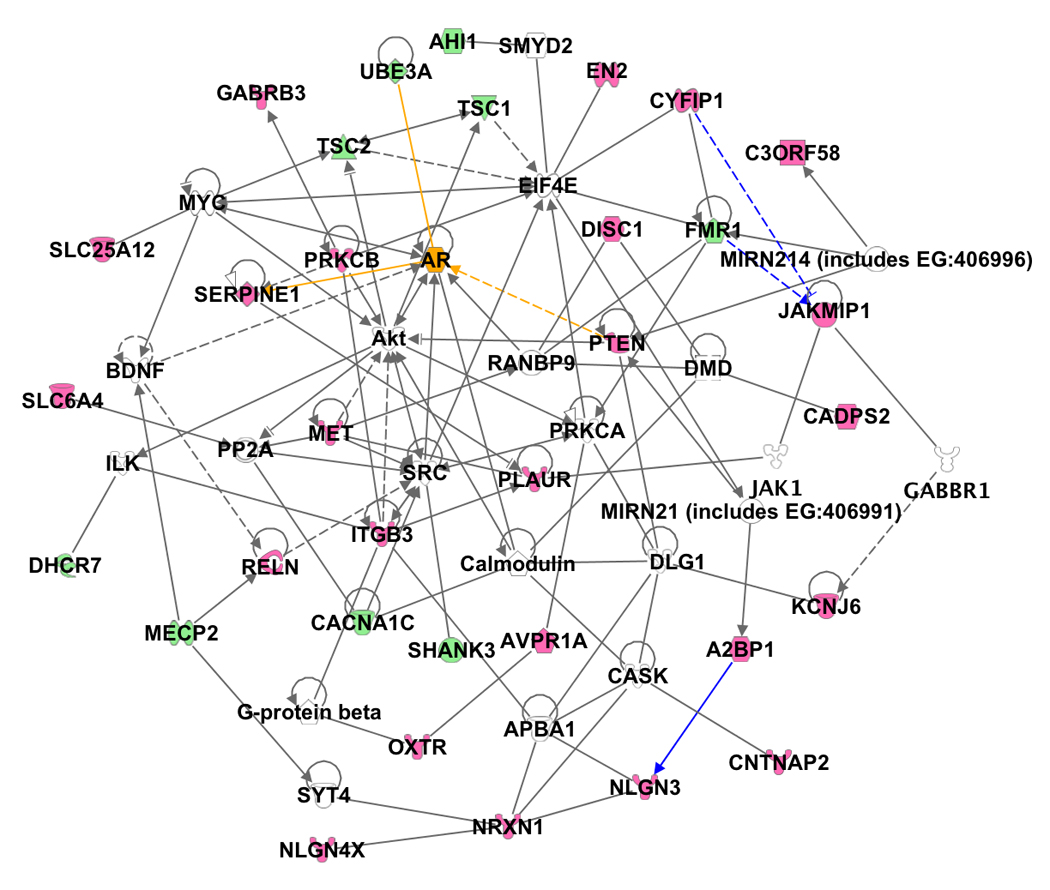
Analysis of the relationships between 33 ASCG (pink emblems) and associated syndrome genes (green emblems) were analyzed using the Ingenuity Pathway Analysis (http://www.ingenuity.com). Direct (solid lines) and indirect (dashed lines) associations between the ASCG demonstrate the close relationship, less than 2 interactions between each ASCG. For clarity, we did not show all of the associations identified, but focus primarily on direct associations that link ASCG. Recently published interactions linking FMR1, CYFIP1, and JAKMIP1 by gene regulation, and A2BP1 (FOX1) with NLGN3 by splicing regulation were added as custom interactions (blue lines). The androgen receptor (orange emblem) and three of its interactions with ASCG (orange lines) are highlighted to demonstrate the correlation with the extreme male brain hypothesis. This is a first attempt to explore the connectivity between these genes, and therefore is not comprehensive. We do not mean to imply a single pathway as causative, as there may be many pathways that could be implicated.
Continuing Quandaries
Despite great advances in the genetics of ASD, there are still at least two major unanswered quandaries: the basis of the skewed sex ratio and the effect of the environment on ASD. Early studies demonstrated that males are diagnosed with ASD four times as often as females, and gender skewing becomes more pronounced in high-functioning autistic and Aspergers patients as the ratio approaches ten males to every one female [38]. Some of the gender bias might be explained by a larger than average number of ASCG on the X chromosome [12], as is the case with candidate loci for other forms of mental retardation. Marshall and coworkers observe that inherited X-linked CNVs are maternally transmitted and suggest this as a possible mechanism for the gender bias [25]. Sebat and coworkers found equal numbers of de novo CNVs between the genders [24], suggesting the bias is not due to increased mutability of the sex chromosomes, at least in terms of de novo structural variation. A second factor that may lead to the increased gender ratio is influence of autosomal loci that preferentially affect males or females, as originally demonstrated by Stone and coworkers [9]. In the largest linkage study to date, the AGP also found evidence for autosomal, but not X chromosome, sex related autism loci [6].
A non-genetic hypothesis has also been proposed to explain the gender difference. “The extreme male brain hypothesis” [39] is based on the fact that fetal testosterone acts as an environmental agent and alters sex-specific neuronal development in utero causing a decrease in function within the social and communication spectrums, thereby predisposing an individual with a susceptible genotype toward the diagnosis of ASD [39]. Our Ingenuity network, presented here, may be consistent with this hypothesis or at least suggests a role for the androgen receptor as it is regulated by PTEN, directly binds UBE3a, and inhibits the transcription of SERPINE, three ASCG (Figure 2, orange lines). However, there is as of yet no direct evidence for “the extreme male brain hypothesis”, as most of the evidence is correlative in nature.
The second quandary is the role of the environment. Given that the concordance of monozygotic twins is not 100%, and certain perinatal factors increase the risk for ASD, a role for environmental factors needs to be considered, eg. parental age [31], premature birth [40], or immune interaction [41]. Herbert and coworkers overlaid the linkage peaks identified from several ASD full genome scans with environmental, toxicologic, and immune related gene databases, finding an overlap of 135 genes [42]. While this analysis was perhaps overly inclusive in the scope of the ASCG genes selected and lacks rigorous statistical substantiation, it does indicate a potential set of gene-environment interactions to test.
Synthesis of an Integrated Model
We have recently discussed genetic models for the etiology of ASD [43] and provided a neurodevelopmental synthesis of autism that is based on altered connectivity between higher order cortical association areas, especially anterior frontal and temporal lobes [44]. Anatomical evidence suggests that during the first three years of life, the trajectory of brain growth is elevated in ASD, head circumference increasing from approximately normal to 10% larger than age matched peers [45]. During this time period, ages two to four, this growth appears to localize in the frontal cortex, temporal lobes, and amygdala [45]. Patients with Fragile X and co-occurring ASD also have an increase in head circumference size compared to those with Fragile X alone [46]. Diffusion tensor imaging shows that the axon tracts to these areas are disrupted [45], consistent with the notion of a developmental disconnection. It is notable that rare mutations in PTEN cause syndromic forms of macrocephaly and autism in humans [47]. Mouse knockouts of PTEN also display macrocephaly and display differences in axon and dendrite arborization. It is not yet know whether PTEN causes ASD directly via changes in brain size, or via effects on axon and dendritic arborization, and the changes in brain size are secondary.
A second major model for autism pathogenesis is that ASD is caused by a disruption of the formation or maintenance of synaptic connections. This model is driven by the identification of the synaptic proteins NLGN3, NLGN4X, NRXN1, and SHANK3 as ASCG [48]. As predicted by this model, the gene ontology terms appear to suggest a significant enrichment of genes that have a synaptic function annotation (Figure 3a,b). On the other hand, a model proposing that ASD is a synaptic disorder may be too limiting. The distribution of gene ontology functions spanning the entirety of nervous system development and function for the 33 ASCG analyzed suggests that broader biological functions may be involved (Figure 3a,b), and many of these genes cause other neurodevelopmental syndromes. So, the broad notion of synaptic dysfunction while clearly contributory may not be sufficient to account for the specificity autism.
Figure 3. Functional annotation of the autism susceptibility candidate genes.

Annotation was performed using the gene ontology (GO) nomenclature assignments from the National Center for Biotechnology Information (NCBI) for the 33 ASCG used for the Ingenuity analysis (A). GO terminologies are divided into function, process and component. For easier visualization, neuro-specific annotations were compiled using the DAVID Bioinformatics database (http://david.abcc.ncifcrf.gov/) as a histogram (B). The 33 genes from the previous analysis were divided by their GO category (blue bars), and as a percentage of the total genes annotated within the same GO category (green bars). The significance of the enrichment is denoted by p-values, shown adjacent to the GO category. The limitations of the GO terminology are that the categories utilize general descriptors to categorize protein function; some functions are not present in the database, and some protein lack annotation, excluding them from analysis. For example, FMR1 is known to function in dendritic morphogenesis; but this function is not described in its GO annotation. Despite limitations, the GO annotations clearly show that ASD affects multiple processes in the brain suggesting models that address a single aspect of biological function are likely to be too limiting.
Conclusion
In this brief overview, we have outlined how genetic advances have led to a new level in understanding ASD etiologies. Genomic tools allowing for the identification of de novo and heritable CNVs have so far contributed the most to our understanding of ASD, explaining about 10% of sporadic ASD. The analysis of co-morbid phenotypes and endophenotypes also provides a promising avenue of investigation, as evidenced by the association of common variants in CNTNAP2 with language endophenotypes in ASD and SLI. Surprisingly, many of the syndromic or rare ASCG appear to potentially interact at the level of molecular pathways, making it likely that mutations of one ASCG may affect the expression and function of others during development. This also provides hope that common treatments could be developed for those with etiologically distinct genetic forms of ASD. Any useful model of ASD pathogenesis will need to combine data from genetic, neurodevelopmental, and cell biological studies in model systems with functional and anatomical studies in human populations. This is especially true, because the majority of ASCG are not ASD specific and have been implicated in other neurodevelopmental disorders such as intellectual disability, epilepsy or psychiatric conditions [12,23,30,33,49]. The mechanism of the male gender bias and the degree and manner in which environmental influences will contribute to the etiology of ASD remain open questions.
Acknowledgements
We sincerely apologize to our colleagues and scientists who have contributed greatly to the literature discussed here, but due to space restrictions were not cited. We would like to thank Dr. Brett Abrahams, Dr. Brent Fogel, Dr. Shaohong Cheng, Li Hong, and Dr. Genevieve Konopka for the critical reading of this manuscript. This work is supported by the National Institute of Child Health and Human Development (BRB, NIH T-32 HD0703230), Autism Speaks, and the National Institute of Mental Health (DHG, P-50 HD055784-01 and RO-1 MH81754-01). We also would like to extend our deepest gratitude to the families that have participated in the Autism Resource Genetic Exchange (AGRE) for helping to advance the field of autism research.
Works Cited
- 1.Association AP. Diagnostic and Statistical Manual of Mental Disorders edn Fourth. Washington DC: American Psychiatric Association; 1994. [Google Scholar]
- 2.Ronald A, Happe F, Bolton P, Butcher LM, Price TS, Wheelwright S, Baron-Cohen S, Plomin R. Genetic heterogeneity between the three components of the autism spectrum: a twin study. J Am Acad Child Adolesc Psychiatry. 2006;45:691–699. doi: 10.1097/01.chi.0000215325.13058.9d.Demonstrates independent inheritance, with up to 90% heritability, of the three core deficits of ASD in both typically and atypically developing twins.
- 3.Lauritsen MB, Pedersen CB, Mortensen PB. The incidence and prevalence of pervasive developmental disorders: a Danish population-based study. Psychol Med. 2004;34:1339–1346. doi: 10.1017/s0033291704002387. [DOI] [PubMed] [Google Scholar]
- 4.Prevalence of autism spectrum disorders--autism and developmental disabilities monitoring network, 14 sites, United States, 2002. MMWR Surveill Summ. 2007;56:12–28. [PubMed] [Google Scholar]
- 5.International Molecular Genetic Study of Autism Consortium. A full genome screen for autism with evidence for linkage to a region on chromosome 7q. Hum Mol Genet. 1998;7:571–578. doi: 10.1093/hmg/7.3.571. [DOI] [PubMed] [Google Scholar]
- 6.Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–328. doi: 10.1038/ng1985.The Autism Genome Project Consortium performed SNP and CNV analysis of 1,181 families, the largest analysis of its kind for ASD, demonstrating a suggestive linkage peak on chromosome 11p12–13 for all families and CNVs within autism susceptibility candidate loci.
- 7.Geschwind DH, Sowinski J, Lord C, Iversen P, Shestack J, Jones P, Ducat L, Spence SJ. The autism genetic resource exchange: a resource for the study of autism and related neuropsychiatric conditions. Am J Hum Genet. 2001;69:463–466. doi: 10.1086/321292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.International Molecular Genetic Study of Autism Consortium. A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet. 2001;69:570–581. doi: 10.1086/323264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stone JL, Merriman B, Cantor RM, Yonan AL, Gilliam TC, Geschwind DH, Nelson SF. Evidence for sex-specific risk alleles in autism spectrum disorder. Am J Hum Genet. 2004;75:1117–1123. doi: 10.1086/426034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cantor RM, Kono N, Duvall JA, Alvarez-Retuerto A, Stone JL, Alarcon M, Nelson SF, Geschwind DH. Replication of autism linkage: fine-mapping peak at 17q21. Am J Hum Genet. 2005;76:1050–1056. doi: 10.1086/430278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yonan AL, Alarcon M, Cheng R, Magnusson PK, Spence SJ, Palmer AA, Grunn A, Juo SH, Terwilliger JD, Liu J, et al. A genomewide screen of 345 families for autism-susceptibility loci. Am J Hum Genet. 2003;73:886–897. doi: 10.1086/378778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PMA, et al. Common genetic variation in the intergenic region between CDH10 and CDH9 is associated with susceptibility to autism spectrum disorders. Nature. In Press.This is the first significant genome wide association study for ASD with replication. It implicates a much more important role for cell adhesion molecule polymorphisms in ASD.
- 14.Lord C, Rutter M, Goode S, Heemsbergen J, Jordan H, Mawhood L, Schopler E. Autism diagnostic observation schedule: a standardized observation of communicative and social behavior. J Autism Dev Disord. 1989;19:185–212. doi: 10.1007/BF02211841. [DOI] [PubMed] [Google Scholar]
- 15.Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 16.Alarcon M, Yonan AL, Gilliam TC, Cantor RM, Geschwind DH. Quantitative genome scan and Ordered-Subsets Analysis of autism endophenotypes support language QTLs. Mol Psychiatry. 2005;10:747–757. doi: 10.1038/sj.mp.4001666. [DOI] [PubMed] [Google Scholar]
- 17.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 18.Liu XQ, Paterson AD, Szatmari P. Genome-wide linkage analyses of quantitative and categorical autism subphenotypes. Biol Psychiatry. 2008;64:561–570. doi: 10.1016/j.biopsych.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duvall JA, Lu A, Cantor RM, Todd RD, Constantino JN, Geschwind DH. A quantitative trait locus analysis of social responsiveness in multiplex autism families. Am J Psychiatry. 2007;164:656–662. doi: 10.1176/ajp.2007.164.4.656. [DOI] [PubMed] [Google Scholar]
- 20.Alarcon M, Cantor RM, Liu J, Gilliam TC, Geschwind DH. Evidence for a language quantitative trait locus on chromosome 7q in multiplex autism families. Am J Hum Genet. 2002;70:60–71. doi: 10.1086/338241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alarcon M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82:150–159. doi: 10.1016/j.ajhg.2007.09.005.Associates CNTNAP2, a gene expressed within the language circuit of the brain, with a language endophenotype in ASD trios.
- 22.Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, Stephan DA, Morton DH. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354:1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 23.Vernes SC, Newbury DF, Abrahams BS, Winchester L, Nicod J, Groszer M, Alarcon M, Oliver PL, Davies KE, Geschwind DH, et al. A functional genetic link between distinct developmental language disorders. N Engl J Med. 2008;359:2337–2345. doi: 10.1056/NEJMoa0802828.Identifies CNTNAP2 as part of a genetic pathway for language, by demonstrating regulation by FOXP2 and genetic association with Specific Language Impairment.
- 24.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659.Utilizing ROMA-mediated comparative genomic hybridization, they performed a large-scale screen for chromosomal structural variants for 264 families. This paper is the first paper to identify CNVs as a major contributor to the etiology of ASD.
- 25.Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657.Demonstrates that ASD can be caused by a heterogeneous set of rare private mutations utilizing homozygocity mapping.
- 27.Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 28.Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH, Jr, Dobyns WB, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 29.Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veltman MW, Craig EE, Bolton PF. Autism spectrum disorders in Prader-Willi and Angelman syndromes: a systematic review. Psychiatr Genet. 2005;15:243–254. doi: 10.1097/00041444-200512000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Croen LA, Najjar DV, Fireman B, Grether JK. Maternal and paternal age and risk of autism spectrum disorders. Arch Pediatr Adolesc Med. 2007;161:334–340. doi: 10.1001/archpedi.161.4.334. [DOI] [PubMed] [Google Scholar]
- 32.Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet. 2007;39:S43–S47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- 33.Iossifov I, Zheng T, Baron M, Gilliam TC, Rzhetsky A. Genetic-linkage mapping of complex hereditary disorders to a whole-genome molecular-interaction network. Genome Res. 2008;18:1150–1162. doi: 10.1101/gr.075622.107.Utilizing a unique computational approach to overlay interaction analysis and linkage analysis, this paper identifies 24 new autism susceptibility candidate genes in silica. Duplicating this analysis for bipolar and schizophrenic populations, they demonstrate a large overlap in the candidate genes between the three disorders blurring the lines between neurodevelopmental and psychiatric disorders.
- 34.Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, Elia M, Schneider C, Melmed R, Sacco R, et al. A genetic variant that disrupts MET transcription is associated with autism. Proc Natl Acad Sci U S A. 2006;103:16834–16839. doi: 10.1073/pnas.0605296103.Identifies an association between the C allele of the MET promoter with ASD based on a candidate gene approach.
- 35.Campbell DB, Li C, Sutcliffe JS, Persico AM, Levitt P. Genetic evidence implicating multiple genes in the MET receptor tyrosine kinase pathway in autism spectrum disorder. Autism Research. 2008;1:159–168. doi: 10.1002/aur.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishimura Y, Martin CL, Vazquez-Lopez A, Spence SJ, Alvarez-Retuerto AI, Sigman M, Steindler C, Pellegrini S, Schanen NC, Warren ST, et al. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007;16:1682–1698. doi: 10.1093/hmg/ddm116. [DOI] [PubMed] [Google Scholar]
- 37.Lintas C, Sacco R, Garbett K, Mirnics K, Militerni R, Bravaccio C, Curatolo P, Manzi B, Schneider C, Melmed R, et al. Involvement of the PRKCB1 gene in autistic disorder: significant genetic association and reduced neocortical gene expression. Mol Psychiatry. 2008 doi: 10.1038/mp.2008.21. [DOI] [PubMed] [Google Scholar]
- 38.Fombonne E. The epidemiology of autism: a review. Psychol Med. 1999;29:769–786. doi: 10.1017/s0033291799008508. [DOI] [PubMed] [Google Scholar]
- 39.Knickmeyer RC, Baron-Cohen S. Fetal testosterone and sex differences in typical social development and in autism. J Child Neurol. 2006;21:825–845. doi: 10.1177/08830738060210101601. [DOI] [PubMed] [Google Scholar]
- 40.Limperopoulos C, Bassan H, Sullivan NR, Soul JS, Robertson RL, Jr, Moore M, Ringer SA, Volpe JJ, du Plessis AJ. Positive screening for autism in ex-preterm infants: prevalence and risk factors. Pediatrics. 2008;121:758–765. doi: 10.1542/peds.2007-2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- 42.Herbert MR, Russo JP, Yang S, Roohi J, Blaxill M, Kahler SG, Cremer L, Hatchwell E. Autism and environmental genomics. Neurotoxicology. 2006;27:671–684. doi: 10.1016/j.neuro.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 43.Geschwind DH. Autism: many genes, common pathways? Cell. 2008;135:391–395. doi: 10.1016/j.cell.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 45.Courchesne E, Pierce K, Schumann CM, Redcay E, uckwalter JA, Kennedy DP, Morgan J. Mapping early brain development in autism. Neuron. 2007;56:399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 46.Chiu S, Wegelin JA, Blank J, Jenkins M, Day J, Hessl D, Tassone F, Hagerman R. Early acceleration of head circumference in children with fragile x syndrome and autism. J Dev Behav Pediatr. 2007;28:31–35. doi: 10.1097/01.DBP.0000257518.60083.2d. [DOI] [PubMed] [Google Scholar]
- 47.Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004;36:1008–1013. doi: 10.1038/ng1419. [DOI] [PubMed] [Google Scholar]