

Abstract
Primary immunodeficiencies (PID) are rare diseases; therefore transnational studies are essential to maximize the scientific outcome and to improve diagnosis and therapy. In order to estimate the prevalence of PID in Europe as well as to establish and evaluate harmonized guidelines for the diagnosis and treatment of PID, the European Society for Immunodeficiencies (ESID) has developed an internet-based database for clinical and research data on patients with PID. This database is a platform for epidemiological analyses as well as the development of new diagnostic and therapeutic strategies and the identification of novel disease-associated genes. Within 4 years, 7430 patients from 39 countries have been documented in the ESID database. Common variable immunodeficiency (CVID) represents the most common entity, with 1540 patients or 20·7% of all entries, followed by isolated immunoglobulin (Ig)G subclass deficiency (546 patients, 7·4%). Evaluations show that the average life expectancy for PID patients varies from 1 to 49 years (median), depending on the type of PID. The prevalence and incidence of PID remains a key question to be answered. As the registration progress is far from finished we can only calculate minimum values for PID, with e.g. France currently showing a minimum prevalence of 3·72 patients per 100 000 inhabitants. The most frequently documented permanent treatment is immunoglobulin replacement; 2819 patients (42% of all patients alive) currently receive this form of treatment.
Keywords: epidemiology, ESID, online database, primary immunodeficiency, registry
Introduction
Primary immunodeficiencies (PID) are rare diseases caused by inherited defects of the immune system. Advances in medical research have led to the identification of more than 140 genes which cause more than 200 different forms of PID [1].
Research on PID to date faces a range of difficulties. These are due not only to the low prevalence, but also to the fact that PID were discovered only half a century ago and are still a somewhat new area for common medicine. In countries such as Bulgaria, PID are even not acknowledged as disease entities and therefore cannot be diagnosed and treated accordingly.
In general, PID are under-diagnosed in most countries, and many medical centres care for only few patients with PID. This has made it extremely difficult to establish sufficient data on the morbidity and mortality of PID. Numerous surveys and registries have been set up to date to answer questions on the epidemiology of PID, mainly by national research networks [2–4]. Authors generally emphasize that under-diagnosis and under-reporting as well as lack of participation by hospitals produce shortcomings in the results. These vary greatly, with incidence rates ranging from 1·4 to 10·1 in 100 000 live births [3,5]. The highest prevalence rate measured to date is 12·4 per 100 000 inhabitants in South Australia [4].
It has been suggested that applying an incidence rate from surveys in smaller regions with good documentation and a centralized health-care system could be a way to estimate incidence numbers for larger areas [5]. However, this approach may not produce valid figures representative for other regions. The prevalence of specific PID varies considerably between ethnic groups [6], which implies that morbidity rates can vary greatly between regions, countries and populations.
In addition to these evident difficulties in epidemiology, the diversity of PID poses another challenge for researchers. PID are caused by a large variety of genetic defects, and only a proportion of these have been discovered to date. Some of the known defects are extremely rare, which makes international collaboration necessary to collect sufficient patient numbers for adequate research. In 1994, European immunologists formed a network of clinicians and researchers, the European Society for Immunodeficiencies (ESID; http://www.esid.org), to support research on the cause, mechanism and therapy in immunodeficiencies.
Since 2004 ESID has been running a database for primary immunodeficiencies, the results of which are presented here. The ESID database is a secure, internet-based patient registry, which combines both clinical and laboratory data of PID patients. It provides a common data set for currently 214 different PID entities and is updated regularly, according to progress in research. This core data set includes data on diagnosis, therapy, laboratory data and health outcome indicators.
With the database, ESID is striving to answer the challenging epidemiological questions outlined above. However, unlike many other registries, it is not merely a single time-point survey restricted to epidemiology; the ESID database also serves as a research platform with long-term documentation. Using the database, researchers have the possibility to identify patient cohorts for genetic screening and multi-centre trials. Data sets can be extended flexibly for studies using the database as a platform for their reporting forms.
A first analysis on the data collected from 2004 to 2006 has been published in this journal [7]. Since then, the number of registered cases has more than tripled, from 2386 to 7430. Although this figure is still far from presenting a complete picture of PID in Europe, the current results give valuable insights into the epidemiology of PID, in particular in countries with a high documentation rate.
With the current results, we want to emphasize the enormous potential of this database network, in particular for conducting laboratory research and clinical trials, leading finally to improvement of diagnosis, classification, prognosis and therapy of primary immunodeficiency disorders.
Materials and methods
The system structure of the ESID online database has been described by Perner et al.[7] and Guzman et al.[8]. Currently (December 2008) 76 documenting centres and national registries from 30 countries have signed a contract and obtained logins for the database system, which are necessary to participate in the online documentation. Most centres are located in Europe, but there are also centres in Egypt and Iran, which show that the efforts of ESID go beyond the geographical borders of Europe. Some of the documenting centres co-ordinate national networks with numerous subcentres, such as France and Italy. Therefore, the actual total number of centres contributing patients to the database is 158.
The ESID database for PID currently consists of 214 disease-specific registries, which are grouped within eight main categories and 73 subcategories (see Table 1). The categorization is based on the latest classification established by the International Union of Immunological Societies (IUIS) in 2007 [9].
Table 1.
Distribution of cases registered in the European Society for Immunodeficiencies (ESID) online database by primary immunodeficiency (PID) diseases. Some entities are subsumed under a common heading for layout reasons.
| PID entity | Total | % of total |
|---|---|---|
| Predominantly antibody disorders | 4073 | 54·82 |
| Agammaglobulinaemias | 518 | 6·97 |
| X-linked (BTK) | 436 | 5·87 |
| Other known genetic defects | 4 | 0·05 |
| Genetic defect unknown | 78 | 1·05 |
| Hypogammaglobulinaemias | 3326 | 44·76 |
| Common variable immunodeficiency (CVID) | 1540 | 20·73 |
| Isolated immunoglobulin (Ig)G subclass deficiency | 546 | 7·35 |
| IgA deficiency | 507 | 6·82 |
| Transient hypogammaglobulinaemia of infancy | 322 | 4·33 |
| Deficiency of specific IgG | 47 | 0·63 |
| Selective IgM deficiency | 49 | 0·66 |
| Thymoma with immunodeficiency | 19 | 0·26 |
| Secondary hypogammaglobulinaemia | 32 | 0·43 |
| Secondary selective IgA deficiency | 16 | 0·22 |
| ICOS deficiency | 6 | 0·08 |
| CD19 antigen deficiency | 3 | 0·04 |
| Other hypogammaglobulinaemias | 239 | 3·22 |
| Class switch recombination defects/HIGM syndromes | 229 | 3·08 |
| CD40L deficiency/X-linked hyper-IgM | 86 | 1·16 |
| CD40 antigen deficiency | 7 | 0·09 |
| Other genetic defects | 27 | 0·36 |
| Genetic defect unknown | 109 | 1·47 |
| Predominantly T cell deficiencies | 671 | 9·03 |
| T–B+ severe combined immunodeficiency (SCID) | 227 | 3·06 |
| X-linked SCID | 75 | 1·01 |
| Janus kinase 3 deficiency (JAK3) | 18 | 0·24 |
| Interleukin 7 receptor deficiency (IL-7R) | 10 | 0·13 |
| Interleukin 2 receptor alpha deficiency (CD25) | 6 | 0·08 |
| Genetic defect unknown | 118 | 1·59 |
| T–B- severe combined immunodeficiency (SCID) | 212 | 2·85 |
| Recombination-activating gene deficiency (RAG1/2) | 62 | 0·83 |
| Adenosine deaminase deficiency (ADA) | 42 | 0·57 |
| DNA cross-link repair protein 1C deficiency (Artemis) | 21 | 0·28 |
| Reticular dysgenesia | 3 | 0·04 |
| Genetic defect unknown | 84 | 1·13 |
| HLA class II deficiency | 57 | 0·77 |
| MHC2TA deficiency | 11 | 0·15 |
| Other genetic defects | 14 | 0·19 |
| Genetic defect unknown | 32 | 0·43 |
| CD3 deficiency | 10 | 0·13 |
| Selective CD4 cell deficiency | 39 | 0·52 |
| Other genetic T cell defects | 14 | 0·19 |
| Other unclassified T cell disorders | 112 | 1·51 |
| Phagocytic disorders | 932 | 12·54 |
| Chronic granulomatous disease (CGD) | 322 | 4·33 |
| X-linked (CYBB) | 182 | 2·45 |
| AR cytochrome b positive type 1 (NCF1) | 24 | 0·32 |
| Other genetic defects | 14 | 0·19 |
| Genetic defect unknown | 102 | 1·37 |
| Severe congenital neutropenia (cyclic and non-cyclic) | 209 | 2·81 |
| Elastase 2 deficiency (ELA2) | 9 | 0·12 |
| Other genetic defects | 5 | 0·07 |
| Genetic defect unknown | 195 | 2·62 |
| Kostmann syndrome (HAX1) | 78 | 1·05 |
| Schwachman–Diamond syndrome | 93 | 1·25 |
| SBDS deficiency | 7 | 0·09 |
| Genetic defect unknown | 86 | 1·16 |
| Defects with susceptibility to mycobacterial infection | 29 | 0·39 |
| Interleukin 12 receptor beta-1 deficiency (IL-12RB1) | 16 | 0·22 |
| Other genetic defects | 9 | 0·12 |
| Genetic defect unknown | 4 | 0·05 |
| Familial haemophagocytic lymphohistiocytosis syndromes | 89 | 1·20 |
| Perforin 1 deficiency (PRF1) | 14 | 0·19 |
| Familial haemophagocytic lymphohistiocytosis 3 (UNC13D) | 15 | 0·20 |
| Genetic defect unknown | 60 | 0·81 |
| Griscelli syndrome | 16 | 0·22 |
| Leucocyte adhesion deficiency (LAD) | 33 | 0·44 |
| LAD type 1 (ITGB2) | 28 | 0·38 |
| LAD type 3 | 5 | 0·07 |
| WHIM syndrome | 15 | 0·20 |
| Glucose-6-phosphate dehydrogenase deficiency (G6PD) | 16 | 0·22 |
| Chediak–Higashi syndrome, genetic defect unknown | 15 | 0·20 |
| Partial albinism and immunodeficiency syndrome | 2 | 0·03 |
| Other phagocytic disorders | 15 | 0·20 |
| Complement deficiencies | 151 | 2·03 |
| Hereditary angioedema (C1inh) | 81 | 1·09 |
| Complement component 2 deficiency | 20 | 0·27 |
| Mannose-binding lectin deficiency (MBL) | 18 | 0·24 |
| Other complement deficiencies | 32 | 0·43 |
| Other well-defined PIDs | 1292 | 17·39 |
| DNA-breakage disorders | 510 | 6·86 |
| Ataxia telangiectasia (ATM) | 405 | 5·45 |
| Nijmegen breakage syndrome (NBS1) | 74 | 1·00 |
| Fanconi anaemia | 15 | 0·20 |
| Other DNA-breakage disorders | 16 | 0·22 |
| DiGeorge syndrome | 247 | 3·32 |
| Wiskott–Aldrich syndrome (WAS) | 289 | 3·89 |
| Mutations in WASP | 217 | 2·92 |
| X-linked thrombocytopenia with mutations in WASP | 2 | 0·03 |
| Genetic defect unknown | 70 | 0·94 |
| X-linked lymphoproliferative syndrome (XLP) | 37 | 0·50 |
| X-linked (SH2D1A) | 18 | 0·24 |
| Genetic defect unknown | 19 | 0·26 |
| Hyper-IgE syndromes | 120 | 1·62 |
| Mutations in STAT3 | 15 | 0·20 |
| Genetic defect unknown | 105 | 1·41 |
| Chronic mucocutaneous candidiasis (CMC) | 26 | 0·35 |
| Autoimmune polyendocrinopathy syndrome type I (AIRE) | 2 | 0·03 |
| Other CMC | 24 | 0·32 |
| Osteopetrosis | 13 | 0·17 |
| Asplenia syndrome | 1 | 0·01 |
| Cartilage hair hypoplasia | 14 | 0·19 |
| Defects of TLR/NFkappa-B signalling | 11 | 0·15 |
| Fc receptor deficiencies | 2 | 0·03 |
| Hermansky–Pudlak syndrome 3 | 2 | 0·03 |
| Hoyeraal–Hreidarsson syndrome | 8 | 0·11 |
| Immunodeficiency centromeric instability facial anomalies syndrome (ICF) | 9 | 0·12 |
| Netherton syndrome | 1 | 0·01 |
| Schimke disease | 2 | 0·03 |
| Autoimmune and immunedysregulation syndromes | 98 | 1·32 |
| Autoimmune lymphoproliferative syndrome (ALPS) | 79 | 1·06 |
| TNFRS member 6 deficiency (ALPS IA-type) | 46 | 0·62 |
| Other genetic defects | 3 | 0·04 |
| ALPS with unknown genetic cause | 30 | 0·40 |
| Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) | 12 | 0·16 |
| Immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) | 7 | 0·09 |
| Autoinflammatory syndromes | 76 | 1·02 |
| Familial mediterranean fever defect | 60 | 0·81 |
| TNF receptor-associated periodic syndrome (TRAPS) | 10 | 0·13 |
| Hyper-IgD syndrome (MVK) | 5 | 0·07 |
| CINCA syndrome | 1 | 0·01 |
| Unclassified immunodeficiencies | 137 | 1·84 |
CINCA: chronic infantile neurological cutaneous articular syndrome; MVK: mevalonate kinase; TNF: tumour necrosis factor; TNFR: tumour necrosis factor receptor; TLR: Toll-like receptor; NF: nuclear factor; STAT: signal transducer and activator of transcription 3; SBDS: Schwachman–Bodan–Diamond syndrome; BTK: Bruton agammablobulinaemia tyrosine kinase; ICOS: inducible co-stimulatory molecule; HIGM: hyper-IgM; CYBB: cytochromosome b588 β; WHIM: Wart, Hypogammaglobulinemia, Infection and Myelokathexis; NCF1: neutrophil cytosolic factor 1; HLA: human leucocyte antigen.
All disease-specific registries share a common core data set. This core data set was designed to bring together basic data for all PID patients in order to make general evaluations possible. It contains information on diagnosis, therapy and quality of life as well as the most important laboratory data for PID [blood and lymphocyte count, immunoglobulin (Ig) levels, complement values and data on important cell surface markers]. The core data set has been extended recently by fields asking for concomitant diseases and infectious episodes, as well as family history.
In addition to the core data, extended disease-specific data sets have been implemented currently for 40 diseases, allowing the documentation of an exhaustive set of parameters that are relevant only for a specific PID, thus making more detailed analyses possible.
Documentation and quality checks
The database is designed to be used for long-term documentation. It offers the possibility to add any number of visit dates for a given patient. Users are asked to update their patients' data at least once a year. On average, first-time data entry takes 10–15 min per patient for the core data set, depending on the availability of the patient data, and longer if additional fields are documented. Regular updates take considerably less time, usually less than 5 min per patient.
Some centres and national networks are maintaining pre-existent local databases for their PID patients. Their data are transferred electronically to the ESID system at regular intervals.
The database has an inbuilt automatic quality assurance system including field type, range and plausibility checks (e.g. date of death must be greater than date of birth, blood counts cannot sum to more than 100%). In addition, data sets are checked regularly for plausibility and completeness by the database administration.
Some fields are mandatory, which means that data cannot be stored unless these fields are completed. However, many fields are open to be completed, taking into account that the data are sometimes not known or currently not available to the documentalist. This explains why, in evaluations, the number of evaluated cases does not always equal the total number of registered cases in the database. Furthermore, the database records both a patient's country of residence as well as the attending centre that entered the patient's data. European health policies allow that patients may be seen in centres outside their country of residence, which explains the difference between the total number of patients documented by centres in a given country and the number of patients living in this country.
Analysis of data and studies
The ESID database offers a platform for different types of studies on primary immunodeficiencies. European researchers have the opportunity to learn from other patients with a specific diagnosis throughout Europe and can contact appropriate documenting centres. The database has been constructed so that users can access data on patients automatically from their centre only. From the beginning of 2007, ESID offers a reporting tool which enables users to query and analyse their centre's data in more detail themselves.
The ESID database has already been used in surveys on X-linked thrombocytopenia (XLT) patients and neuroendocrine carcinoma in patients with CD40L deficiency. Multi-centre clinical trials on common variable immunodeficiency (CVID) and X-linked agammaglobulinaemia (XLA) are currently under way.
Results
The total number of registered patients was 7430 (November 2008). Of these cases, 6706 patients were alive, while 724 were deceased or lost to follow-up; 4312 patients (58%) had one or more follow-up documentations in addition to the initial registration.
Of all patients, 2911 (39·2%) were female and 4519 (60·8%) were male. We see a similar gender distribution both among children and adults. Among the 6706 living patients, 3575 were aged 15 years or younger and 3131 patients were aged 16 years or older. In the first group (15 years or younger), 2309 (64·6%) were male while 1266 (35·4%) were female. Compared to this gender distribution, the female share was slightly higher in the second group (aged 16 years or older): 1776 (56·7%) were male and 1355 (43·3%) were female.
Predominantly antibody deficiencies formed the largest PID group with 4073 patients (54·8%), while CVID was by far the most common single disease with 1540 patients entered (20·7%) (Table 1).
Epidemiology
Using the number of documented patients, a minimum prevalence of PID in the current total population can be calculated for each participating country. As shown in Table 2, this number is relatively low in many countries, compared to results of earlier registries. This is due to the fact that in most countries documentation is far from complete. However, with 2804 registered patients, France serves as the best example of a good coverage of PID patients. Therefore, we have calculated the minimal incidence of PID in newborns for France from 1995 to 2006, grouped into 4-year periods (Fig. 1a).
Table 2.
Minimum primary immunodeficiency (PID) prevalence based on reported cases in the European Society for Immunodeficiencies (ESID) database: patients per 100 000 inhabitants by countries. Only countries with a prevalence >0·2 are displayed.
| Country | Alive PID patients documented | Population (millions inhabitants) | Documented PID patients per 100 000 inhabitants |
|---|---|---|---|
| France | 2399 | 64·47 | 3·72 |
| Ireland | 76 | 4·24 | 1·79 |
| Turkey | 1083 | 70·59 | 1·53 |
| United Kingdom | 878 | 60·59 | 1·45 |
| Estonia | 15 | 1·34 | 1·12 |
| Italy | 655 | 59·13 | 1·11 |
| Belgium | 98 | 10·53 | 0·93 |
| Poland | 352 | 38·12 | 0·92 |
| Czech Republic | 88 | 10·31 | 0·85 |
| Greece | 89 | 11·17 | 0·8 |
| Germany | 552 | 82·24 | 0·67 |
| Serbia | 47 | 7·27 | 0·65 |
| Switzerland | 38 | 7·59 | 0·5 |
| Slovakia | 22 | 5·43 | 0·41 |
| Sweden | 32 | 9·18 | 0·35 |
| Slovenia | 6 | 2·02 | 0·3 |
| Portugal | 27 | 10·95 | 0·25 |
Total populations source: Wikipedia.
Fig. 1.
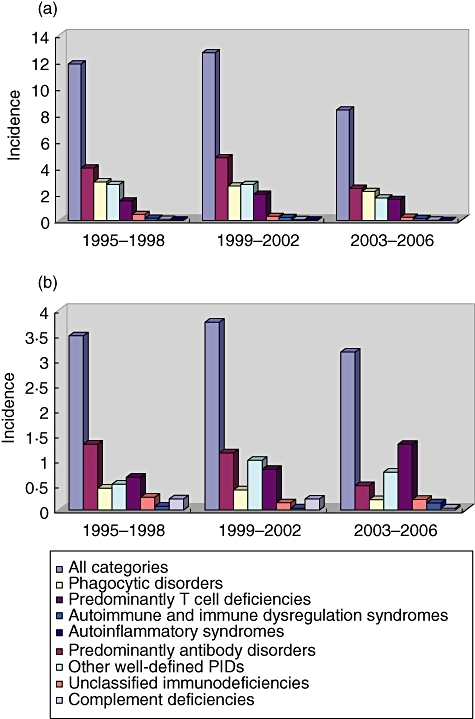
Minimum incidence of primary immunodeficiency (PID) cases in 100 000 live births, grouped in 4-year periods, and distribution in main PID categories. (a) France: n (1995–2006) = 1110, demographics: INSEE (http://www.insee.fr); (b) United Kingdom, n (1995–2006) = 288, demographics: UK Office for National Statistics (http://www.statistics.gov.uk).
The distribution of the PID categories shows that predominantly antibody disorders were the most common PID in newborns. However, they were followed closely by phagocytic disorders and other well-defined PIDs. An analysis of the data from the United Kingdom, where 907 patients have been registered, gives us a similar picture, although phagocytic disorders were recorded much less frequently (Fig. 1b).
To reach a better understanding of these numbers, we must also take into account that many PID patients are diagnosed only after they have reached adulthood. For example, in the ESID database, more than 21% of all registered patients were diagnosed at 16 years of age or later. The share is even greater in antibody disorders, where 33·9% were diagnosed in adulthood (Table 3).
Table 3.
Number of patients diagnosed in childhood and adulthood, by primary immunodeficiency (PID) category.
| Main PID category | Patients diagnosed by age 15 or younger | Patients diagnosed by age 16 or older | Share of diagnosis in adulthood (%) |
|---|---|---|---|
| All categories | 5607 | 1533 | 21·47 |
| Predominantly antibody disorders | 2594 | 1332 | 33·93 |
| Predominantly T cell deficiencies | 643 | 17 | 2·58 |
| Phagocytic disorders | 852 | 29 | 3·29 |
| Other well-defined PIDs | 1185 | 62 | 4·97 |
| Complement deficiencies | 89 | 43 | 32·58 |
| Autoinflammatory syndromes | 67 | 9 | 11·84 |
| Autoimmune and immunedysregulation syndromes | 89 | 6 | 6·32 |
| Unclassified immunodeficiencies | 88 | 35 | 28·46 |
Therefore, when looking at the time-span we selected to calculate incidences (1995–2006), we must assume that at least a fifth of the PID patients born in these years have not yet been diagnosed, which means that both the total incidence as well as the incidence for specific categories is actually higher than shown in Fig. 1. In particular, the incidence rate for PID with a large share of late-onset and late-diagnosis patients such as antibody disorders and complement deficiencies is probably remarkably higher.
Mortality
Data were evaluated for 641 deceased patients with information available on the date of death. Life expectancy varies considerably between the types of PID. While the oldest antibody-deficient patient documented so far had lived as long as 85 years, other PID categories show much lower average ages at death (Fig. 2). However, with improved diagnosis and treatment, life expectancy for PID patients has increased. For example, of 290 living patients with chronic granulomatous disease (CGD), 24 were currently 30 years and older. In contrast, among the deceased CGD patients, the oldest had reached an age of only 27 years.
Fig. 2.
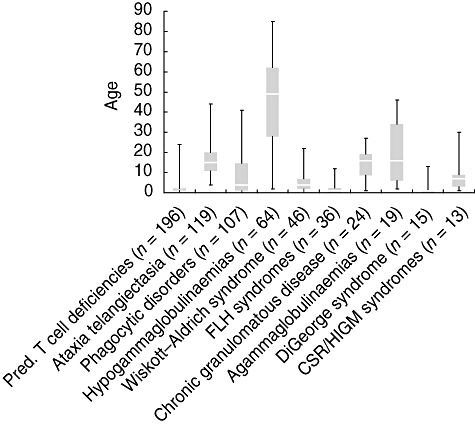
Age at death of patients from selected subcategories and diseases. Box and whisker chart with whiskers representing the maximum and minimum values; the box represents the upper and lower quartile, the white line represents the median. The number of evaluated patients is given in brackets.
Therapy
Ig replacement represents one of the mainstay therapeutic regimens in PID. Of the 6706 alive patients, 2819 (42%) were receiving Ig replacement. Antibiotics were the second most frequent drugs prescribed, and were taken on a regular basis by 1516 patients (22·6%); 1410 patients (21%) were currently not receiving any permanent medication. A total of 632 patients (8·5% of all patients) had undergone bone marrow transplantation.
The rate of Ig replacement was highest in the group of antibody-deficient patients, where 2273 (57·4%) of 3960 living patients received this form of therapy. There was a high rate of Ig replacement (1257 of 1479; 85%) in CVID patients and 382 of 415 (92%) XLA patients on Ig replacement (Table 4).
Table 4.
Percentage of alive patients receiving immunglobulin replacement within the largest primary immunodeficiency (PID) subgroups in the European Society for Immunodeficiencies (ESID) database.
| PID subcategory | Alive patients | % receiving immunoglobulin (Ig) replacement (%) |
|---|---|---|
| Agammaglobulinaemias | 494 | 91 |
| Common variable immunodeficiency (CVID) | 1479 | 85 |
| Class switch recombination defects (CSR)/HIGM syndromes | 215 | 57 |
| X-linked lymphoproliferative syndrome (XLP) | 33 | 51 |
| Other unclassified T cell disorders | 91 | 46 |
| T–B- severe combined immunodeficiency (SCID) | 138 | 45 |
| HLA class II deficiency | 32 | 43 |
| Wiskott–Aldrich syndrome (WAS) | 234 | 43 |
| T–B+ severe combined immunodeficiency (SCID) | 153 | 40 |
| CD4-deficiency | 35 | 31 |
| Chronic mucocutaneous candidiasis (CMC) | 26 | 30 |
| Immunodeficiencies of unknown cause | 129 | 25 |
| Hypogammaglobulinaemias (without CVID) | 1772 | 24 |
| Hyper-IgE syndromes | 112 | 23 |
| DNA-breakage disorder | 351 | 22 |
| Familial haemophagocytic lymphohistiocytosis syndromes (FLH) | 51 | 17 |
| Autoimmune lymphoproliferative syndrome (ALPS) | 73 | 15 |
| Leucocyte adhesion deficiency (LAD) | 28 | 7 |
| Kostmann syndrome | 61 | 6 |
| Chronic granulomatous disease (CGD) | 290 | 3 |
| DiGeorge syndrome | 227 | 3 |
| Severe congenital neutropenia (cyclic and non-cyclic) | 198 | 3 |
HIGM: hyper-IgM; HLA: human leucocyte antigen.
Immunoglobulins were administered intravenously (IVIG) in 2137 patients (75·8%), while 674 patients (23·9%) made use of home therapy with subcutaneous Ig infusions (SCIG) and two patients received intramuscular immunoglobulins. In the remaining six patients, the route of application was not documented.
The use of SCIG as home therapy is available in only some European countries. Figure 3 shows that among the documented patients, Germany showed the highest rate of SCIG, with 53·9% receiving SCIG and 46·1% receiving IVIG.
Fig. 3.
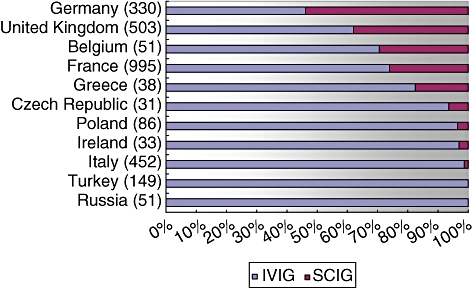
Shares of intravenous immunoglobulin (IVIG) and subcutaneous immunoglobulin (SCIG) replacement in selected European countries as documented in the European Society for Immunodeficiencies (ESID) database. The total number of documented patients receiving immunoglobulin replacement in each country is given in brackets.
There are also differences in the mean interval of IVIG administration between countries. However, the results suggest that applying IVIG every 3–4 weeks is common in many countries (Fig. 4).
Fig. 4.
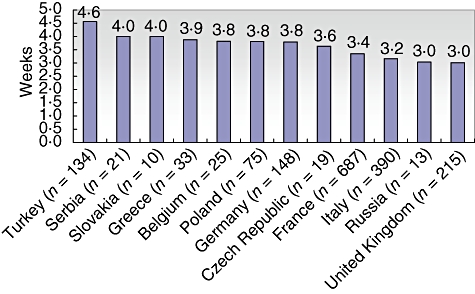
Mean interval of intravenous immunoglobulin (IVIG) replacement by countries as documented in the European Society for Immunodeficiencies (ESID) database. The total number of patients currently treated with IVIG for whom the interval has been documented is given in brackets. Only countries with 10 or more cases containing data on intervals are displayed.
A key target in Ig replacement is achieving an adequate IgG trough level in the patient's blood. The trough serum level, which is measured immediately before Ig is administered, should be at least 5 g/l, and ideally within the normal range of healthy individuals (7–16 g/l) [10,11]. The mean IgG trough level in the ESID database was 6·85 g/l for CVID patients (720 patients receiving Ig replacement with information on the trough level available) and 7 g/l for XLA patients (178 patients).
Discussion
Our evaluations show that the documentation progress in the ESID online database has gained momentum. While the average number of newly reported cases per year was 954 in October 2006, the time of our first publication, the average increase since then has more than doubled to 2000 patients per year, resulting in a total of 7430 documented patients.
However, documentation still lags behind in many countries, and there is a bias in the documentation due to the limited number of institutions that document patients. For example, complement deficiencies have risen from 0·6% to 2% of all cases since 2006, but this is still far from the 6% share they had in the old ESID registry (1994–2002).
Documentation has proved most successful in countries with a national PID network that draws on local funds for documentation. This is very notably the case in France, where the national centre CEREDIH (http://www.ceredih.fr) has already registered 2821 PID patients nationwide by employing data entry personnel who travel to local hospitals to document patient data. Several countries, such as Germany, Switzerland, the United Kingdom and Sweden, are currently in the process of setting up similar networks with the aim of achieving, ideally, a complete nationwide documentation.
It is relatively problematic to obtain valid data on the epidemiology of PID. From our data set, we were able to calculate a minimum prevalence which is merely an indicator of how good is the documentation in each country. However, with 2399 living patients documented in the heterogeneous population of France, the overall prevalence of PID will not be less than 3·72 per 100 000.
Furthermore, it would be interesting to apply the very promising approach which the Australasian Society for Clinical Immunology and Allergy (ASCIA) has undertaken recently to estimate PID morbidity [4]. Data on the number of patients receiving Ig replacement for the indication of PID from national health authorities was used to approximate the rate of under-reporting. The United Kingdom is currently undertaking efforts to acquire similar statistical data.
The ESID online database represents an outstanding achievement, which was realized with limited financial resources and thanks to the active contributions from PID clinical teams and researchers. In view of the current achievements, we are confident that the database is becoming a valuable source for the management of and the research on PID in all European countries.
Acknowledgments
We are most grateful to the staff at all the medical centres and national registries participating in the database project for their continuous contribution: J. Beauté, R. Micol, Y. Dudoit, L. Benslama (Paris); A. Plebani, L. Notarangelo (Brescia); C. Pignata (Naples); C. Bangs (Manchester); M. Lucas (Oxford); P. Tierney (Newcastle); C. Core, J. Dempster, V. Knerr (London); A. Exley, D. Kumararatne (Cambridge); O. Paschenko, I. Kondratenko, Anna Shcherbina (Moscow); S. Velbri (Tallinn); P. Ciznar (Bratislawa); R. Duobiene (Vilnius); S. Kilic (Bursa-Görükle); N. Kütükcüler (Bornova-Izmir); Ö. Sanal (Ankara); I. Reisli (Konya); O. Yegin (Antalya); M. Kanariou (Athens); E. Papadopoulou-Alataki, M. Trachana, M. Hatzistilianou (Thessaloniki); C. M. Farber (Bruxelles); I. Meyts (Leuven); S. Pasic (Belgrade); D. Richter (Zagreb); L. Marodi (Debrecen); I. Touitou (Montpellier); M. Abuzakouk, C. Feighery (Dublin); V. Thon, J. Litzman (Brno); M. Cucuruz (Timisoara); B. Wolska (Warsaw); A. Szaflarska (Krakow); S. Reda (Cairo); P. Soler, I. Caragol (Barcelona); P. Llobet (Granollers); I. Savchak (Lviv); L. Marques (Porto); A. Koren (Ljubljana); M. Hörnes (Zurich); S. Shchebet (Minsk); S. Goldacker, H. Ritterbusch (Freiburg); M. Faßhauer (Leipzig); F. Sollinger (Munich); T. Witte, U. Baumann (Hannover); H. Wittkowski, D. Viemann (Münster); T. Niehues (Krefeld); H. Stimm (Frankfurt); N. Brodszki (Lund). The complete list of documenting centres is available at http://www.esid.org/centers.php. This work was supported by EU grants SP23-CT-2005-006411 and HEALTH-F2-2008-201549 and by PPTA Europe (http://www.pptaglobal.org) sponsorship of ESID.
Conflicts of interest
B. Gathmann and G. Kindle have received funding from Grifols S.A. to prepare this publication. The team of authors have received funds from PPTA and the European Union to facilitate data collection.
References
- 1.Fischer A. Trends in primary immunodeficiencies. Keynote lecture at the ESID meeting in 's-Hertogenbosch. October.
- 2.Stray-Pedersen A, Abrahamsen T, Froland S. Primary immunodeficiency diseases in Norway. J Clin Immunol. 2000;20:477–85. doi: 10.1023/a:1026416017763. [DOI] [PubMed] [Google Scholar]
- 3.Al-Herz W. Primary immunodeficiency disorders in Kuwait: first report from Kuwait National Primary Immunodeficiency Registry (2004–2006) J Clin Immunol. 2008;28:186–93. doi: 10.1007/s10875-007-9144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirkpatrick P, Riminton S. Primary immunodeficiency diseases in Australia and New Zealand. J Clin Immunol. 2007;27:517–24. doi: 10.1007/s10875-007-9105-z. [DOI] [PubMed] [Google Scholar]
- 5.Matamoros N, Llambi JM, Espanol T, et al. Primary immunodeficiency syndrome in Spain: first report of the National Registry in children and adults. J Clin Immunol. 1997;17:333–9. doi: 10.1023/a:1027382916924. [DOI] [PubMed] [Google Scholar]
- 6.Hammarström L, Smith CIE. Genetic approach to common variable immunodeficiency. In: Ochs HD, Smith CIE, Puck JM, editors. Primary immunodeficiency diseases. A molecular and genetic approach. Oxford: Oxford University Press; 1999. pp. 3–12. [Google Scholar]
- 7.Eades-Perner AM, Gathmann B, Knerr V, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2004–06. Clin Exp Immunol. 2007;147:306–12. doi: 10.1111/j.1365-2249.2006.03292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guzman D, Veit D, Knerr V, et al. The ESID online database network. Bioinformatics. 2007;23:654–5. doi: 10.1093/bioinformatics/btl675. [DOI] [PubMed] [Google Scholar]
- 9.Geha RS, Notarangelo LD, Casanova JL, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120:776–94. doi: 10.1016/j.jaci.2007.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sneller MC, Strober W, Eisenstein E, Jaffe JS, Cunningham-Rundles C. NIH conference. New insights into common variable immunodeficiency. Ann Intern Med. 1993;118:720–30. doi: 10.7326/0003-4819-118-9-199305010-00011. [DOI] [PubMed] [Google Scholar]
- 11.Gardulf A, Andersen V, Björkander J, et al. Subcutaneous immunoglobulin replacement in patients with primary antibody deficiencies: safety and costs. Lancet. 1995;345:365–9. doi: 10.1016/s0140-6736(95)90346-1. [DOI] [PubMed] [Google Scholar]