

Abstract
We constructed an oncolytic adenovirus, Adeno-hTERT-E1A, with deletions of the viral E1B, E3A, and E3B regions and insertion of a human telomerase reverse transcriptase (hTERT) promoter-driven early viral 1A (E1A) cassette that confers high transcriptional activity in multiple human tumor cell lines. The oncolytic potential of Adeno-hTERT-E1A was characterized in comparison with that of the E1B-55kDa- and E3B-region-deleted oncolytic adenovirus ONYX-015. Tumor cells infected with Adeno-hTERT-E1A expressed dramatically higher levels of E1A oncoprotein, underwent enhanced lysis, and displayed an earlier and higher apoptotic index than cells infected with ONYX-015. Despite the increase in virus-induced apoptotic death, Adeno-hTERT-E1A replicated and produced functional progeny leading to viral spread, but with reduced efficiency compared with ONYX-015, in particular in A549 cells. Virus-induced E1A expression, host cell apoptosis, viral hexon protein production, and DNA synthesis were markedly reduced in primary human hepatocytes after infection with Adeno-hTERT-E1A as compared with ONYX-015. The strong oncolytic activity of Adeno-hTERT-E1A in tumor cell culture translated into superior antitumor activity in vivo in an MDA-MB-231 solid tumor xenograft model. Adeno-hTERT-E1A thus has strong therapeutic potential and an improved safety profile compared with ONYX-015, which may lead to reduced toxicity in the clinic.
Introduction
Replication-conditional adenoviruses offer a unique therapeutic platform for cancer treatment owing to their strong intrinsic oncolytic activity in addition to their potential for therapeutic gene delivery (Bauerschmitz et al., 2002; Jounaidi et al., 2007). Novel oncolytic adenoviruses have been developed to increase antitumor activity and to improve the tumor specificity of viral replication in an effort to reduce the risk of viral spread in primary host tissues. Nonessential viral genes may be deleted to increase the capacity for delivery of therapeutic genes, including suicide genes, immunomodulatory genes, and prodrug activation enzyme genes (Jounaidi et al., 2007). In another approach, novel cancer/tissue-specific promoter elements may be used to control expression of the early viral 1A (E1A) oncoprotein, which is critical for the transcriptional activation and replication of adenoviruses (Nevins, 1981; Everts and van der Poel, 2005; Jounaidi et al., 2007). This strategy yields a tumor cell-selective, conditionally replicating adenovirus and is an effective way to increase cancer-specific replication while decreasing viral toxicity to primary host tissues. However, although the anticancer activity of these viruses is well established in preclinical studies, their use has resulted in limited success in the clinic, indicating a need for further improvements in oncolytic viral activity, specificity, and tumor cell delivery.
Telomerase, which is highly expressed in the vast majority of human cancers but not in most host tissues (Kim et al., 1994; Mo et al., 2003), presents an ideal tumor-specific trait for regulation of oncolytic viruses. Telomerase is a ribonucleic complex designed to prevent telomere shortening and senescence, thereby conferring immortality to malignant cells (Hanahan and Weinberg, 2000). The telomerase complex is composed of an RNA template component, a reverse transcriptase (human telomerase protein/enzyme [hTERT]), and telomerase-associated proteins (Stewart and Weinberg, 2006). Telomerase activity requires only the RNA component and hTERT. A 320-bp hTERT promoter fragment was previously shown to contain the sequences required to recapitulate high telomerase expression in cancer cells. This proximal promoter contains five SP1-binding sequences, which in normal somatic cells repress telomerase expression (Won et al., 2002); two E-boxes, containing c-Myc- and Max-binding sites (Oh et al., 1999); and an activating enhancer-binding protein-2 (AP-2)-binding site, which induces hTERT expression (Kirkpatrick et al., 2003; Ma et al., 2003). AP-2β, a member of the AP family, was shown to be sufficient to support tumor-specific reactivation of hTERT in human lung cancer cells (Deng et al., 2007). In addition, hTERT transcription could be upregulated by hypoxia, suggesting telomerase levels may vary within the same tumor (Anderson et al., 2006). The hTERT promoter has been used successfully in several gene therapy models (Wirth et al., 2005).
Many telomerase promoter-regulated adenoviral vectors that retain one or both of the E1B gene products, E1B-55 kDa and/or E1B-19 kDa, have been described (Wirth et al., 2005). The E1B-55 kDa gene product interacts directly with p53 and mediates its inactivation, translocation to the cytoplasm, ubiquitination, and degradation (Steegenga et al., 1998; Grand et al., 1999). E1B-19 kDa inhibits the downstream apoptotic actions of p53 (White et al., 1992). E1B-19 kDa functions like the antiapoptotic factor Bcl-2 and can inactivate the proapoptotic factor Bax (Han et al., 1996). E1B-19 kDa can also antagonize the proapoptotic functions of E1A by binding to the powerful transcription repressor Btf, which promotes cell death (Kasof et al., 1999; Hale and Braithwaite, 1999). E1B gene deletions yield highly oncolytic adenoviruses with improved antitumor effects (Duque et al., 1999; Harrison et al., 2001; Kim et al., 2007). We investigated the potential of combining the increased oncolytic activity conferred by deletion of both E1B genes with the tumor cell specificity imparted by the use of an hTERT core promoter regulating the adenoviral E1A gene. Tumor cells infected with the resulting adenovirus, Adeno-hTERT-E1A, expressed dramatically higher levels of E1A oncoprotein, underwent enhanced lysis, and displayed an earlier and higher apoptotic index than cells infected with ONYX-015, a widely studied oncolytic virus that has undergone extensive clinical evaluation (Crompton and Kirn, 2007). A substantial increase in specificity compared with ONYX-015 was observed, with a dramatic decrease in Adeno-hTERT-E1A genome replication, E1A expression, and viral cycle completion seen in primary human hepatocytes. Finally, the strong oncolytic activity of Adeno-hTERT-E1A in cultured tumor cells translated into superior antitumor activity in vivo compared with ONYX-015, suggesting Adeno-hTERT-E1A has strong therapeutic potential with reduced toxicity to the patient.
Materials and Methods
Cell lines and reagents
Cyclophosphamide was purchased from Sigma-Aldrich (St. Louis, MO). Fetal bovine serum (FBS) and Dulbecco's modified Eagle's medium (DMEM) were purchased from Invitrogen (Frederick, MD). Human tumor cell lines MCF-7 (breast), MDA-MB-231 (breast), ZR-75 (breast), BT-20 (breast), BT-549 (breast), U251 (brain), and A549 (lung) were obtained from D. Scudiero (National Cancer Institute [NCI], Bethesda, MD). MCF-7/4HC cells, which display ~6-fold resistance to activated cyclophosphamide (4-hydroxycyclophosphamide, 4-OH-CPA), are described elsewhere (Chen and Waxman, 1995). Human tumor cells were grown at 37°C in a humidified, 5% CO2 atmosphere in RPMI 1640 culture medium containing 5% FBS, penicillin (100 units/ml), and streptomycin (100 μg/ml). MDA-MB-231 cells used in this study (designated MDA-MB-231v) were derived from the parental MDA-MB-231 tumor cell line grown as a solid subcutaneous tumor in a male scid mouse, extracted, and then made into a single-cell suspension using a collagenase protocol (Jounaidi and Waxman, 2001). Tumor cells implanted in scid mice (see Evaluation of Antitumor Activity in Human Tumor Xenografts) were grown in DMEM–10% FBS and antibiotics. The conditionally replicating adenovirus ONYX-015 (Bischoff et al., 1996), which contains a wild-type viral E3B region and an E1B-55 kDa gene deletion, was obtained from Onyx Pharmaceuticals (Richmond, CA). Adeno-β-galactosidase (Adeno-β-Gal) is an E1 and E3 region-deleted, replication-deficient adenovirus that contains a β-galactosidase reporter under the control of a cytomegaloviral (CMV) promoter (Chen et al., 1996).
hTERT promoter activity assays
The hTERT core promoter fragment (−371 to +27) was amplified by polymerase chain reaction (PCR) from the plasmid pBT-3915 (a generous gift from I. Horikawa, NCI) (Horikawa et al., 2002), using primers 5′-tagctagcTTAGGCCGATTCGACCTCTC-3′ and 5′-ggctcgagGGCTTCCCACGTGCGCA-3′, where lower-case letters designate engineered restriction enzyme sites. The TATA-less hTERT core promoter was verified by sequencing and subcloned into the reporter plasmid pGL3-Basic (Promega, Madison, WI) upstream of the luciferase gene, using the restriction enzymes NheI and XhoI (New England BioLabs, Ipswich, MA). The resulting luciferase reporter vector, pGL3-hTERT-Luc, was transfected with TransIT-LT1 (cat. no. MIR 2300; Mirus, Madison, WI) into individual tumor cell lines followed by dual luciferase analysis (cat. no. E1910; Promega). Briefly, 30,000 cells were seeded per well of a 48-well plate and 24 hr later the cells were transfected with 100 ng of pGL3-hTERT-Luc, 5 ng of phRL-CMV (Promega), and 145 ng of salmon sperm DNA. Hoechst 33258 dye (final concentration, 100 μM) was used to increase transfection efficiency. The cells were lysed 48 hr later and luciferase activity was measured. Luciferase activity was normalized to the protein concentration of each sample because of large variations in phRL-CMV reporter activity between cell lines.
Quantitative PCR analysis for hTERT and human telomerase RNA
The expression of hTERT and human telomerase RNA (hTER) mRNA was assayed by quantitative PCR (qPCR). Briefly, total RNA for each cell line was isolated with TRI-zol (Invitrogen) according to the manufacturer's instructions for monolayer cells in a 6-well plate format. Reverse transcription of 1 μg of total RNA in a volume of 20 μl was carried out with a high-capacity cDNA reverse transcription kit (cat. no. 4368814; Applied Biosystems, Foster City, CA). cDNA (4 μl; 1:50 dilution) in a total volume of 16 μl (including SYBR green and PCR primers) was amplified by qPCR with the following primers: 5′-ACGGCGACATGGAGAACAA-3′ (hTERT sense), 5′-CACTGTCTTCCGCAAGTTCAC-3′ (hTERT antisense), 5′-GAAGGGCGTAGGCGCCGTGCTTTTGC-3′ (hTER sense), and 5′-GTTTGCTCTAGAATGAACGGTGGAAGG-3′ (hTER antisense). Samples were incubated at 95°C for 10 min followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min in an ABI PRISM 7900HT sequence detection system (Applied Biosystems). Results were analyzed by the comparative cycle threshold CT (ΔΔCT) method as described by the manufacturer. Data are presented as relative levels of each RNA compared with the RNA level in MCF-7 cells after normalization to the 18S RNA content of each sample.
Construction and preparation of Adeno-hTERT-E1A
Luciferase cDNA was excised from pGL3-hTERT-Luc, using HindIII and XbaI, and the vector was religated to generate pGL3-hTERT. The viral E1A gene was next amplified by PCR from the plasmid pXC1 (Microbix, Toronto, ON, Canada), using the primers 5′-tagagatctGCCACTCTTGAGTGCCAGCGAG-3′ (forward) and 5′-tagagatctGTTAACCACACACGCAATCACAGG-3′ (reverse). The PCR product was digested with BglII and then ligated into the BglII site of pGL3-hTERT to create pGL3-hTERT-E1A. This vector was sequenced to verify E1A gene integrity and orientation. In addition, pGL3-hTERT-E1A was tested for E1A functionality by transient transfection into a panel of four tumor cell lines on a 6-well plate scale (1 μg of plasmid per ~300,000 cells per well), followed by protein extraction at 100°C with sodium dodecyl sulfate (SDS) total lysis buffer (10 mM Tris-HCl [pH 7.4], 1% SDS) plus protease inhibitors ([1 mM phenylmethylsulfonyl fluoride, 50 mM NaF, 0.5 mM Na3VO4] and 1× Complete [cat. no. 11 697 498 001; Roche, Mannheim, Germany]). E1A protein levels were determined by Western blotting with mouse monoclonal anti-E1A antibody (1:300 dilution, mouse anti-E1A, cat. no. V10148; Biomeda, Foster City, CA).
To generate the final adenoviral construct, Adeno-hTERT-E1A, the entire hTERT-E1A cassette including a simian virus 40 (SV40) late poly(A) signal was excised by digestion with MluI and BamHI, blunt ended, and then inserted into the EcoRI-opened site of pShuttle (Clontech, Palo Alto, CA). The entire IceuI–PISceI-excised cassette was then ligated into the Adeno-X expression system adenoviral genome as per the manufacturer's instructions (Clontech). Adenoviral stocks were propagated, amplified, and purified as previously described (Jounaidi and Waxman, 2004). Viral titers were quantified with an Adeno-X rapid titer kit (Clontech). Viral aliquots were stored at −80°C.
Cell line infectivity and oncolysis
The intrinsic adenoviral infectivity of each cell line was determined with the replication-deficient Adeno-β-Gal. Cells were seeded at 30,000 per well in 12-well plates in duplicate. Twenty-four hours later the cells were infected for a 4-hr period with Adeno-β-Gal at multiplicities of infection (MOIs) of 0, 10, 25, and 100 in RPMI containing 5% FBS (400 μl/well). The culture medium in each well was then replaced with 1 ml of fresh medium (RPMI–5% FBS). The cells were incubated for a further 48 hr followed by staining with 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal). β-Galactosidase activity was quantified with a microtiter plate reader (A650) after eluting the X-Gal stain in 1 ml of dimethyl sulfoxide per well. To compare the cytolytic activity of Adeno-hTERT-E1A with that of ONYX-015, cells were plated overnight in triplicate at 14,000 cells per well of a 24-well plate. After 24 hr the cells were infected with virus in a 200-μl volume of culture medium containing the indicated MOI of Adeno-hTERT-E1A or ONYX-015 for a 4-hr period, after which 1 ml of fresh medium was added to each well. Cells remaining 3 or 6 days later, as indicated, were stained and quantified according to a crystal violet–alcohol extraction assay (Jounaidi et al., 1998). Data are expressed as cell number (A595) relative to uninfected cell controls, mean ± SD for triplicate samples.
Western blotting
Cells infected with either Adeno-hTERT-E1A or ONYX-015 were lysed in SDS total lysis buffer containing protease inhibitors (as described previously) and boiled at 100°C for 10 min. Lysates (50–100 μg of protein by the Lowry method) were electrophoresed on SDS–7.5% polyacrylamide gels and then blotted onto nitrocellulose membranes (cat. no. 1620115; Bio-Rad, Hercules, CA). Blots were probed with anti-E1A antibody (diluted 1:300, mouse anti-E1A monoclonal antibody [mAb], cat. no. V10148; Biomeda) followed by horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody (diluted 1:3000, cat. no. NA931, ECL; GE Healthcare Life Sciences, Piscataway, NJ) or with anti-PARP [poly(ADP-ribose) polymerase] p85 antibody (diluted 1:1000, rabbit anti-PARP p85 mAb, cat. no. 1074-1; Epitomics, Burlingame, CA) followed by HRP-conjugated anti-rabbit secondary antibody (diluted 1:3000, cat. no. NA934, ECL; GE Healthcare Life Sciences). Antibody–antigen complexes were visualized with Amersham ECL detection reagent (cat. no. RPN2106; GE Healthcare Life Sciences) or SuperSignal substrate (cat. no. 34096; Pierce Biotechnology, Rockford, IL).
Apoptotic index/terminal deoxytransferase-mediated dUTP nick-end labeling
To assay virus-induced apoptosis, U251 cells were seeded at 14,000 cells per chamber in 8-chamber slides. The next day, cells in each chamber were infected with Adeno-hTERT-E1A or ONYX-015 at MOIs of 0, 5, 10, and 25. Slides were fixed with 4% paraformaldehyde after infection for 12, 24, or 48 hr and then processed with the DeadEnd colorimetric TUNEL (terminal deoxytransferase-mediated dUTP nickend labeling) system (cat. no. G7360; Promega).
Hexon staining assay for completion of adenoviral replication and release of functional viral particles into culture supernatant
Adenoviral hexon protein staining was used as a marker for late-stage adenoviral replication and to assay the release of functional virus from adenovirus-infected cells into the culture supernatant, as determined by hexon staining of A549 cells incubated for 30 hr with the culture supernatant from adenovirus-infected cells (A549 indicator cells). Cells were seeded at 200,000 cells per well in 12-well plates and, 24 hr later, duplicate wells were infected with Adeno-β-gal, Adeno-hTERT, or ONYX-015 at MOIs of 1 and 3. Viral infections took place in a minimal volume (400 μl/well) for an initial 4-hr period, after which each well was aspirated, washed with 1 ml of fresh RPMI–5% FBS growth medium, refreshed with 1 ml of RPMI–5% FBS, and incubated for the indicated times, at which point the supernatant was removed and applied to uninfected A549 cells. This washing procedure resulted in complete removal of virus present from the initial infection (data not shown). A separate plate of cells was seeded and infected for 1, 3, or 5 days for U251 cells, and for 3, 5, or 7 days for A549 cells (three plates per cell line). At each time point, supernatants from each well (~1 ml) were used to infect 12-well plates containing 200,000 A549 cells per well (A549 indicator cells, seeded 24 hr earlier) as described previously, that is, a 4-hr incubation with 1 ml of transferred supernatant, followed by the addition of 1 ml of fresh RPMI–5% FBS medium per well. The A549 indicator cells were incubated for a further 26 hr (total incubation, 30 hr) and then methanol fixed, stored at −20°C, and processed by hexon staining. Hexon protein was visualized by immunohistochemical staining with mouse anti-hexon monoclonal antibody (anti-adenovirus 1,2,5,6 hexon, diluted 1:1000; Chemicon International/Millipore, Bedford, MA) followed by HRP-conjugated anti-mouse secondary antibody (diluted 1:500, cat. no. NA931, ECL; GE Healthcare Life Sciences) as detailed in the Adeno-X rapid titer kit protocol (Clontech). The same protocol was used for viral titration of supernatants isolated over various days from the initially infected U251 and A549 cells, serially diluted, and applied to 293 cells (100,000 cells per well; 24-well plate). Hexon staining was also performed on A549 indicator cells infected with cell lysates taken from A549 cells initially infected in the same manner to assay functional intracellular virions 30 hr postinfection.
Adenoviral infection and replication in human primary hepatocytes
Primary human hepatocytes preseeded in 12-well plates (obtained from S. Strom, University of Pittsburgh, Pittsburgh, PA) were infected at ~80% confluency with either Adeno-hTERT-E1A or ONYX-015 at MOIs of 0, 1, and 10. After 48 hr the cells were scraped into 400 μl of phosphate-buffered saline (PBS) containing protease inhibitors (as described above). Cell extracts were prepared by freeze–thawing the scraped cells three times, after which an aliquot was analyzed by Western blotting with antibody to E1A and PARP p85 (30 μg of protein per lane). The cell extracts were spun at 14,000 rpm for 5 min to sediment cellular debris, and adenoviral genomic DNA present in the supernatant was then assayed by qPCR, using E1A-specific primers: 5′-ACCTGCCACGAGGCTGG-3′ (sense) and 5′-CCCACTGCCCATAATTTTCACT-3′ (antisense). To assay for completion of adenoviral replication, monitored by adenoviral hexon protein production, the hepatocytes were infected with Adeno-β-gal, Adeno-hTERT-E1A, or ONYX-015 at MOIs of 1 and 3 (each in duplicate) in a volume of 400 μl/well for an initial 4-hr incubation, after which 1 ml of fresh hepatocyte growth medium was added to each well. Forty-eight hours later, hexon protein was visualized by immunohistochemistry as described previously.
Evaluation of antitumor activity in human tumor xenografts
Five-week-old (19 to 24 g) male ICR/Fox Chase scid mice (Taconic Farms, Germantown, NY) were housed in the Boston University Laboratory of Animal Care Facility (Boston, MA) and treated in accordance with approved protocols and federal guidelines. Mice were injected subcutaneously via each posterior flank with 6 × 106 MDA-MB-231v cells in 0.2 ml of serum-free DMEM, using a 0.5-in. 27.5-gauge needle and a 1-ml insulin syringe. Tumor areas (length × width) were measured twice weekly with Vernier calipers (cat. no. 62379-531; VWR International, West Chester, PA) and tumor volumes were calculated as follows: Volume = (π/6) × (L × W)3/2. When the tumors reached ~125 mm3 in volume, the mice were divided into three groups (6 mice, 12 tumors per group):
Group 1: PBS (vehicle) injected intratumorally, 50 μl/tumor (control)
Group 2: Adeno-hTERT-E1A injected intratumorally, 1 × 109 plaque-forming units (PFU) injected in a volume of 50 μl/tumor (hTERT)
Group 3: ONYX-015 injected intratumorally, 1 × 109 PFU injected in a volume of 50 μl/tumor (ONYX)
Eighteen days after the first treatment, a second viral or vehicle injection was administered to each tumor. Tumor growth rates before viral administration were similar in all three groups. Tumor growth delay was calculated from the difference in time (in days) required for each tumor in the Adeno-hTERT-E1A or ONYX-015 treatment group to increase in size by 2-fold or by 5-fold, as compared with the control tumors. Thus, growth delay (2×) = t2× (treated) − t2× (control), and growth delay (5×) = t5× (treated) − t5× (control), where t2× and t5× are the times required for each tumor to increase in size 2-fold and 5-fold, respectively, relative to the day 0 tumor volume. Percent tumor growth inhibition was calculated by comparing tumor volumes for the Adeno-hTERT-E1A and ONYX-015 treatment groups with that of control tumors on days 18 and 46 after the initial treatment (day 0; see Fig. 7) for n = 12 tumors per group. Oneway analysis of variance (ANOVA), using Bonferroni multiple comparison, was carried out with GraphPad Prism 4 software (GraphPad Software, San Diego, CA).
FIG. 7.

Antitumor activity of Adeno-hTERT-E1A and ONYX-015 in an MDA-MB-231 tumor xenograft model. MDA-MB-231v tumor xenografts were implanted in the flanks of scid mice and treated with 1 × 109 PFU of each virus by intratumoral injection (arrows) as described in Materials and Methods. Shown in (A) are relative tumor volumes, normalized to the tumor volume on the first day of viral injection (day 0). (B) Mouse body weight data. Values shown represent means and SE for n = 6 mice and n = 12 tumors per treatment group. Oneway analysis of variance of tumor volume data with Bonferroni multiple test correction yielded the following significance values: p < 0.001 for PBS vehicle injected (control) versus Adeno-hTERT-E1A treated (Ad-hTERT), p < 0.01 for control versus ONYX-015 treated, and p < 0.05 for Ad-hTERT versus ONYX-015 treated. See Table 2 for tumor growth delay and volume growth inhibition analysis.
Results
Evaluation of core hTERT promoter element for cancer-specific expression of E1A
hTERT promoter fragments ranging from 3.8 kb to 255 bp have been used to drive the expression of E1A in oncolytic adenoviruses (Wirth et al., 2005). We evaluated an hTERT core promoter fragment (Poole et al., 2001), nucleotides −371 to +27 relative to the transcription start site, which contains two E-boxes (Horikawa et al., 2002), an AP2-β-binding site (Deng et al., 2007), and five SP1 sites required for hTERT repression in normal tissues (Won et al., 2002). To ascertain whether this hTERT promoter fragment could confer strong expression in tumor cells, a luciferase reporter plasmid driven by this promoter was transfected into a panel of eight human tumor cell lines (Fig. 1A). Endogenous hTERT and hTER transcript levels were determined by qPCR analysis (Fig. 1B and C). hTERT promoter-regulated luciferase activity had a cell line expression profile similar, but not identical, to that of endogenous hTERT RNA, suggesting that the core hTERT promoter activity is an important determinant of cellular hTERT RNA levels. hTERT RNA varied considerably between cell lines (Fig. 1B), whereas the RNA-primer component, hTER, was expressed at similar levels across the panel (Fig. 1C). hTERT RNA was not detectable in human hepatocytes (Fig. 1B) (Poole et al., 2001). To assess the utility of the core hTERT promoter for transcription of the adenoviral E1A gene, E1A was inserted into an expression plasmid downstream of the core hTERT promoter. Transfection into MCF-7/4HC, MDA-MB-231, U251, and A549 cells followed by E1A Western blot analysis (Fig. 1D) verified the functionality of the hTERT-E1A cassette and revealed detectable E1A protein expression in the four cell lines tested.
FIG. 1.

Telomerase promoter characterization. (A) Luciferase reporter activity of the hTERT core promoter cloned upstream of the luciferase gene and transfected into a panel of eight human tumor cell lines. qPCR analysis of endogenous hTERT RNA (B) and hTER RNA (C) is shown for the same panel of cell lines as in (A), with the addition of human hepatocytes. RNA levels were set relative to that of MCF-7 cells. hTERT mRNA was undetectable in BT-20 cells and in primary human hepatocytes. Data shown represent means + SD for n = 3 replicates. (D) Western blot showing E1A expression in MCF-7/4HC, MDA-MB-231, U251, and A549 cells after transfection with pGL3-hTERT-E1A. Fifteen micrograms of protein was loaded for the 293T cell E1A-positive control; 50 μg was loaded for each of the transfected cell lines. The three bands reflect multiple transcripts. No E1A was present in untransfected cell line controls (data not shown).
Oncolytic profile of Adeno-hTERT-E1A in comparison with ONYX-015
We constructed Adeno-hTERT-E1A, which contains the previously described hTERT core promoter-driven E1A cassette, deletions of the adenoviral E1B-55 kDa and E1B-19 kDa genes, and the entire E3A and E3B gene regions (Fig. 2). The lytic profile of Adeno-hTERT-E1A was compared with that of ONYX-015, an E1B-55 kDa-deleted tumor cell replicating adenovirus with E3B region deletions, which has been extensively evaluated in clinical studies (Crompton and Kirn, 2007). Tumor cell lines were infected with Adeno-hTERT-E1A or with ONYX-015 over a range of MOIs and then incubated for time periods up to 6 days followed by staining with crystal violet to visualize and quantify cell lysis (Fig. 3). In U251 cells, Adeno-hTERT-E1A infection resulted in massive cell death as early as 24–48 hr, whereas another 24–48 hr was required for ONYX-015 to elicit cell killing (data not shown). By day 3, and also on day 6, Adeno-hTERT-E1A exhibited improved lysis compared with ONYX-015 in U251 cells (Fig. 3A), as well as in MCF-7/4HC and MDA-MB-231 cells (Fig. 3C and D). Adeno-hTERT-E1A and ONYX-015 induced comparable cell death in A549 cells 48–72 hr post-infection (Fig. 3B, day 3). However, over time, Adeno-hTERT-E1A infection, in particular at lower MOIs, became static and was counterbalanced by continued growth of surrounding, uninfected A549 cells. In contrast, ONYX-015 infection led to complete killing of the A549 cell population. Infection of the panel of four cell lines with Adeno-β-gal revealed high intrinsic adenoviral infectivity in A549 and U251 cells; MCF-7/4HC and MDA-MB-231 cells were more difficult to infect as indicated by lower X-Gal staining (Fig. 3E). These results reflect differences in the intrinsic infectivity of each cell line, which is determined primarily by cell surface levels of the coxsackievirus–adenovirus receptor (Wood et al., 1999). The more extensive lysis of A549 and U251 cells by day 3 (Fig. 3A and B) may thus be attributable to this higher level of intrinsic infectivity compared with MCF-7/4HC and MDA-MB-231 cells.
FIG. 2.

Schematic depiction of Adeno-hTERT-E1A genome construction. Transcription factor-binding sites in the hTERT core promoter fragment (−371 to +27) are indicated in our core hTERT promoter (step 1), which was cloned upstream of the viral E1A gene in the pGL3 vector. Next, the hTERT-E1A-polyA cassette was excised with MluI and BamHI, blunt ended, and then ligated into the EcoRI site of an intermediate pShuttle vector (step 2) for the ultimate viral genome-cloning step (step 3), using the Adeno-X system genome. All cloning details are described in Materials and Methods.
FIG. 3.

Oncolytic activity of Adeno-hTERT-E1A and ONYX-015. Cell lines seeded 24 hr earlier were infected with Adeno-hTERT-E1A or ONYX-015 at the indicated MOIs. Three or 6 days later the cells were stained with crystal violet to quantify remaining cells in each of the indicated human tumor cells lines: (A) U251, (B) A549, (C) MCF-7/4HC, and (D) MDA-MB-231. Data shown in (A–D) represent means + SD, based on n = 3 replicates, with the uninfected cell controls set to 100%. (E) The replication-deficient adenovirus Adeno-β-gal was used to assay the intrinsic adenoviral infectivity of each cell line, as determined by X-Gal staining 48 hr postinfection. β-Galactosidase activity was quantified with a microtiter plate reader (A650) after eluting the X-Gal stain in 1 ml of dimethyl sulfoxide per well. Data shown represent A650 values normalized to crystal violet staining (means + SD, n = 2).
Elevated E1A production in Adeno-hTERT-E1A-infected tumor cells
E1A protein production, a prerequisite for adenoviral replication and an indicator of the viral potential to lyse infected cells, was monitored in Adeno-hTERT-E1A-infected tumor cells. In MCF-7/4HC cells, which showed moderate levels of hTERT promoter activity (Fig. 1A), E1A protein levels were substantially higher 48 hr after infection with Adeno-hTERT-E1A as compared with ONYX-015 (Fig. 4A), in which the endogenous adenoviral E1A promoter regulates E1A transcription. Higher E1A protein levels were also obtained after Adeno-hTERT-E1A infection in the case of U251, A549, and MDA-MB-231 cells 24 and 48 hr after viral infection (Fig. 4B and C). In U251 and A549 cells, E1A expression peaked early, perhaps reflecting their high intrinsic adenoviral infectivity (Fig. 3E) and high apparent hTERT promoter activity (Fig. 1A). By contrast, E1A accumulation was delayed in MDA-MB-231 cells, in which adenoviral infectivity and hTERT promoter activity were both lower.
FIG. 4.
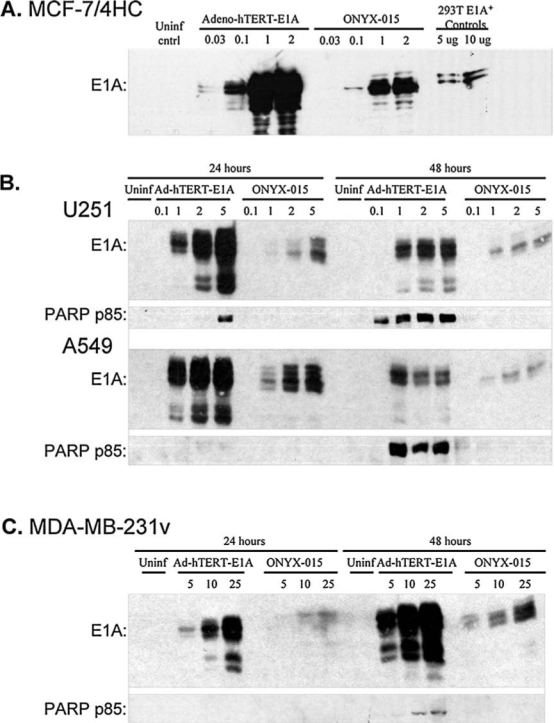
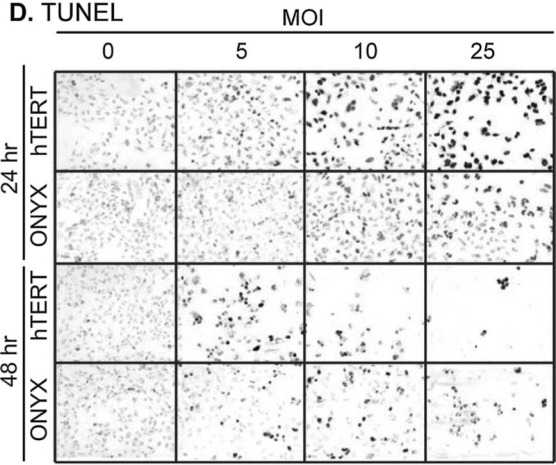
E1A protein levels and apoptosis induction in virus-infected cells. (A) Western blot of E1A protein from total cell lysates (100 μg/lane) of MCF-7/4HC cells infected for 48 hr with either Adeno-hTERT-E1A or ONYX-015. In the last two lanes, a 293T cell extract was loaded as an E1A-positive control. Corresponding Western blots for E1A protein, followed by reprobing for PARP p85 protein (50 μg of cell lysate per lane), are shown for U251, A549, and MDA-MB-231 cells (B and C) 24 and 48 hr after viral infection at the indicated MOIs. For each cell line, 300,000 cells were seeded per well in a 6-well plate 24 hr before infection. Western blots for U251 and MDA-MB-231 extracts were exposed in parallel; the signal intensities shown are thus directly comparable. (D) TUNEL staining of U251 cells infected with Adeno-hTERT-E1A (hTERT) or ONYX-015 (ONYX) at the MOIs indicated at both 24 and 48 hr postinfection, seen as dark stained cells in this panel. Less stain is visible in Adeno-hTERT-E1A-infected U251 cells by 48 hr, because many cells have already died and few remained for staining. No TUNEL activity was seen 12 hr postinfection (data not shown).
Enhanced tumor cell apoptosis after Adeno-hTERT-E1A infection
The early peak in E1A protein levels seen in Adeno-hTERT-E1A-infected U251 cells is consistent with our observation that U251 cells start dying 24–48 hr after infection with Adeno-hTERT-E1A as compared with 48–72 hr after infection with ONYX-015. This suggests that Adeno-hTERT-E1A may kill tumor cells by an E1A-induced apoptotic mechanism (Rao et al., 2004). To test this hypothesis, the virus-infected cells were assayed for cleavage of poly(ADP-ribose) polymerase (PARP), an early apoptotic event. In both U251 cells and A549 cells, strong PARP cleavage was seen after infection with Adeno-hTERT-E1A but not ONYX-015 (Fig. 4B and C). In U251 cells, the PARP cleavage product p85 was detected 24 hr after infection with 5 MOI of Adeno-hTERT-E1A, and at MOIs as low as 0.1 by 48 hr postinfection. In A549 cells, PARP cleavage was detected at 48 hr at MOIs as low as 1 (Fig. 4B). The increase in PARP cleavage correlated with the decline of E1A protein at higher MOIs at 48 hr, suggesting that the dying U251 and A549 cells are not able to sustain viral replication. In MDA-MB-231 cells, which showed lower intrinsic adenoviral infectivity and lower hTERT promoter activity than did U251 and A549 cells, a higher range of MOIs was required to elicit E1A expression and PARP cleavage (Fig. 4C). The ability of Adeno-hTERT-E1A to induce apoptosis in the infected cells was further investigated by TUNEL staining, which reflects terminal apoptotic events culminating in cellular DNA degradation. U251 cells infected with Adeno-hTERT-E1A underwent massive cell death, as evidenced by the earlier, more widespread TUNEL staining as compared with cells infected with ONYX-015 (Fig. 4D).
Impact of enhanced host cell apoptosis on Adeno-hTERT-E1A viral spread
We investigated next whether the early and high levels of Adeno-hTERT-E1A-induced apoptosis interfere with the ability of the virus-infected cells to release active, functional virus and thereby spread the oncolytic infection within a tumor cell population. Supernatants from A549 and U251 cells infected with Adeno-hTERT-E1A or ONYX-015 were collected on various days postinfection and then were titered for released, functional virions via an end-point dilution assay using 293 cells and hexon immunostaining. In parallel, Adeno-β-gal was used as a replication-deficient virus control. In A549 cells, which are preferentially lysed by ONYX-015, hexon staining revealed infection in all of the cells by day 3, leading to complete cell lysis 5–7 days after the initial infection. In contrast, Adeno-hTERT-E1A required more time for widespread hexon staining and lysis to occur (Fig. 5A, left). In addition, ONYX-015-infected A549 cells were substantially more efficient in terms of the release of functional virions into the supernatant, as detected by titration in 293 cells infected with serial dilutions of the harvested viral supernatants (Fig. 5A, right). In U251 cells, in which Adeno-hTERT-E1A induces apoptosis and extensive cell death (see the substantial decrease in cell density from days 1 to 3 and from 3 to 5; Fig. 5B, hTERT, left), Adeno-hTERT-E1A was still able to complete its viral life cycle and release functional virions (Fig. 5B, right). Extensive hexon staining was first seen in ONYX-015-infected U251 cells on day 3, at which time nearly all of the Adeno-hTERT-E1A-infected U251 cells were already lysed. Limited cell loss was seen by day 5 in the ONYX-015 infected U251 cells, in which the amount of virus released increased from day 3 yet was still low compared with Adeno-hTERT-E1A (Fig. 5B, right). We investigated next whether the delayed accumulation of Adeno-hTERT-E1A in the supernatant of infected A549 cells is due to impeded viral release, for example, due to the absence of the adenoviral death protein (ADP), or to a block in viral replication. To investigate this question, cell lysates prepared from A549 cells 30 hr after infection with Adeno-hTERT-E1A or ONYX-015 were applied to uninfected A549 indicator cells. Hexon staining 30 hr later revealed few functional virions from the Adeno-hTERT-E1A-infected A549 cells (Fig. 5C, hTERT, lysate reapplication), whereas ONYX-015 produced much greater amounts of functional virus, to the point that widespread lysis of the A549 indicator cells was already apparent (Fig. 5C, ONYX, lysate reapplication). Thus, in A549 cells, the production of functional virions is substantially impaired with Adeno-hTERT-E1A as compared with ONYX-015.
FIG. 5.

Adenoviral replication and spread. Left: Hexon immunohistochemical staining (IHC) of A549 cells (A) or U251 cells (B) either 3, 5, and 7 days after infection (A) or 1, 3, and 5 days after infection (B) with either Adeno-βgal, Adeno-hTERT-E1A, or ONYX-015 as indicated, at an MOI of 3. Right: Supernatants from each well of an experiment similar to that shown on the left side of (A) and (B) were harvested and serial dilutions were applied to fresh 293 cells for 40 hr followed by hexon staining to titer functional adenovirus released from the initially infected cells (log scale, base 10). The decreases in A549 cell hexon staining on days 5 and 7 and U251 hexon staining on days 3 and 5 reflect massive cell death induced by ONYX-015 and Adeno-hTERT-E1A, respectively. (C) Left: Hexon staining of A549 cells 30 hr postinfection with either Adeno-βgal, Adeno-hTERT-E1A, or ONYX-015 at MOIs of 1 and 3. Right: Corresponding samples, in which cell lysates were applied to A549 indicator cells, which were stained for hexon protein 30 hr later. Adeno-β-gal is replication defective and serves as a negative control. All images were taken at a magnification of ×40.
Viral replication in primary human hepatocytes
Human hepatocytes, for which adenovirus has high intrinsic infectivity (Jaffe et al., 1992), were investigated for their ability to support Adeno-hTERT-E1A infection and replication. Human hepatocytes were highly infectable with adenovirus, as determined by X-Gal staining of hepatocytes from three individual donors 48 hr after infection with the replication-deficient Adeno-β-gal (Fig. 6A, left; and data not shown). Next, we investigated whether the regulation of E1A by the hTERT core promoter would limit replication of Adeno-hTERT-E1A in human hepatocytes, given the apparent absence of hTERT promoter activity in these cells (Fig. 1B). Total cell extracts prepared 48 hr postinfection were analyzed for viral genomic DNA by qPCR using E1A gene-specific primers (Fig. 6A, right). ONYX-015 is reported to replicate minimally in primary hepatocytes as compared with cancer cells (Au et al., 2007), but nevertheless, ONYX-015 DNA was readily detectable in infected human hepatocytes (Fig. 6A, right). Adeno-hTERT-E1A replication proceeded at a ~26-fold lower level than ONYX-015 in hepatocytes from donor liver 1 (L1) and at a ~4- to 7-fold lower level in liver 3 (L3) (Fig. 6A, right). These findings were verified by assaying functional adenoviral production in the infected cells, as determined by hexon protein production. Hexon protein levels were ~24- to 29-fold lower in Adeno-hTERT-E1A-infected human hepatocytes compared with ONYX-015-infected hepatocytes in the case of liver donor L2 and ~18- to 21-fold lower in the case of donor L3 (Fig. 6B and Table 1). As expected, no hexon staining was observed with the replication-deficient Adeno-β-gal, which served as a negative control for residual hexon coat protein derived from the infecting viral particles. Higher E1A protein was observed in the ONYX-015-infected hepatocyte lysates, consistent with the more permissive replication of ONYX-015 as compared with Adeno-hTERT-E1A in human hepatocytes (Fig. 6C, top). Reprobing the same blot for the PARP cleavage product p85 indicated higher apoptosis in the ONYX-015-infected hepatocytes (Fig. 6C, bottom).
FIG. 6.

Adenoviral infectivity and replication in primary human hepatocytes. (A) Left: Adenoviral infectivity of primary human hepatocytes determined by Adeno-β-gal infection at MOIs of 1 and 10 followed by X-Gal staining 48 hr later. Data are shown for hepatocytes from two individual liver donors, designated L1 and L3. Right: Viral replication in primary human hepatocytes infected with Adeno-hTERT-E1A or ONYX-015, donors L1 and L3. Cell lysates were assayed for genomic E1A DNA by qPCR, using an intronic reverse primer. (B) Hexon staining of human hepatocytes (donors L2 and L3) infected with Adeno-β-gal, Adeno-hTERT-E1A, or ONYX-015 at MOIs of 1 and 3. See Table 1 for quantification of hexon staining. (C) Western blots showing lower E1A protein and PARP p85 cleavage product in Adeno-hTERTE1A-infected human hepatocytes (donor L3) compared with ONYX-015-infected human hepatocytes (MOIs of 1 and 10). The 293T cell positive control for E1A shown in the last lane exhibited a lower apparent protein mobility, as in Fig. 1D.
Table 1.
Adenoviral Hexon Staining in Infected Human Hepatocytesa
| |
Liver 2 |
Liver 3 |
||
|---|---|---|---|---|
| Virus | MOI of 1 | MOI of 3 | MOI of 1 | MOI of 3 |
| Adeno-β-gal | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| Adeno-hTERT-E1A | 0.58 ± 0.78b | 0.96 ± 1.08b | 1.30 ± 0.19b | 3.30 ± 0.13b |
| ONYX-015 | 13.75 ± 4.91 | 27.83 ± 14.19 | 22.94 ± 2.85 | 67.99 ± 7.70 |
| Ratio (ONYX-015/Adeno-hTERT-E1A) | 23.7 | 28.9 | 17.6 | 20.6 |
Hexon staining counts averaged from 20 to 50 fields of primary human hepatocytes from liver donors 2 and 3, stained 48 hr after infection with Adeno-β-gal. Adeno-hTERT-E1A, or ONYX-015 at MOIs of 1 and 3. Data shown represent means ± SD for n = 20–50 fields per well, 2 wells per MOI, at ×200 magnification.
p < 0.0001 for hexon staining in cells infected with Adeno-hTERT-E1A compared with ONYX-015 (two-tailed paired t test, 95% confidence intervals).
In vivo antitumor activity in a human breast cancer xenograft model
The antitumor activity of Adeno-hTERT-E1A was investigated in scid mice bearing subcutaneous human breast cancer MDA-MB-231 xenografts. MDA-MB-231 cells show moderate intrinsic infectivity by adenovirus (Fig. 3E) and low to moderate hTERT promoter activity (Fig. 1A). Thirteen days after tumor cell inoculation, the tumor-bearing mice were randomized into three treatment groups (6 mice and 12 tumors per group), each having an average tumor volume of ~125 mm3. Mice were injected intratumorally with PBS (vehicle) or with PBS containing either Adeno-hTERT-E1A or ONYX-015 at 1 × 109 PFU/tumor. Tumor volumes, normalized to the first day of viral injection, were determined twice per week (Fig. 7A). After the first viral injection, Adeno-hTERT-E1A showed greater antitumor activity, inducing a tumor volume-doubling delay of 10.5 days and an overall tumor volume growth inhibition of 45% compared with controls. This can be compared with a tumor volume-doubling delay of 2.5 days and 26% tumor volume growth inhibition for ONYX-015. There was also an increase in the time required for the tumor volumes to increase 5-fold (~24 days) and an overall volume growth inhibition of ~64% with Adeno-hTERT-E1A compared with the control group, versus only 11 days and 42% with ONYX-015 (Table 2). The tumor growth trends for each virus were significantly different from each other (p < 0.05 for Adeno-hTERT-E1A vs. ONYX-015; one-way ANOVA analysis with Bonferroni multiple comparison), and both viral treatment groups were significantly different from the virus-free control (p < 0.001 for control vs. Adeno-hTERT-E1A; and p < 0.01 for control vs. ONYX-015). No signs of host toxicity were observed with either viral treatment, as determined by monitoring body weights (Fig. 7B).
Table 2.
Tumor Growth Delay and Tumor Volume Growth Inhibition in Adenovirus-Infected MDA-MB-231 Tumorsa
| A. Tumor Growth Delay | ||||
|---|---|---|---|---|
| Tumor growth delay data | Days to 2× | 2× tumor growth delay (days) | Days to 5× | 5× tumor growth delay (days) |
| Control | 7.08 ± 0.88 | N/A | 22.67 ± 2.46 | N/A |
| Adeno-hTERT-E1A | 17.58 ± 2.22b,c | 10.5 ± 3.10 | 46.00 ± 2.60b,c | 23.33 ± 5.06 |
| ONYX-015 | 9.58 ± 1.30 | 2.5 ± 2.18 | 33.75 ± 2.36d | 11.08 ± 4.82 |
| B. Tumor Growth Inhibition | ||
|---|---|---|
| Tumor growth inhibition data | Virus | Tumor growth inhibition (%) |
| Treatment period 1 (days 0 to 18) | Adeno-hTERT-E1A | 44.9 ± 3.2b,c |
| ONYX-015 | 25.8 ± 5.6e | |
| Treatment period 2 (days 0 to 46) | Adeno-hTERT-E1A | 63.5 ± 4.6b,f |
| ONYX-015 | 42.1 ± 10.8g | |
Abbreviation: N/A, Not applicable.
All p values determined by one-way ANOVA with Bonferroni multiple comparison.
Tumor-bearing mice were treated with Adeno-hTERT-E1A or ONYX-015 by intratumoral injection as described in Fig. 7. Tumor growth delay (A) and tumor growth inhibition (B) were calculated in comparison with control tumors as described in Materials and Methods. Inhibition data were normalized by setting the last measurement of the control group (days 18 and 46 for treatment periods 1 and 2, respectively) to 100%. Tumor growth delay and growth inhibition data are presented as percent means ± SE based on n = 12 tumors per group.
p < 0.001 (control vs. Adeno-hTERT-E1A).
p < 0.01 (Adeno-hTERT-E1A vs. ONYX-015).
p < 0.05 (control vs. ONYX-015).
p < 0.001 (control vs. ONYX-015).
p < 0.05 (Adeno-hTERT-E1A vs. ONYX-015).
p < 0.01 (control vs. ONYX-015).
Discussion
Placement of viral regulatory genes under the control of the hTERT promoter, which is active in a majority of human cancers (Kim et al., 1994), is increasingly regarded as an ideal way to restrict the replication of oncolytic adenoviruses to malignant tissues. Several tumor cell-replicating, hTERT-driven adenoviruses have been described (Wirth et al., 2005); however, they differ most notably from Adeno-hTERT-E1A, the oncolytic adenovirus described in the present study, insofar as they contain either one or both E1B region genes: E1B-19kDa and E1B-55kDa. Several of these hTERT promoter-regulated viruses include upstream hTERT regulatory sequences, which may contain repressor sequences absent from the hTERT core promoter. The core hTERT promoter fragment presently used to regulate E1A expression in Adeno-hTERT-E1A was chosen to maximize activity in cancer cell lines based on earlier promoter deletion analysis (Horikawa et al., 1999; Poole et al., 2001). Evaluation of the hTERT-E1A expression cassette demonstrated functionality, and the hTERT-luciferase reporter cassette showed a cell line-specific pattern of reporter activity that was qualitatively similar to the endogenous hTERT activity. Thus, the hTERT core promoter sequence appears to confer high levels of expression in a cell line-dependent manner, with the truncated promoter behaving qualitatively similar to the endogenous full-length promoter.
The E1B-55 kDa-deleted, oncolytic adenovirus ONYX-015 has been evaluated in multiple phase II and phase III clinical trials (Crompton and Kirn, 2007). E1B-55 kDa protein is known to bind, sequester, and facilitate the degradation of p53 (Steegenga et al., 1998; Grand et al., 1999), and as such, ONYX-015 was first believed to replicate only in p53-deficient cells, imparting tumor cell specificity to viral replication (Bischoff et al., 1996). However, ONYX-015 was later shown to replicate in certain cells bearing a wild-type p53 gene (Dix et al., 2001), suggesting the possible risk of permissive replication in normal tissues. E1B-55 kDa protein also plays a role in host cell protein synthesis shutoff and in late adenoviral mRNA export, thus restricting replication to tumor cells, as well as normal cells, where this transport is phenocopied by other factors (O'Shea et al., 2004). These observations indicate a need to improve the safety of E1B-55 kDa-deleted oncolytic adenoviruses. The second E1B region gene product, E1B-19 kDa, functions like the antiapoptotic mitochondrial protein Bcl-2: it binds to and represses the proapoptotic transcription repressor Btf (Imazu et al., 1999; Kasof et al., 1999), and it inactivates the proapoptotic Bcl2 family member Bax (Han et al., 1996). Both E1B genes delay adenovirus-induced cell death long enough for completion of viral replication and repackaging. In many cancer cells, where apoptosis is already suppressed, these antiapoptotic genes are not required for viral replication (Rao et al., 2004, 2006), enabling these genes to be deleted in the construction of oncolytic adenoviruses (Duque et al., 1999; Harrison et al., 2001). The present deletion of both E1B genes in the context of high hTERT promoter-regulated E1A protein expression yielded a highly oncolytic virus that retains high cancer cell selectivity with greatly reduced replication in primary hepatocytes.
The high levels of E1A protein achieved in Adeno-hTERT-E1A-infected cells also likely contributed to the increased oncolytic activity seen with Adeno-hTERT-E1A as compared with ONYX-015 in all tumor cell lines tested except for A549. E1A, an immediate-early viral protein, is typically expressed within 6 hr of infection and is crucial for the onset of viral replication and subsequent expression of further downstream early and late viral genes (Nevins, 1981). As such, Adeno-hTERT-E1A infection led to oncolysis that was more rapid and more complete than after ONYX-015 infection in the case of MCF-7/4HC (breast), U251 (brain), and MDA-MB-231 (breast) cancer cells. E1A itself has substantial intrinsic cytotoxic activity (Hale and Braithwaite, 1999; Zhou et al., 2003; Rao et al., 2004) and its high-level expression in the absence of both anti-apoptotic E1B proteins in Adeno-hTERT-E1A-infected cells resulted in early and high levels of apoptosis, which most likely also contributed to cell death. In our oncolysis and hexon-staining studies, cells infected with ONYX-015 did not appear to go through apoptotic cell death as evidenced by lack of PARP cleavage and TUNEL staining (Fig. 4). They appear to have two discrete morphologies, one of a healthy cell bearing normal cell-line specific characteristics and one of a rounded-up cell about to lyse, whereas those infected with Adeno-hTERT-E1A typically appear as sickly, rounded-up cells, which were shown to undergo apoptosis.
Adeno-hTERT-E1A and ONYX-015 differ with respect to five adenoviral gene products, in addition to their distinct E1A promoter regulatory elements, making it difficult to definitively identity the factors responsible for the unique properties of each virus. However, ONYX-015 was initially chosen to assess the oncolytic potential of Adeno-hTERT-E1A while comparing it with a clinically evaluated adenovirus. ONYX-015, but not Adeno-hTERT-E1A, encodes the anti-apoptotic factor E1B-19 kDa, as well as the E3 region adenoviral death protein (ADP), which facilitates late-stage lysis and release of mature viral progeny from infected A549 cells (Doronin et al., 2003). The similar oncolytic profiles of both viruses in A549 cells at earlier time points might be explained by efficient viral lysis in the ONYX-015-infected cells as compared with the early apoptotic death seen in Adeno-hTERT-E1A-infected cells. We considered the possibility that the viral ADP protein might explain why the long-term lysis and spread of ONYX-015 in A549 cells is superior, outpacing Adeno-hTERT-E1A in its ability to kill the cells, in particular at low MOIs. However, it does not appear that the ADP protein is responsible for the increased lysis and spread of ONYX-015 compared with Adeno-hTERT-E1A in A549 cells, as indicated by the much higher levels of functional virus seen in lysates of ONYX-015-infected cells as compared with Adeno-hTERT-E1A-infected cells. Indeed, in other studies, E1B-19 kDa-deleted adenoviruses were equally able to produce viral plaques and lyse A549 cells, independent of whether ADP was present (Subramanian et al., 2006). Plaque formation and disruption of cell lysis were impeded only in the presence of E1B-19 kDa, which stabilizes the nuclear lamin structure, and only in the absence of ADP, which counteracts this added nuclear stability late in the viral life cycle to instigate efficient cell lysis (Subramanian et al., 2006). Accordingly, in the case of Adeno-hTERT-E1A, in which both proteins in the antagonist pair, E1B-19 kDa and ADP, are absent, viral release per se is not the problem. The more extensive lysis of A549 cells by ONYX-015 as compared with Adeno-hTERT-E1A must therefore result from other factors unique to ONYX-015 that confer its ability to efficiently replicate and produce high functional viral titers in A549 cells, in spite of the lower levels of E1A. One possibility is that the high levels of E1A protein produced in A549 cells infected with Adeno-hTERT-E1A kill many of the host cells prematurely via apoptosis, thereby decreasing the efficiency of successive rounds of viral replication and reinfection.
E1A can be used as a therapeutic gene to induce antitumoral responses, including inhibition of angiogenesis (Zhou et al., 2003). E1A also can sensitize tumor cells to chemotherapy and radiation (Sanchez-Prieto et al., 1996; Martin-Duque et al., 1999). Therefore, the high level of E1A expression achieved in Adeno-hTERT-E1A-infected tumor cells may not only provide for tumor-specific regulation of viral replication but may induce other E1A-dependent antitumor mechanisms. Differences in apoptosis induction between Adeno-hTERT-E1A and ONYX-015 may reflect the absence of both antiapoptotic genes (E1B-55 kDa and E1B-19 kDa) in Adeno-hTERT-E1A, as compared with ONYX-015, which retains the anti-apoptotic Bcl-2 analog E1B-19 kDa, as well as the elevated expression of E1A, which can induce p53 and stimulate apoptosis (Hale and Braithwaite, 1999; Kasof et al., 1999). Indeed, high E1A levels correlated with a high apoptotic index in U251, A549, MDA-MB-231, and MCF-7 cells infected with Adeno-hTERT-E1A (data not shown). The similar A549 cell oncolytic profiles exhibited by both viruses at higher MOIs at earlier but not later time points might be explained by the delivered dose of Adeno-hTERT-E1A initially inducing high levels of cell death via E1A-dependent apoptosis. ADP-containing replication-competent adenoviruses, such as ONYX-015, induce cell death via a necrosis-like pathway that is apoptotic machinery independent (Abou El Hassan et al., 2004), which is not the case for Adeno-hTERT-E1A. As such, the cell death induced by Adeno-hTERT-E1A may best be described as telomerase-regulated E1A suicide gene therapy using a replication-conditional virus.
Adeno-hTERT-E1A displayed superior antitumor activity compared with ONYX-015 against breast tumor MDA-MB-231 xenografts, with no apparent increase in host toxicity. Moreover, the genetic modifications introduced into Adeno-hTERT-E1A greatly decreased its replication competence in primary human hepatocytes. This can be explained by the absence of hTERT expression in human hepatocytes, which precludes E1A expression and all downstream events in the adenoviral replication cycle. The use of hTERT promoter sequences to regulate Adeno-hTERT-E1A replication likely contributes to its substantially decreased replication in hepatocytes compared with ONYX-015, where E1A induction, genome replication, and viral protein production were readily detected, as were viral completion/repackaging and capsid assembly, as indicated by hexon staining. Adenoviral E1A protein has been linked to acute hepatotoxicity via induction of tumor necrosis factor-α and transaminitis in mice (Engler et al., 2004). Although hTERT RNA was not detectable in hepatocytes, we could still detect E1A and hexon protein in a few cells infected with Adeno-hTERT-E1A. These cells may be hepatocytes undergoing abortive replication, cells with a mutated hTERT and an apoptosis status that reflects oncogenic potential, or perhaps the presence of other liver-associated cell types in the human hepatocyte preparations. The absence of both antiapoptotic proteins in Adeno-hTERT-E1A should allow primary host cells to apoptose and thereby abort any instances of viral infection that could otherwise lead to viral replication. The increased oncolytic response associated with complete deletion of the E1B region therefore does not infringe on the replication specificity of Adeno-hTERT-E1A and its safety profile against primary tissues. These findings were confirmed in primary hepatocytes isolated from three independent donors, highlighting the improved safety and replication specificity of Adeno-hTERT-E1A, and further substantiating the use of the hTERT core promoter to simultaneously improve cancer selectivity and minimize host toxicity.
While this study was in progress, Bilsland and coworkers described the use of an hTERT promoter-regulated E1A cassette in an oncolytic adenovirus lacking the entire E1B gene region, and reported a phenotype similar to that of our Adeno-hTERT-E1A virus, in particular elevated E1A expression and a modified oncolytic profile (Bilsland et al., 2007). Their results support our finding that deletion of the E1B region enhances early oncolysis, which may diminish replication completion and virion maturation. The present study extends those findings to include an investigation of the effects of Adeno-hTERT-E1A on apoptosis induction, viral release, hepatocyte safety, and antitumor activity in vivo.
In conclusion, we have engineered an oncolytic adenovirus, Adeno-hTERT-E1A, which displays an improved oncolytic profile in many cancer cell lines and substantially reduced replication in human hepatocytes compared with ONYX-015, an oncolytic virus that has undergone extensive testing in clinical trials. The diminished propensity of Adeno-hTERT-E1A to replicate in a primary organ such as the liver, which is highly susceptible to adenoviral infection (Wood et al., 1999), may allow for an increase in virus dose to improve therapeutic activity without increasing hepatic toxicity. Future studies will examine potential combination therapies in which Adeno-hTERT-E1A will be used with anticancer prodrug-activating enzymes, such as cytochrome P-450 2B11 (Jounaidi et al., 2006), to further enhance therapeutic potential.
Acknowledgments
Supported in part by NIH grant CA49248 (to D.J.W.) and by Susan G. Komen Breast Cancer Foundation grant BCTR0504032 (to Y.J.).
Author Disclosure Statement
No competing financial interests exist.
References
- Abou El Hassan M.A. van der Meulen-Muileman I. Abbas S. Kruyt F.A. Conditionally replicating adenoviruses kill tumor cells via a basic apoptotic machinery-independent mechanism that resembles necrosis-like programmed cell death. J. Virol. 2004;78:12243–12251. doi: 10.1128/JVI.78.22.12243-12251.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson C.J. Hoare S.F. Ashcroft M. Bilsland A.E. Keith W.N. Hypoxic regulation of telomerase gene expression by transcriptional and post-transcriptional mechanisms. Oncogene. 2006;25:61–69. doi: 10.1038/sj.onc.1209011. [DOI] [PubMed] [Google Scholar]
- Au T. Thorne S. Korn W.M. Sze D. Kirn D. Reid T.R. Minimal hepatic toxicity of Onyx-015: Spatial restriction of coxsackie–adenoviral receptor in normal liver. Cancer Gene Ther. 2007;14:139–150. doi: 10.1038/sj.cgt.7700988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauerschmitz G.J. Barker S.D. Hemminki A. Adenoviral gene therapy for cancer: From vectors to targeted and replication competent agents [review] Int. J. Oncol. 2002;21:1161–1174. [PubMed] [Google Scholar]
- Bilsland A.E. Merron A. Vassaux G. Keith W.N. Modulation of telomerase promoter tumor selectivity in the context of oncolytic adenoviruses. Cancer Res. 2007;67:1299–1307. doi: 10.1158/0008-5472.CAN-06-3000. [DOI] [PubMed] [Google Scholar]
- Bischoff J.R. Kirn D.H. Williams A. Heise C. Horn S. Muna M. Ng L. Nye J.A. Sampson-Johannes A. Fattaey A. McCormick F. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells [see comments] Science. 1996;274:373–376. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- Chen G. Waxman D.J. Identification of glutathione S-transferase as a determinant of 4-hydroperoxycyclophosphamide resistance in human breast cancer cells. Biochem. Pharmacol. 1995;49:1691–1701. doi: 10.1016/0006-2952(95)00079-f. [DOI] [PubMed] [Google Scholar]
- Chen L. Waxman D.J. Chen D. Kufe D.W. Sensitization of human breast cancer cells to cyclophosphamide and ifosfamide by transfer of a liver cytochrome P450 gene. Cancer Res. 1996;56:1331–1340. [PubMed] [Google Scholar]
- Crompton A.M. Kirn D.H. From ONYX-015 to armed vaccinia viruses: The education and evolution of oncolytic virus development. Curr. Cancer Drug Targets. 2007;7:133–139. doi: 10.2174/156800907780058862. [DOI] [PubMed] [Google Scholar]
- Deng W.G. Jayachandran G. Wu G. Xu K. Roth J.A. Ji L. Tumor-specific activation of human telomerase reverses transcriptase promoter activity by activating enhancer-binding protein-2β in human lung cancer cells. J. Biol. Chem. 2007;282:26460–26470. doi: 10.1074/jbc.M610579200. [DOI] [PubMed] [Google Scholar]
- Dix B.R. Edwards S.J. Braithwaite A.W. Does the antitumor adenovirus ONYX-015/dl1520 selectively target cells defective in the p53 pathway? J. Virol. 2001;75:5443–5447. doi: 10.1128/JVI.75.12.5443-5447.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doronin K. Toth K. Kuppuswamy M. Krajcsi P. Tollefson A.E. Wold W.S. Overexpression of the ADP (E3-11.6K) protein increases cell lysis and spread of adenovirus. Virology. 2003;305:378–387. doi: 10.1006/viro.2002.1772. [DOI] [PubMed] [Google Scholar]
- Duque P.M. Alonso C. Sanchez-Prieto R. Lleonart M. Martinez C. De Buitrago G.G. Cano A. Quintanilla M. Ramon y Cajal S. Adenovirus lacking the 19-kDa and 55-kDa E1B genes exerts a marked cytotoxic effect in human malignant cells. Cancer Gene Ther. 1999;6:554–563. doi: 10.1038/sj.cgt.7700077. [DOI] [PubMed] [Google Scholar]
- Engler H. Machemer T. Philopena J. Wen S.F. Quijano E. Ramachandra M. Tsai V. Ralston R. Acute hepatotoxicity of oncolytic adenoviruses in mouse models is associated with expression of wild-type E1a and induction of TNF-α. Virology. 2004;328:52–61. doi: 10.1016/j.virol.2004.06.043. [DOI] [PubMed] [Google Scholar]
- Everts B. van der Poel H.G. Replication-selective oncolytic viruses in the treatment of cancer. Cancer Gene Ther. 2005;12:141–161. doi: 10.1038/sj.cgt.7700771. [DOI] [PubMed] [Google Scholar]
- Grand R.J. Parkhill J. Szestak T. Rookes S.M. Roberts S. Gallimore P.H. Definition of a major p53 binding site on Ad2E1B58K protein and a possible nuclear localization signal on the Ad12E1B54K protein. Oncogene. 1999;18:955–965. doi: 10.1038/sj.onc.1202358. [DOI] [PubMed] [Google Scholar]
- Hale T.K. Braithwaite A.W. The adenovirus oncoprotein E1a stimulates binding of transcription factor ETF to transcriptionally activate the p53 gene. J. Biol. Chem. 1999;274:23777–23786. doi: 10.1074/jbc.274.34.23777. [DOI] [PubMed] [Google Scholar]
- Han J. Sabbatini P. Perez D. Rao L. Modha D. White E. The E1B 19K protein blocks apoptosis by interacting with and inhibiting the p53-inducible and death-promoting Bax protein. Genes Dev. 1996;10:461–477. doi: 10.1101/gad.10.4.461. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Harrison D. Sauthoff H. Heitner S. Jagirdar J. Rom W.N. Hay J.G. Wild-type adenovirus decreases tumor xenograft growth, but despite viral persistence complete tumor responses are rarely achieved—deletion of the viral E1b-19-kD gene increases the viral oncolytic effect. Hum. Gene Ther. 2001;12:1323–1332. doi: 10.1089/104303401750270977. [DOI] [PubMed] [Google Scholar]
- Horikawa I. Cable P.L. Afshari C. Barrett J.C. Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res. 1999;59:826–830. [PubMed] [Google Scholar]
- Horikawa I. Cable P.L. Mazur S.J. Appella E. Afshari C.A. Barrett J.C. Downstream E-box-mediated regulation of the human telomerase reverse transcriptase (hTERT) gene transcription: Evidence for an endogenous mechanism of transcriptional repression. Mol. Biol. Cell. 2002;13:2585–2597. doi: 10.1091/mbc.E01-11-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imazu T. Shimizu S. Tagami S. Matsushima M. Nakamura Y. Miki T. Okuyama A. Tsujimoto Y. Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene. 1999;18:4523–4529. doi: 10.1038/sj.onc.1202722. [DOI] [PubMed] [Google Scholar]
- Jaffe H.A. Danel C. Longenecker G. Metzger M. Setoguchi Y. Rosenfeld M.A. Gant T.W. Thorgeirsson S.S. Stratford-Perricaudet L.D. Perricaudet M. Pavirani A. Lecocq J.-P. Crystal R.G. Adenovirus-mediated in vivo gene transfer and expression in normal rat liver. Nat. Genet. 1992;1:372–378. doi: 10.1038/ng0892-372. [DOI] [PubMed] [Google Scholar]
- Jounaidi Y. Waxman D.J. Frequent, moderate-dose cyclophosphamide administration improves the efficacy of cytochrome P-450/cytochrome P-450 reductase-based cancer gene therapy. Cancer Res. 2001;61:4437–4444. [PubMed] [Google Scholar]
- Jounaidi Y. Waxman D.J. Use of replication-conditional adenovirus as a helper system to enhance delivery of P450 prodrug-activation genes for cancer therapy. Cancer Res. 2004;64:292–303. doi: 10.1158/0008-5472.can-03-1798. [DOI] [PubMed] [Google Scholar]
- Jounaidi Y. Hecht J.E. Waxman D.J. Retroviral transfer of human cytochrome P450 genes for oxazaphosphorine-based cancer gene therapy. Cancer Res. 1998;58:4391–4401. [PubMed] [Google Scholar]
- Jounaidi Y. Chen C.S. Veal G.J. Waxman D.J. Enhanced antitumor activity of P450 prodrug-based gene therapy using the low Km cyclophosphamide 4-hydroxylase P450 2B11. Mol. Cancer Ther. 2006;5:541–555. doi: 10.1158/1535-7163.MCT-05-0321. [DOI] [PubMed] [Google Scholar]
- Jounaidi Y. Doloff J.C. Waxman D.J. Conditionally replicating adenoviruses for cancer treatment. Curr. Cancer Drug Targets. 2007;7:285–301. doi: 10.2174/156800907780618301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasof G.M. Goyal L. White E. Btf, a novel death-promoting transcriptional repressor that interacts with Bcl-2-related proteins. Mol. Cell. Biol. 1999;19:4390–4404. doi: 10.1128/mcb.19.6.4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. Kim J.H. Choi K.J. Kim P.H. Yun C.O. E1A- and E1B-double mutant replicating adenovirus elicits enhanced oncolytic and antitumor effects. Hum. Gene Ther. 2007;18:773–786. doi: 10.1089/hum.2006.167. [DOI] [PubMed] [Google Scholar]
- Kim N.W. Piatyszek M.A. Prowse K.R. Harley C.B. West M.D. Ho P.L. Coviello G.M. Wright W.E. Weinrich S.L. Shay J.W. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick K.L. Ogunkolade W. Elkak A.E. Bustin S. Jenkins P. Ghilchick M. Newbold R.F. Mokbel K. hTERT expression in human breast cancer and non-cancerous breast tissue: Correlation with tumour stage and c-Myc expression. Breast Cancer Res. Treat. 2003;77:277–284. doi: 10.1023/a:1021849217054. [DOI] [PubMed] [Google Scholar]
- Ma H. Urquidi V. Wong J. Kleeman J. Goodison S. Telomerase reverse transcriptase promoter regulation during myogenic differentiation of human RD rhabdomyosarcoma cells. Mol. Cancer Res. 2003;1:739–746. [PubMed] [Google Scholar]
- Martin-Duque P. Sanchez-Prieto R. Romero J. Martinez-Lamparero A. Cebrian-Sagarriga S. Guinea-Viniegra J. Dominguez C. Lleonart M. Cano A. Quintanilla M. Ramon y Cajal S. In vivo radiosensitizing effect of the adenovirus E1A gene in murine and human malignant tumors. Int. J. Oncol. 1999;15:1163–1168. doi: 10.3892/ijo.15.6.1163. [DOI] [PubMed] [Google Scholar]
- Mo Y. Gan Y. Song S. Johnston J. Xiao X. Wientjes M.G. Au J.L. Simultaneous targeting of telomeres and telomerase as a cancer therapeutic approach. Cancer Res. 2003;63:579–585. [PubMed] [Google Scholar]
- Nevins J.R. Mechanism of activation of early viral transcription by the adenovirus E1A gene product. Cell. 1981;26:213–220. doi: 10.1016/0092-8674(81)90304-4. [DOI] [PubMed] [Google Scholar]
- Oh S. Song Y.H. Kim U.J. Yim J. Kim T.K. In vivo and in vitro analyses of Myc for differential promoter activities of the human telomerase (hTERT) gene in normal and tumor cells. Biochem. Biophys. Res. Commun. 1999;263:361–365. doi: 10.1006/bbrc.1999.1366. [DOI] [PubMed] [Google Scholar]
- O'Shea C.C. Johnson L. Bagus B. Choi S. Nicholas C. Shen A. Boyle L. Pandey K. Soria C. Kunich J. Shen Y. Habets G. Ginzinger D. McCormick F. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6:611–623. doi: 10.1016/j.ccr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Poole J.C. Andrews L.G. Tollefsbol T.O. Activity, function, and gene regulation of the catalytic subunit of telomerase (hTERT) Gene. 2001;269:1–12. doi: 10.1016/s0378-1119(01)00440-1. [DOI] [PubMed] [Google Scholar]
- Rao X.M. Tseng M.T. Zheng X. Dong Y. Jamshidi-Parsian A. Thompson T.C. Brenner M.K. McMasters K.M. Zhou H.S. E1A-induced apoptosis does not prevent replication of adenoviruses with deletion of E1b in majority of infected cancer cells. Cancer Gene Ther. 2004;11:585–593. doi: 10.1038/sj.cgt.7700739. [DOI] [PubMed] [Google Scholar]
- Rao X.M. Zheng X. Waigel S. Zacharias W. McMasters K.M. Zhou H.S. Gene expression profiles of normal human lung cells affected by adenoviral E1B. Virology. 2006;350:418–428. doi: 10.1016/j.virol.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Sanchez-Prieto R. Quintanilla M. Cano A. Leonart M.L. Martin P. Anaya A. Ramon y Cajal S. Carcinoma cell lines become sensitive to DNA-damaging agents by the expression of the adenovirus E1A gene. Oncogene. 1996;13:1083–1092. [PubMed] [Google Scholar]
- Steegenga W.T. Riteco N. Jochemsen A.G. Fallaux F.J. Bos J.L. The large E1B protein together with the E4orf6 protein target p53 for active degradation in adenovirus infected cells. Oncogene. 1998;16:349–357. doi: 10.1038/sj.onc.1201540. [DOI] [PubMed] [Google Scholar]
- Stewart S.A. Weinberg R.A. Telomeres: Cancer to human aging. Annu. Rev. Cell Dev. Biol. 2006;22:531–557. doi: 10.1146/annurev.cellbio.22.010305.104518. [DOI] [PubMed] [Google Scholar]
- Subramanian T. Vijayalingam S. Chinnadurai G. Genetic identification of adenovirus type 5 genes that influence viral spread. J. Virol. 2006;80:2000–2012. doi: 10.1128/JVI.80.4.2000-2012.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. Sabbatini P. Debbas M. Wold W.S. Kusher D.I. Gooding L.R. The 19-kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor α. Mol. Cell. Biol. 1992;12:2570–2580. doi: 10.1128/mcb.12.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth T. Kuhnel F. Kubicka S. Telomerase-dependent gene therapy. Curr. Mol. Med. 2005;5:243–251. doi: 10.2174/1566524053586536. [DOI] [PubMed] [Google Scholar]
- Won J. Yim J. Kim T.K. Sp1 and Sp3 recruit his-tone deacetylase to repress transcription of human telomerase reverse transcriptase (hTERT) promoter in normal human somatic cells. J. Biol. Chem. 2002;277:38230–38238. doi: 10.1074/jbc.M206064200. [DOI] [PubMed] [Google Scholar]
- Wood M. Perrotte P. Onishi E. Harper M.E. Dinney C. Pagliaro L. Wilson D.R. Biodistribution of an adenoviral vector carrying the luciferase reporter gene following intravesical or intravenous administration to a mouse. Cancer Gene Ther. 1999;6:367–372. doi: 10.1038/sj.cgt.7700090. [DOI] [PubMed] [Google Scholar]
- Zhou Z. Zhou R.R. Guan H. Bucana C.D. Kleinerman E.S. E1A gene therapy inhibits angiogenesis in a Ewing's sarcoma animal model. Mol. Cancer Ther. 2003;2:1313–1319. [PubMed] [Google Scholar]