

Abstract
Transition paths are a uniquely single-molecule property not yet observed for any molecular process in solution. The duration of transition paths is the tiny fraction of the time in an equilibrium single-molecule trajectory when the process actually happens. Here, we report the determination of an upper bound for the transition path time for protein folding from photon-by-photon trajectories. FRET trajectories were measured on single molecules of the dye-labeled, 56-residue 2-state protein GB1, immobilized on a glass surface via a biotin-streptavidin-biotin linkage. Characterization of individual emitted photons by their wavelength, polarization, and absolute and relative time of arrival after picosecond excitation allowed the determination of distributions of FRET efficiencies, donor and acceptor lifetimes, steady state polarizations, and waiting times in the folded and unfolded states. Comparison with the results for freely diffusing molecules showed that immobilization has no detectable effect on the structure or dynamics of the unfolded protein and only a small effect on the folding/unfolding kinetics. Analysis of the photon-by-photon trajectories yields a transition path time <200 μs, >10,000 times shorter than the mean waiting time in the unfolded state (the inverse of the folding rate coefficient). Szabo's theory for diffusive transition paths shows that this upper bound for the transition path time is consistent with previous estimates of the Kramers preexponential factor for the rate coefficient, and predicts that the transition path time is remarkably insensitive to the folding rate, with only a 2-fold difference for rate coefficients that differ by 105-fold.
Keywords: Alexa 488, fluorescence, FRET, maximum likelihood function, protein GB1
A detailed description and understanding of mechanisms of protein folding has been one of the great challenges to biophysical science. The simplest system to study, and the one that has produced the most insights, is a protein exhibiting 2-state behavior (1–7). A 2-state protein has only 2-populations of molecules in equilibrium and at all times in kinetic experiments—folded and unfolded. In ensemble folding experiments kinetics are studied by rapidly changing solution conditions, e.g., the temperature or denaturant concentration, and monitoring the relaxation of the 2 populations to their new equilibrium ratio with probes such as fluorescence, circular dichroism or infrared spectroscopy. Single molecule kinetics, however, can be studied at equilibrium. As can be seen from the schematic of a trajectory in Fig. 1, the dynamical nature of equilibrium is dramatically demonstrated when observing Förster resonance energy transfer (FRET) in a single-molecule fluorescence experiment. There are fluctuations due to shot noise about a mean value in each state, interrupted by what appear to be instantaneous jumps in FRET efficiency signaling folding or unfolding. The residence or waiting times in each state are exponentially distributed, with the mean time in the unfolded and folded segments of the trajectories corresponding to the inverse of the folding and unfolding rate coefficients, respectively.
Fig. 1.

Schematics of a transition path (TP) (A) and an equilibrium single-molecule FRET efficiency (E) trajectory (B). The transition path time is the duration of a successful folding trajectory that passes x0 and reaches x1 for the first time, without visiting values of x < x0 (8). Compared with the waiting times in each state, the transition path (TP) appears as an instantaneous jump in the FRET efficiency.
Rate coefficients can, albeit with assumptions, be much more easily obtained from a combination of ensemble kinetic and equilibrium experiments, where the former measure the sum of the rate coefficients and the latter their ratio. The unique information in a single-molecule experiment is contained in the very rapid transitions between the 2 states when the protein is either folding or unfolding. Indeed, all mechanistic information about folding and unfolding is contained in these so-called transition paths (Fig. 1), which can only be observed for single molecules. The duration of the transition path is the tiny fraction of the time in a trajectory that it takes for a protein to fold or unfold when it actually happens (8). With the possible exception of one study of RNA folding (9), transition path times have not been measured for any molecular process in solution.
A realistic goal for single-molecule FRET experiments is to measure transition path times for protein folding and unfolding and, ultimately, to obtain distance versus time trajectories during the transition paths. The distribution of transition path times and of distance versus time trajectories will be totally new kinds of demanding tests for atomistic molecular dynamics simulations of folding (10), which, if accurate, contain everything one would ever want to know about a protein folding mechanism. If more than one distance could be measured simultaneously, e.g., by using 3 or more dyes (11–13), model-independent information on the width of the microscopic pathway distribution could be derived from correlations among the distances (14). In this work we take a major step toward these important goals by determining an upper bound for the transition path time from single-molecule FRET trajectories of the 56 residue 2-state protein GB1, immobilized on a glass surface by a biotin-streptavidin-biotin linkage (Fig. 2).
Fig. 2.

Immobilization of dye labeled proteins via biotin (protein)-streptavidin-biotin (surface) linkage to PEG-coated glass coverslip. Donor (Alexa Fluor 488) and acceptor (Alexa Fluor 594) dyes are labeled at the cysteine residues at positions 10 and 57 (Avi-GB1K10C/C57). Spacer and AviTag sequences with a biotin molecule were added at the N terminus of the protein.
Although the idea that much could be learned about protein folding mechanisms from such trajectories has been apparent since the very early days of single-molecule spectroscopy, an indication of the difficulty in measuring reliable trajectories is evidenced by the fact that there have been only 3 additional studies since the first measurements on single-immobilized proteins by Hochstrasser and coworkers almost 10 years ago (15–18). The practical problem has been to immobilize the protein and measure long FRET trajectories of the protein folding and unfolding, without spurious effects from dye photophysics or from the immobilization method, until one of the dyes “bleaches,” i.e., ceases to emit photons because of an irreversible photochemical change. To overcome this hurdle we have characterized individual emitted photons by their wavelength, polarization, and absolute and relative time of arrival after picosecond excitation. These experiments allowed us to determine distributions of FRET efficiencies, donor and acceptor lifetimes, steady state polarizations, and waiting times in the folded and unfolded states. Acquisition of single-molecule spectra, in addition to just classifying photons as being emitted from the donor or acceptor based on their “color” (i.e., as determined by dichroic mirrors and filters), enabled a clear distinction between jumps in the FRET efficiency due to folding or unfolding transitions of the polypeptide and those corresponding to a previously unknown photophysical change of the commonly used donor dye, Alexa Fluor 488. As a result, no trajectories were discarded and we could understand in detail >95% of the several thousand recorded folding/unfolding trajectories. Comparison with the results for freely diffusing molecules showed that immobilization has no detectable effect on the structure or dynamics of the unfolded protein and only a small effect on the folding/unfolding kinetics.
By analyzing photon-by-photon trajectories free of any spurious effects from dye photophysics, in which the arrival time of a photon is known to 100 ns accuracy, we can place an upper bound on the transition path time for each folding or unfolding transition. This upper bound is discussed in terms of an analytic expression of Szabo (Eq. 3) for the transition path time in a simple barrier-crossing model. It makes the interesting prediction that the transition path time is remarkably insensitive to free energy barrier heights, scaling as the logarithm of the barrier height, in contrast to the overall folding or unfolding time that depends on the exponential of the barrier height.
Results
FRET Trajectories and Efficiency Distributions.
Fig. 3 shows single-molecule fluorescence trajectories at 3 different urea concentrations, which are representative of trajectories that contain one or more transitions between folded and unfolded states and no spurious photophysics. They were measured at sufficiently low excitation intensity that data could be collected for an average duration of 10 seconds before either the donor or acceptor bleached. In each of the 3 trajectories only 2 levels of the FRET efficiency are observed, as expected for a 2-state protein, one close to 1.0 corresponding to the folded state where the dyes are in close proximity, and a second at ≈0.6 corresponding to the unfolded state where the average distance between the dyes is considerably increased [after using lifetime data to correct for differences in quantum yields and detection efficiency of photons from the 2 dyes (19), the true FRET efficiency for the unfolded state is closer to 0.35 (see Table S1)]. The emission from the green-fluorescing donor and red-fluorescing acceptor do not show the expected anti-correlation because both donor and acceptor fluorescence are partially quenched in the folded state (Fig. S2), with the result that there is not a large increase in the acceptor emission when the protein folds. The quenching arises from a transient interaction between the dyes, as demonstrated by the lack of quenching when the protein is singly labeled with either a donor or acceptor dye and the finding that the fluorescent lifetime of the acceptor in the folded molecule is ≈2/3 that of the unfolded molecule in a free diffusion experiment (see Fig. 5B). As we shall see in the following, this partial quenching in the folded protein introduces only a minor complication in the analysis of the data.
Fig. 3.

Representative FRET trajectories. FRET trajectories in Right were calculated from the donor and acceptor trajectories in Left. The photons were collected into 10-ms bins at 3 M urea and 20-ms bins at other urea concentrations.
Fig. 5.

Photophysics of dyes. (A) Distribution of FRET efficiency differences (ΔEU) between 2 unfolded states separated by a folded state in the same trajectory. Blue dashed line is the distribution calculated from a Gaussian fit to the unfolded peak in Fig. 4 at 6 M urea (see Fig. S4 for data at other urea concentrations), and the red dashed line is the maximum width because of shot noise for nA + nD > 1000 photons. The narrower width of the red distribution compared with the blue distributions suggests some heterogeneity in either the surface or the streptavidin. (B) Lifetime distribution of the acceptor in the folded and unfolded states. For trajectories showing multiple transitions, photons of all folded (unfolded) states in the same trajectory were added up to calculate the lifetimes. Trajectories containing >6,000 photons (donor + acceptor) were considered. Vertical dashed line indicates the lifetime in the unfolded state obtained from the free diffusion experiment. (C) Trajectories in Upper demonstrate an abrupt decrease and increase in the signals in donor and acceptor channels, respectively, for Alexa Fluor 488 labeled on Avi-GB1C57 because of the formation of a red-shifted donor emission spectrum. We call the form of the dye with the red shifted spectrum Alexa Fluor 488R. The lifetime and the fraction of donor photons detected in the acceptor channel (the donor “leak”) were calculated for the individual segments of the trajectories consisting of >3,000 photons. Dashed lines in the histogram plots are the lifetime and the leak obtained from the free diffusion experiment. Low- and high-leak states corresponding to Alexa Fluor 488 and Alexa Fluor 488R emission, respectively, are indicated with a red rectangular box and a green ellipse. (D) Ensemble fluorescence (solid) and absorption (dashed) spectra of dyes labeled on Avi-GB1C57 and transmission windows of the filter set (shaded area). λEx = 470 nm is indicated with an arrow. (E) Complex trajectory resulting from change of donor emission spectrum. Each spectrum and fluorescence decay were obtained from different segments in a single-trajectory: folded state (1), unfolded state and Alexa Fluor 488 (2), unfolded state and Alexa Fluor 488R (3), and acceptor bleached state and Alexa Fluor 488R (4). The vertical dashed lines indicate the peak positions of the spectra in F–H. (F–H) Donor and acceptor fluorescence spectra averaged over segments distinguished by the fraction of donor photons detected in the acceptor channel. (F) Alexa Fluor 488 labeled on Avi-GB1C57 (0 M urea). (G) Alexa Fluor 488 in the donor-only (inactive acceptor) state of Avi-GB1K10C/C57 (5 M urea). (H) Alexa Fluor 488 and Alexa Fluor 594 labeled on Avi-GB1K10C/C57 for the unfolded state. Spectra are scaled to the height of the acceptor spectra for an emphasis on the insensitivity of the acceptor spectrum to the protein and the donor. (I) Blinking kinetics of dyes. (J) Anisotropy of Alexa Fluor 488 labeled on Avi-GB1K10C/C57 either in donor-only (inactive acceptor) or in the unfolded states.
Comparing the mean values and widths of the FRET efficiency distributions with those of freely diffusing molecules is the first test for determining spurious effects from immobilization. Fig. 4 shows that the mean values for the unfolded molecules are virtually identical in the 2 types of experiments for the unfolded state. The width of the FRET efficiency distribution for the freely diffusing molecules is only slightly larger than what is expected from shot noise for individual bursts containing 30 or more detected photons. This small excess width most probably arises from small differences in the FRET efficiency for molecules with donor at position 10 and acceptor at position 57 and those with the reversed positions (Fig. 2), presumably because of small differences in the acceptor lifetime (see Fig. 5B, below). A detailed comparison cannot be made for folded molecules, because the partial quenching produces bursts with too few photons in the free diffusion experiment.
Fig. 4.

FRET efficiency distributions. FRET efficiency distributions of immobilized proteins were obtained from the efficiencies of the initial segments of the trajectories. The ranges for folded (red), unfolded (yellow), and donor-only (green) states are 0.85 ≤ E, 0.4 ≤ E < 0.85, and E < 0.4, respectively. The equilibrium constant is equal to the ratio of the fractions belonging to the folded and unfolded states. (K = fF/fU) The Gaussian distribution in the unfolded peak (red dashed lines) was estimated from the distribution of the acceptor lifetime (τA) in Fig. 5B and does not include the small (<10%) contribution to the width from the shot noise (see Fig. 5A). Orange dashed lines are the mean FRET efficiencies of the unfolded state measured by free diffusion experiment (Inset). Yellow dashed line in the distribution from free diffusion experiment is the upper bound of the width because of shot noise (σ = [E(1 − E)/(nA + nD)]1/2, nA + nD = 30).
For the immobilized molecules, the FRET efficiency distribution is more complex, but also illuminating (Fig. 4). Shot noise accounts for less than half the width, because the FRET efficiency was calculated from segments of the trajectory in the folded or unfolded states that were sufficiently long (≥1 s) to result in >1,000 detected photons. The excess width can, however, be quantitatively explained by small variations in the lifetime of acceptor molecules (Fig. 5B). For the freely diffusing molecule, the lifetime is 5 ns at 5 M urea for unfolded molecules, whereas for the immobilized molecule it varies between ≈4 and 5 ns. Assuming that the radiative lifetime (1/krad) is unaffected as suggested by the constant spectra in Fig. 5H, the FRET efficiency distribution, directly obtained from the acceptor lifetime (τA) distribution via the acceptor quantum yield (QY = kradτA) distribution, superimposes on the observed FRET efficiency distribution (variation in the donor quantum yield (Fig. 5C) has no effect on the observed FRET efficiency; see SI Text, Eq. S5).
In addition to the distributions for folded and unfolded immobilized molecules, there are 2 much smaller populations with FRET efficiencies centered at ≈0.25 and ≈0.8. Trajectories of molecules labeled with donor only, together with measurements of emission spectra and lifetimes, showed that these peaks result from a previously unknown photochemical change of the donor dye, Alexa Fluor 488, that results in a large red shift (≈25 nm) in its emission spectrum and a larger percentage increase in lifetime, as expected from τ ∼ λ3 (20) (Fig. 5 C and F). We call this form of the dye Alexa Fluor 488R. No spectral changes were observed for the acceptor dye. As a consequence of the red shift (Fig. 5F), there is an increase in the FRET efficiency from the increase in Förster radius R0 because of the increased donor-acceptor spectral overlap, and a further increase in the measured FRET efficiency because a larger fraction of donor photons are not blocked by the long pass filter at 600 nm and are detected in the acceptor channel (Fig. 5 D and F). Trajectories of molecules labeled with donor only show segments with apparent FRET efficiencies of ≈0.25 because of leakage of donor photons into the acceptor channel (Fig. 5C).
The peak at ≈0.8, then, corresponds to unfolded molecules with a red-shifted donor spectrum and the peak at ≈0.25 to molecules with both a red-shifted donor spectrum and either a bleached acceptor or an acceptor in a transient nonemitting state known as “blinking” (Fig. 5I). An example of a complex trajectory (Fig. 5E), which was initially quite puzzling, is now fully explained because of emission spectra measured for each segment. In the first segment of the trajectory the molecule is folded; in the second it is unfolded; in the third it is unfolded with the donor altered to Alexa Fluor 488R; in the fourth the acceptor is bleached and the donor remains Alexa Fluor 488R; finally the donor dye bleaches.
The important result from these experiments is that immobilization does not appear to affect the structure distribution in the unfolded state, as measured by the mean value of the FRET efficiency, which for a Gaussian chain is completely determined by the mean squared distance between the dyes attached to cysteines separated by 46 residues in the sequence. For comparison with other proteins, the mean-squared distance can be calculated from the mean efficiency, which can be accurately determined from the lifetime measurements in the free diffusion experiment (Table S1) (19, 21).
Trajectories and Transition Statistics.
Having discovered important photophysical properties of the dyes, it is now possible to interpret almost all of the observed trajectories in terms of folding and unfolding, a red-shifted donor spectrum, and blinking, unblinking, and bleaching of either donor or acceptor dyes. Table S2 summarizes the results for all 2,003 measured trajectories. Approximately 20% of the trajectories show blinking of either the donor or acceptor (Fig. 5I) and ≈17% of the trajectories contain segments with an Alexa Fluor 488R donor. Trajectories in which the protein remains in either the folded state or unfolded state before bleaching of one of the dyes varies from 30% to 3% and from 5% to 36%, respectively, as the urea concentration increases from 3 M to 7 M. The transition statistics are given in Table S3, where a procedure (see SI Text) similar to that of Nienhaus and coworkers was used to define a transition (18). Only ≈2% correspond to transitions between high FRET efficiency values that would be assigned to folded molecules, which might be caused by quantum yield changes of the acceptor resulting from association or dissociation of a donor-acceptor complex.
A transition map at 6 M urea is given in Fig. 6, which shows the initial and final FRET efficiencies for every transition (see Fig. S3 for transition maps at all concentrations). In the idealized experiment, only 2 clusters of points would appear before bleaching of the acceptor dye, corresponding to the transitions: unfolded → folded and folded → unfolded. Many more clusters appear because of the spectral shift of the donor dye and blinking and unblinking of the acceptor dye. Overall 94% of all observed transitions are interpretable in terms of either these photochemical changes (9%) or unfolded → folded (37%) and folded → unfolded (48%) transitions. Of the 6% un-interpretable transitions, some may arise from the detection of 2 molecules at the same time (see SI Text) and others from sudden changes in κ2 due to sticking or unsticking of the dyes, as revealed by anisotropy measurements (Fig. 5J and Fig. S7). In most cases, the anisotropy is very low (≈0.05) indicating free rotation of the dye, but a small fraction (0.12) of the distribution shows relatively high anisotropy (> 0.15). The FRET efficiency of the unfolded state with the high anisotropy will be different from that of low anisotropy, which results in broadening the FRET efficiency distribution, and the transition between low and high anisotropy states will appear as a transition between unfolded states (see SI Text for further details).
Fig. 6.

Transition map at 6M urea. Transitions are categorized into 3 types according to the FRET efficiencies before and after the transition: between folded and unfolded states (red), within folded or unfolded states (blue), and acceptor bleaching or blinking (green). The ranges of FRET efficiencies for folded (F) and unfolded (U) states are determined from the E histogram in Fig. 4. Ellipses indicate interpretable transitions in terms of Alexa Fluor 488 (red) and red-shifted (light blue) Alexa Fluor 488R fluorescence. The widths of the ellipses are determined by the width of the distributions in Fig. 4. (± 2.6 σ obtained from the Gaussian fits).
Dynamics of Unfolded Molecules.
An important property of the unfolded state for investigating the effect of immobilization is the dynamics of the polypeptide chain. Fig. 7 shows the cross-correlation of the donor and the acceptor photons calculated as
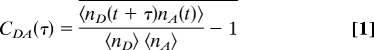 |
where, nD(t) and nA(t) are the number of donor and acceptor photons in a given bin at time t, respectively; 〈… .〉, is an average in a given segment and the upper bar is an average over unfolded state segments >5 s. Despite the noise in the microsecond range due to the small number of photons per bin, the cross correlation is flat compared with curves with decay times of 1, 5, and 20 microseconds with an amplitude of −0.53 as τ → 0, which was estimated from the end-to-end distribution of a Gaussian chain (see SI Text). The very small amplitude for the observed correlation function shows that the dynamics are occurring on a submicrosecond time scale.
Fig. 7.

Donor and acceptor intensity cross correlation averaged over unfolded segments longer than 5 seconds (continuous curves). Dashed curves are simulated decays of cross correlations with correlation times of 1 μs (light blue), 5 μs (green) and 20 μs (red) and the correlation amplitude at τ → 0 (CDA (0) = −0.53) calculated from the E distribution of a Gaussian chain (black).
Kinetics of Folding and Unfolding.
Fig. 8 shows that the waiting times in either the unfolded or folded state are exponentially distributed, as expected for a 2-state system, with the average waiting times corresponding to the reciprocal of the folding and unfolding rate coefficients, respectively. For a 2-state system the ensemble relaxation rate to the new equilibrium following a perturbation of any size is the sum of the folding and unfolding rate coefficients. Fig. 9 compares the ensemble relaxation rates measured in stopped flow experiments with the sum of the rate coefficients from single-molecule trajectories at 4 different urea concentrations [a small correction was made to account for trajectories interrupted by dye bleaching (see SI Text)].
Fig. 8.

Folding and unfolding rates obtained from the exponential fits of the waiting time histograms. Errors were calculated from the fit with 95% confidence level. Rates corrected for the finite length of trajectories are indicated in red.
Fig. 9.

Comparison of single-molecule folding kinetics (blue squares uncorrected with 2σ fitting error, blue crosses corrected) with ensemble kinetics (red circles with 2σ calculated from average of 4–7 experiments) from stopped flow measurement with dye labeled proteins (λEx = 488 nm). (Inset) Fluorescence decay from the stopped flow measurement exhibits a single-exponential decay (red).
Transition Path Times.
At the laser intensities used in the experiments described so far (≈300 W/cm2), the frequency of detected photons from the folded state is between 1 and 5 photons per millisecond, with the FRET efficiency calculated from photons collected for the most part in 20-ms bins. With this detection frequency and binning, the transitions between unfolded and folded states appear to be instantaneous, occurring within one bin; and therefore their duration is <20 ms. To observe transitions with better time resolution, the laser intensity was increased by a factor of 10. The detected photon frequency increased to between 10 and 50 photons per millisecond, but instead of an average of 1.3 folding or unfolding transitions per trajectory, now only 1 in 20 trajectories showed a transition because of the dramatically shortened length of trajectories before donor (58%) or acceptor (42%) bleaching. At the higher excitation laser intensity of ≈3,000 W/cm2 the average duration of a trajectory before bleaching of one of the dyes is only 130 ms compared with 10 s at the lower excitation intensity. Examination of 1151 molecules resulted in 78 transitions between folded and unfolded states (see Tables S2 and S3 and Fig. S3). Fig. 10 shows 4 of these trajectories with high photon detection frequency with 2 different binnings, and photon-by-photon trajectories of arrival times known to 100 ns accuracy.
Fig. 10.

Estimation of the window time within which transition occurs. FRET trajectories with 2-ms binning show instantaneous transitions (first row). Red and green circles are time tagged acceptor and donor photons (second row). The transition interval found by Eq. S17 (of SI Text) is indicated with blue vertical dashed line in the photon strings. Likelihood values are shown in cyan, which are normalized to the maximum. Interval in which transition occurs with 95% confidence level estimated by Eq. 4 are shown as dashed orange lines. Panels in the third row show FRET trajectories near the transition, which are binned by the width of the transition window (orange numbers in row 2 and orange vertical dashed lines). Horizontal dashed lines in the binned trajectories show the range of ± 2σ, where σ is the mean standard deviation in the folded (red) and unfolded (green) parts of the trajectory (σ = [E(1 − E)/(nA + nD)]1/2).
With 2-ms binning, the transitions still are clearly instantaneous, placing a more restrictive upper bound for the transition path time of 2 ms. One way of reducing the detectable upper bound further would be to bin the FRET trajectories in successively smaller and smaller bins, with varying start times, and examine the trajectory to determine the smallest bin time that yields a transition between states in a single bin. Instead, we used a much less cumbersome and more objective method by analyzing the photon-by-photon trajectories, using statistical methods to obtain the minimum bin size. The basic idea of the method, described in Materials and Methods and SI Text in detail, is that it is highly improbable for a green photon to be emitted by a molecule in the folded state, because the average FRET efficiency is 0.97. Similarly, it is highly unlikely to observe a long string of red photons in the unfolded state where the average FRET efficiency is 0.6. For example, if 6 consecutive red photons are detected, the probability of being in the unfolded state is 0.66/(0.66 + 0.976) = 0.05 (Eq. 4). A second factor that must be considered is that quenching in the folded state reduces the frequency of detected photons in the folded state by ≈2-fold. To divide the trajectory into folded and unfolded segments, we first assumed that the transition is instantaneous, and used a maximum likelihood method (22) to determine the photon interval at which the transition occurs. Simulations show that the accuracy of determining the correct interval is 50% and is 81% for determining the correct interval to within 2 photons (see SI Text and Fig. S9). We then constructed a time window surrounding this transition point in which it is known with 95% confidence that the string of photons outside this window correspond to the FRET efficiency of either the folded or unfolded state. From this analysis we conclude that the transition must have occurred within this window time. This conclusion is supported by observing that transitions appear instantaneous when the bin size for the photons is the window time (Fig. 10).
The analysis of all 46 transitions for photon detection rates of >10/ms for the folded state is given in Fig. S10, whereas Fig. 11 presents a summary of these transitions as plots of window time versus reciprocal of the average photon frequency within the window on the folded side of the assumed instantaneous transition point. There is a significant correlation in these plots, indicating that the apparent upper bound for the transition path time depends on the photon frequency. We therefore take the window times determined with the highest photon frequency as the upper bound for the transition path time ≈200 μs.
Fig. 11.

Time window (τwindow) within which unfolded → folded (squares) or folded → unfolded (circles) transitions occur as a function of the inverse of the photon counting rate on the folded side of the transition point within the time window (νF). τwindow is the time interval defined by the dashed orange lines in Fig. 10 and Fig. S10) estimated using Eq. 4 with a 95% confidence level in the low (green, light green) and high (red, orange) excitation intensity. For comparison, the same method was applied to the photon string presented in figure 2c of ref. 17 after digitizing the data, which results in τwindow = 2.2 ms (purple x).
Discussion
As pointed out in the Introduction, all of the mechanistic information on how a protein folds or unfolds is contained in the transition paths (Fig. 1)—the rapid transitions between the 2 states that appear instantaneous compared with the waiting times in each state (Figs. 3 and 10). To proceed to investigate the properties of transition paths, our initial objective in this study has been to measure trajectories of single-molecules folding and unfolding, without interference from spurious effects caused by dye photophysics or by linking the protein to a surface. We chose the 56 residue α/β protein GB1 (Fig. 2), because it is a 2-state protein for which kinetic studies have been carried out on a large number of site-directed mutants (23, 24). From the initial measurements it was clear that a significant fraction of the trajectories would not show the idealized trajectory of the FRET efficiency shown in Fig. 1. To fully understand the origin of possible artifacts we decided not to discard any trajectory, but to try to understand every trajectory by determining the wavelength, polarization, and relative and absolute time of arrival of photons at the detector. The net result of our analysis (Tables S2 and S3) is that we understand 95% of the trajectories in detail.
Approximately 60% of the trajectories measured at the lower of the 2 illumination intensities used show the expected simple 2-state behavior (Fig. 3), with only 2 values of the FRET efficiency, one near 1.0 and another near 0.6 (Table S2). The remaining 40% of the trajectories showed blinking or at least one transition in which either the initial or final value of the FRET efficiency differed considerably from 1.0 or 0.6. By measuring lifetimes and spectra, and not just classifying photons as being emitted by the donor or acceptor, we discovered that almost all of the remaining 40% of trajectories could be explained in terms of dye photophysics—transient periods in which the dyes do not emit photons (“blinking”) and a previously unknown form of the commonly used donor dye, Alexa Fluor 488, with an emission spectrum shifted ≈25 nm to longer wavelength (Alexa Fluor 488R).
To determine the extent of any effect on the properties of the protein resulting from the biotin-streptavidin-biotin linkage to the glass surface, we compared the results for the immobilized protein with those measured for the freely diffusing molecules. The mean FRET efficiency for the immobilized and freely diffusing molecules is virtually identical, showing that the root mean-squared distance between the dyes (Table S1) in the unfolded state is the same. This distance increases with increasing denaturant concentration as observed for several other proteins (19, 25–29).
To ascertain whether immobilization influences the dynamics of the unfolded molecules, as judged by the fluctuations in the interdye distance, the donor-acceptor cross-correlation function was calculated from 1 μs to 1 s from the trajectories using Eq. 1. There is no significant amplitude, indicating that there are no dynamics in this time regime, as also observed for the unfolded cold shock protein, CspTm, for which the correlation time was found to be ≈50 nanoseconds (30, 31). Submicrosecond dynamics are also expected from several other types of studies on end-to-end diffusion constants for freely diffusing unstructured polypeptides (32–34). Thus, our finding of submicrosecond dynamics indicates that the interactions of the unfolded protein with the surface and linker are weak.
The most important comparison to be made in assessing the effect of immobilization is between the folding/unfolding kinetics for freely diffusing and immobilized molecules. In single-molecule experiments, the reciprocal of the average waiting time in the unfolded and folded states corresponds to the folding and unfolding rate coefficients, respectively. For a perfect 2-state system the waiting times should be exponentially distributed, as we observe (Fig. 8). Because the observation time in the single-molecule free diffusion experiment is only ≈1 ms, we measured the kinetics for freely diffusing, dye-labeled molecules in ensemble experiments in which the denaturant concentration was rapidly changed in a stopped flow instrument. If the kinetics are unaffected, the sum of the rate coefficients from the single-molecule trajectories should be identical to the relaxation rate measured in the stopped flow experiment. The maximum difference between the 2 rates is less than a factor of 2, indicating that there is some, albeit small, effect of immobilization.
Having achieved our initial goal of being able to objectively select trajectories free of spurious dye photophysics, we then proceeded to determine what we could learn about transition path times. To improve the time resolution in the experiment, the illumination laser intensity was increased by 10-fold, with the result that the average duration of a trajectory before one of the dyes bleached was reduced from 10 s to 130 ms. Using 2-ms instead of 20-ms bins for calculating trajectories of the FRET efficiency, the transitions still appeared instantaneous (Fig. 10). To further improve the time resolution we took advantage of our measurement of the absolute arrival time of individual photons at the detector, which is known with an accuracy of 100 ns, and analyzed these photon-by-photon trajectories with statistical methods (Fig. 10). We determined with 95% confidence the time window around the photon interval that divides the trajectory into folded and unfolded segments, where this interval was obtained from a maximum likelihood analysis (see SI Text). Because the transition occurs at some point within this time window, the window time represents an upper bound for the apparent transition path time. This conclusion is supported by the observation that transitions still appear to be instantaneous in FRET efficiency trajectories using the window time from the photon trajectory as the bin size (Fig. 10 and Fig. S10). The apparent upper bound for the transition time correlates with the inverse of the photon detection frequency, suggesting that the true upper bound for the transition path time is the calculated window time at the highest photon frequencies, and is therefore ≈200 μs (Fig. 11). If either the unfolded or folded states have FRET efficiencies indistinguishable from the transition state, then the upper bound is approximately twice as long, and might be longer if there is slow annealing of side chains as suggested by the Monte Carlo simulations of Shakhnovich and coworkers (35).
Haran and coworkers (17) performed the only previous study of immobilized single-molecule folding/unfolding trajectories for a 2-state protein, CspTm, which has been extensively studied in free diffusion experiments (19, 29–31, 36, 37) (see Schuler and Eaton (38) for a discussion of the more complex folding/unfolding trajectories of non-two state proteins measured by Rhoades et al. and Kuzmenkina et al. (17, 18)). To isolate and immobilize the protein they encapsulated CspTm in lipid vesicles, and linked the vesicles to a glass surface. They reported an upper bound of 100–200 μs for the transition path time, and showed an example of a photon trajectory from which this upper bound was calculated. However, the average interval between detected photons was 500 μs, which the data in Fig. 11 and our analysis method indicate is closer to an upper bound of 2 ms. It appears that the upper bound for the transition path time was obtained by using a maximum likelihood method to determine the most likely interval between photons at which an folding or unfolding transition occurs, assuming that the transition is instantaneous, and simply assigning the upper bound as the length of this interval.*
Having determined an upper bound for the transition path time for protein GB1, it would be important to know how much must the time resolution in our experiment be increased to resolve a transition path and, eventually, to measure distance versus time trajectories during the transition path. Although it has been appreciated for quite some time that transition path times for barrier crossing processes are much shorter than the times given by the inverse of the rate coefficients (39), 2 important questions have remained: exactly how much shorter and how do transition path times depend on barrier heights? Our present experiments indicate that the transition path time (< 200 μs) is at least 10,000-fold shorter than the folding time (2 s).
A theory of diffusive barrier crossings can be used to provide potential answers to these questions and guide future experiments. For transition paths that begin at x0 on the unfolded side of a free energy barrier and end at x1 on the folded side, without crossing x0, the mean time for transit from x0 to x1 (Fig. 1), the average transition path time 〈tTP〉 can be evaluated from (8):
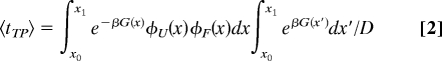 |
where, G(x) is the free energy as a function of the reaction coordinate x, β = 1/kBT, φU and φF are the fractions of trajectories starting from x that reach U(< x0) and F (> x1) first, respectively (i.e., splitting probabilities), and D is the diffusion coefficient assumed to be independent of the reaction coordinate. The value of 〈tTP〉 is the same for the reverse transition path, i.e., x1 to x0. For an harmonic barrier of height ΔG‡ >2 kBT, Eq. 2. yields (Szabo, personal communication):
where γ = 0.577 … is Euler's constant and (ω‡)2 is the curvature of the barrier. Assuming equal curvatures of the barrier and the reactant well, (ω)2, 〈tTP〉, can be calculated from the Kramers preexponential factor, k0 = Dωω‡β/2π, and the rate coefficient, k = k0exp(−βΔG‡). Eq. 3. can be used to estimate a transition path time of 0.6 to 6 μs from previous estimates for k0 of 105 to 106 s−1 (40) and our measured rate coefficients of ≈0.5 s−1 at the midpoint denaturant concentration (Fig. 8). Consequently, it should be possible in future experiments to time-resolve the transition path by a combination of increased viscosity to slow diffusion across the free energy barrier and higher excitation laser intensity to increase the signal/noise in the FRET trajectories by increasing the photon detection frequency. This is, however, a nontrivial task because of the nonlinear relation between the laser illumination intensity and length of trajectories before dye bleaching, so that both alterations in the experiment will decrease the fraction of trajectories (currently ≈0.05) that contain folding or unfolding transitions. With higher photon count rates, moreover, it would also be possible to determine whether or not there are any transitions involving short-lived intermediates, as suggested by the atomistic Monte Carlo simulations of protein GB1 by Shimada and Shakhnovich (41). The simulations also predict 3 distinct, 3-state pathways, which is just the kind of issue that can only be investigated with single-molecule methods.
The interesting prediction of Eq. 3 is the remarkable insensitivity of the transition path time to the free energy barrier height. Using the preexponential factor of 106 s−1, the transition path time for a protein that folds in 2 s, such as protein GB1 studied here, is only 2-fold longer than a protein that folds 105 times faster. An important assumption in Eq. 3, however, is that the preexponential factor, k0 be the same for slow- and fast-folding proteins, which requires that the free energy barrier region not contain significant minima (41), and that the curvatures and the diffusion coefficient, which measures the roughness of the underlying energy landscape, also be approximately the same. It will be very interesting, therefore, to not only resolve transition path times in future experiments, but also to compare these times for proteins that fold with widely different rates.
Materials and Methods
Materials.
Because the N and C termini are on opposite sides of the protein GB1 (PDB 3GB1) molecule (Fig. 2), the difference in the FRET efficiency for the folded and unfolded protein was maximized by substituting a lysine at position 10 with cysteine and adding a cysteine at the C terminus. Details of the protein engineering, purification, and dye attachment to these 2 cysteines are given in SI Text. The midpoint unfolding denaturation concentrations of the unlabeled and dye-labeled protein are the same to within 0.5 M urea (see Figs. 4 and 9 and Fig. S2).
Single-Molecule Instrument.
Single molecule measurements were performed using a Picoquant Microtime 200 confocal fluorescence microscope (Berlin, Germany). A 470-nm pulsed diode laser (20-MHz repetition rate, 80 ps FWHM, 0.9 μW or 9 μW average power) was used to excite the donor chromophore, and donor and acceptor fluorescence were detected by single-photon avalanche diodes (SPAD). A TimeHarp200 card was used to record the detection channel (donor, acceptor), the absolute arrival time (100-ns resolution with ≈350 ps jitter) and the time interval between excitation and detection (37-ps resolution) of each photon. Single molecule spectra were measured with a spectrograph (Shamrock 303i, Andor Technology) and CCD (DV887, Andor Technology).
Method for Estimating an Apparent Upper Bound for Transition Path Times.
The first step in estimating an upper bound for the transition path time was to locate the interval between photons, which divides the trajectory into folded and unfolded segments, assuming that the transition is instantaneous. This could be done by visual inspection, because it corresponds to the point at which there is a clear change in the donor emission rate. A more objective and automated procedure was used to determine this interval from the maximum point of a likelihood function, which is proportional to the probability that an instantaneous transition occurs at a given photon interval (see SI Text). An upper bound for the transition path time of each transition was then estimated by finding the narrowest time window surrounding the most probable point for an instantaneous transition point, in which the probability, with a confidence level greater than some value α, for the strings of photons before and after this transition point to be in either the folded or unfolded states. A conditional probability for n (> 1) photons before or after the transition point to belong to the folded or the unfolded states is
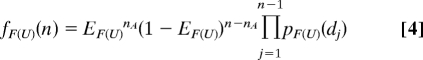 |
where fF(U)(n) is the probability of n photons belonging to the folded (unfolded) molecule, nA is the number of acceptor photons, EF and EU are the FRET efficiencies of folded and unfolded states, which are same as the probabilities for a given photon to be red in the folded and unfolded states, respectively, pF(U)(d) (= νF(U)exp(−νF(U)d)) is the distribution of time intervals d between 2 photons detected in the folded (unfolded) state. dj is the time interval between photon j and j + 1, and νF and νU are the photon counting rates in the folded and unfolded states. Then, the smallest ns satisfying PF(U) > α1/2 are found for photon strings before and after the transition point, where α1/2 is 0.975 for a confidence level of 0.95. The length of the combined photon string is the apparent upper bound of the transition path time.
Supplementary Material
Acknowledgments.
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health. We thank Attila Szabo and Irina Gopich for many helpful discussions and for sharing their unpublished theoretical results, and Annie Aniana for technical assistance with protein preparation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 11823.
This article contains supporting information online at www.pnas.org/cgi/content/full/0901178106/DCSupplemental.
We are aware of only one other study, Lee et al. (9), that addresses the problem of measuring transition path times for a barrier crossing process. Lee et al. (9) measured folding/unfolding FRET trajectories with an average interphoton interval of 200 μs for single-RNA molecules. They carried out an extensive statistical analysis of the acceptor-donor cross-correlation function for segments of the trajectory containing a folding or unfolding transition using a theoretical correlation function based on a simple model, and extracted a transition path time of 240 μs (with a 50% error) for folding. However, the difference in the intercept of the correlation function, from which this time was calculated, and the intercept in their model equations for a transition too fast to resolve is only 3% compared with our calculation of 2σ for the intercept of 10%. Furthermore, the authors report that the unfolding transition path time could not be resolved, without any suggestion for why folding and unfolding transition path times might differ.
References
- 1.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein-folding—a synthesis. Proteins-Struct Funct Gen. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 2.Dobson CM, Sali A, Karplus M. Protein folding: A perspective from theory and experiment. Angew Chem Int Ed. 1998;37:868–893. doi: 10.1002/(SICI)1521-3773(19980420)37:7<868::AID-ANIE868>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 3.Thirumalai D, Klimov DK, Dima RI. Insights into specific problems in protein folding using simple concepts. Adv Chem Phys. 2002;120:35–76. [Google Scholar]
- 4.Fersht AR, Daggett V. Protein folding and unfolding at atomic resolution. Cell. 2002;108:573–582. doi: 10.1016/s0092-8674(02)00620-7. [DOI] [PubMed] [Google Scholar]
- 5.Oliveberg M, Wolynes PG. The experimental survey of protein-folding energy landscapes. Quart Rev Biophys. 2005;38:245–288. doi: 10.1017/S0033583506004185. [DOI] [PubMed] [Google Scholar]
- 6.Shakhnovich E. Protein folding thermodynamics and dynamics: Where physics, chemistry, and biology meet. Chem Rev. 2006;106:1559–1588. doi: 10.1021/cr040425u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dill KA, Ozkan SB, Shell MS, Weikl TR. The protein folding problem. Ann Rev Biophys Biomolec Struct. 2008;37:289–316. doi: 10.1146/annurev.biophys.37.092707.153558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hummer G. From transition paths to transition states and rate coefficients. J Chem Phys. 2004;120:516–523. doi: 10.1063/1.1630572. [DOI] [PubMed] [Google Scholar]
- 9.Lee T-H, et al. Measuring the folding transition time of single RNA molecules. Biophys J. 2007;92:3275–3283. doi: 10.1529/biophysj.106.094623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snow CD, Sorin EJ, Rhee YM, Pande VS. How well can simulation predict protein folding kinetics and thermodynamics? Ann Rev Biophys Biomol Struct. 2005;34:43–69. doi: 10.1146/annurev.biophys.34.040204.144447. [DOI] [PubMed] [Google Scholar]
- 11.Clamme JP, Deniz AA. Three-color single-molecule fluorescence resonance energy transfer. Chemphyschem. 2005;6:74–77. doi: 10.1002/cphc.200400261. [DOI] [PubMed] [Google Scholar]
- 12.Hohng S, Joo C, Ha T. Single-molecule three-color FRET. Biophys J. 2004;87:1328–1337. doi: 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee NK, et al. Three-color alternating-laser excitation of single molecules: Monitoring multiple interactions and distances. Biophys J. 2007;92:303–312. doi: 10.1529/biophysj.106.093211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Onuchic JN, Wang J, Wolynes PG. Analyzing single molecule trajectories on complex energy landscapes using replica correlation functions. Chem Phys. 1999;247:175–184. [Google Scholar]
- 15.Talaga DS, et al. Dynamics and folding of single two-stranded coiled-coil peptides studied by fluorescent energy transfer confocal microscopy. Proc Natl Acad Sci USA. 2000;97:13021–13026. doi: 10.1073/pnas.97.24.13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhoades E, Gussakovsky E, Haran G. Watching proteins fold one molecule at a time. Proc Natl Acad Sci USA. 2003;100:3197–3202. doi: 10.1073/pnas.2628068100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhoades E, Cohen M, Schuler B, Haran G. Two-state folding observed in individual protein molecules. J Am Chem Soc. 2004;126:14686–14687. doi: 10.1021/ja046209k. [DOI] [PubMed] [Google Scholar]
- 18.Kuzmenkina EV, Heyes CD, Nienhaus GU. Single-molecule Förster resonance energy transfer study of protein dynamics under denaturing conditions. Proc Natl Acad Sci USA. 2005;102:15471–15476. doi: 10.1073/pnas.0507728102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merchant KA, Best RB, Louis JM, Gopich IV, Eaton WA. Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc Natl Acad Sci USA. 2007;104:1528–1533. doi: 10.1073/pnas.0607097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macklin JJ, Trautman JK, Harris TD, Brus LE. Imaging and time-resolved spectroscopy of single molecules at an interface. Science. 1996;272:255–258. [Google Scholar]
- 21.Best RB, et al. Effect of flexibility and cis residues in single-molecule FRET studies of polyproline. Proc Natl Acad Sci USA. 2007;104:18964–18969. doi: 10.1073/pnas.0709567104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gopich IV, Szabo A. Single-molecule FRET with diffusion and conformational dynamics. J Phys Chem B. 2007;111:12925–12932. doi: 10.1021/jp075255e. [DOI] [PubMed] [Google Scholar]
- 23.McCallister EL, Alm E, Baker D. Critical role of β-hairpin formation in protein G folding. Nat Struct Biol. 2000;7:669–673. doi: 10.1038/77971. [DOI] [PubMed] [Google Scholar]
- 24.Krantz BA, Mayne L, Rumbley J, Englander SW, Sosnick TR. Fast and slow intermediate accumulation and the initial barrier mechanism in protein folding. J Mol Biol. 2002;324:359–371. doi: 10.1016/s0022-2836(02)01029-x. [DOI] [PubMed] [Google Scholar]
- 25.Schuler B, Kremer W, Kalbitzer HR, Jaenicke R. Role of entropy in protein thermostability: Folding kinetics of a hyperthermophilic cold shock protein at high temperatures using F-19 NMR. Biochemistry. 2002;41:11670–11680. doi: 10.1021/bi026293l. [DOI] [PubMed] [Google Scholar]
- 26.Laurence TA, Kong XX, Jager M, Weiss S. Probing structural heterogeneities and fluctuations of nucleic acids and denatured proteins. Proc Natl Acad Sci USA. 2005;102:17348–17353. doi: 10.1073/pnas.0508584102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuzmenkina EV, Heyes CD, Nienhaus GU. Single-molecule FRET study of denaturant induced unfolding of RNase H. J Mol Biol. 2006;357:313–324. doi: 10.1016/j.jmb.2005.12.061. [DOI] [PubMed] [Google Scholar]
- 28.Sherman E, Haran G. Coil-globule transition in the denatured state of a small protein. Proc Natl Acad Sci USA. 2006 doi: 10.1073/pnas.0601395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffmann A, et al. Mapping protein collapse with single-molecule fluorescence and kinetic synchrotron radiation circular dichroism spectroscopy. Proc Natl Acad Sci USA. 2007;104:105–110. doi: 10.1073/pnas.0604353104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nettels D, Gopich IV, Hoffmann A, Schuler B. Ultrafast dynamics of protein collapse from single-molecule photon statistics. Proc Natl Acad Sci USA. 2007;104:2655–2660. doi: 10.1073/pnas.0611093104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nettels D, Hoffmann A, Schuler B. Unfolded protein and peptide dynamics investigated with single-molecule FRET and correlation spectroscopy from picoseconds to seconds. J Phys Chem B. 2008;112:6137–6146. doi: 10.1021/jp076971j. [DOI] [PubMed] [Google Scholar]
- 32.Lapidus LJ, Steinbach PJ, Eaton WA, Szabo A, Hofrichter J. Effects of chain stiffness on the dynamics of loop formation in polypeptides. J Phys Chem B. 2002;106:11628–11640. [Google Scholar]
- 33.Buscaglia M, Lapidus LJ, Eaton WA, Hofrichter J. Effects of denaturants on the dynamics of loop formation in polypeptides. Biophys J. 2006;91:276–288. doi: 10.1529/biophysj.105.071167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moglich A, Joder K, Kiefhaber T. End-to-end distance distributions and intrachain diffusion constants in unfolded polypeptide chains indicate intramolecular hydrogen bond formation. Proc Natl Acad Sci USA. 2006;103:12394–12399. doi: 10.1073/pnas.0604748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kussell E, Shimada J, Shakhnovich EI. Side-chain dynamics and protein folding. Proteins-Struct Funct Gen. 2003;52:303–321. doi: 10.1002/prot.10426. [DOI] [PubMed] [Google Scholar]
- 36.Schuler B, Lipman EA, Eaton W. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature. 2002;419:743–747. doi: 10.1038/nature01060. [DOI] [PubMed] [Google Scholar]
- 37.Lipman EA, Schuler B, Bakajin O, Eaton WA. Single-molecule measurement of protein folding kinetics. Science. 2003;301:1233–1235. doi: 10.1126/science.1085399. [DOI] [PubMed] [Google Scholar]
- 38.Schuler B, Eaton WA. Protein folding studied by single-molecule FRET. Curr Opin Struct Biol. 2008;18:16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bennett CH. In: Algortihms for Chemical Computations. Christoffersen EE, editor. New York: Am Chem Soc; 1976. pp. 63–97. [Google Scholar]
- 40.Kubelka J, Hofrichter J, Eaton WA. The protein folding “speed limit”. Curr Opin Struct Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 41.Shimada J, Shakhnovich EI. The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation. Proc Natl Acad Sci USA. 2002;99:11175–11180. doi: 10.1073/pnas.162268099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.