

Summary
The LKB1 – AMPK signaling pathway serves as a critical cellular sensor coupling energy homeostasis to cell growth, proliferation and survival. However, how tumor cells suppress this signaling pathway to gain growth advantage under conditions of energy stress is largely unknown. Here, we show that AMPK activation is suppressed in melanoma cells with the B-RAF V600E mutation and that down-regulation of B-RAF signaling activates AMPK. We find that in these cells LKB1 is phosphorylated by ERK and Rsk, two kinases downstream of B-RAF, and that this phosphorylation compromises the ability of LKB1 to bind and activate AMPK. Furthermore, expression of a phosphorylation-deficient mutant of LKB1 allows activation of AMPK and inhibits melanoma cell proliferation and anchorage-independent cell growth. Our findings provide a molecular linkage between the LKB1-AMPK and the RAF-MEK-ERK pathways and suggest that suppression of LKB1 function by B-RAF V600E plays an important role in B-RAF V600E-driven tumorigenesis.
Introduction
The RAF-MEK-ERK protein kinase signaling cascade is a central pathway that regulates cell growth, proliferation, differentiation and survival in response to extracellular stimuli (Chong et al., 2003; Wellbrock et al., 2004). Somatic mutations in B-RAF, a member of the RAF kinase family, have been found in ∼6% of human cancer (Davies et al., 2002), with the highest incidence in malignant melanoma (50-70%), papilliary thyroid cancer (∼30%), serous ovarian cancer (∼30%) and colorectal cancer (∼15%) (Dhomen and Marais, 2007; Garnett and Marais, 2004; Tuveson et al., 2003). More recently, germline mutations of B-RAF have also been identified in cardio-facio-cutaneous syndrome (Schubbert et al., 2007). More than 90% of the oncogenic B-RAF mutations (Ikenoue et al., 2003) occur as V600E, which induces constitutively active ERK signaling (Wan et al., 2004). The oncogenic B-RAF V600E mutant has been shown to be important for tumor induction, growth, maintenance and progression, but the detailed molecular mechanisms remain to be elucidated (Dhomen and Marais, 2007; Gray-Schopfer et al., 2005).
The tumor suppressor LKB1 is a serine/threonine protein kinase mutated in autosomal dominantly inherited Peutz-Jeghers syndrome (PJS), a disease characterized by increased risk of benign and malignant tumors in multiple tissues, harmartomatous polyps in the gastrointestinal tract and mucocutaneous pigmentation (for reviews, see (Alessi et al., 2006; Katajisto et al., 2007). Somatic mutations in LKB1 have also been observed frequently in sporadic lung adenocarcinomas (Sanchez-Cespedes et al., 2002), and its inactivation in the mouse promotes development of metastatic lung adenocarcinomas (Ji et al., 2007). Genetic studies have shown that LKB1 modulates cell growth, cell proliferation and cell survival in response to stress. Mouse embryonic fibroblasts lacking LKB1 fail to senescence in culture (Bardeesy et al., 2002) but more readily undergo apoptosis in response to energy stress (Shaw et al., 2004b). In addition, LKB1 has been implicated in the control of epithelial cell polarity based on C. elegans and Drosophila genetics and on mammalian cell culture (Baas et al., 2004; Martin and St Johnston, 2003; Watts et al., 2000).
The recently discovered role for LKB1 in activation of AMP-dependent protein kinase (AMPK) (Hawley et al., 2003; Shaw et al., 2004b; Woods et al., 2003) has begun to explain many of the phenomena associated with loss of LKB1. LKB1 directly phosphorylates AMPK at Thr-172 in the activation loop of this enzyme and accumulation of phosphate at this position in response to elevation of cellular AMP is required for the activation of AMPK in most cellular contexts. The failure of AMPK to be activated in response to energy stress has been invoked to explain the failure of LKB1-/- cells to undergo cell cycle arrest and to suppress protein synthesis and other macromolecular syntheses in response to energy stress conditions, such as those observed in tumor growth (Inoki et al., 2003; Jones et al., 2005; Luo et al., 2005; Motoshima et al., 2006; Shaw et al., 2004a). Of particular interest, the phosphorylation of tuberin and RAPTOR by AMPK has been shown to play a role in suppressing mTOR signaling in response to energy stress (Gwinn et al., 2008; Inoki et al., 2003; Shaw et al., 2004a). A host of AMPK substrates have been identified in recent years and many of these play critical roles in regulating macromolecule synthesis and cellular energy (Carling, 2004; Hardie, 2005; Kahn et al., 2005; Motoshima et al., 2006; Shaw, 2006). It is likely that other targets of LKB1, including the AMPK-related MARK family protein kinases (Lizcano et al., 2004), also contribute to the various defects in cellular regulation in cells lacking LKB1.
This recent insight into the critical role played by the LKB1-AMPK axis in suppressing cell growth and cell cycle entry raises interesting possibilities for pharmaceutical intervention to suppress tumor growth through activation of this pathway (Hardie, 2007) and also raises questions about how tumor cells suppress this pathway to allow continued growth under conditions of energy stress. While somatic loss of function mutations in LKB1 are not frequent in human cancers other than lung adenocarcinoma, epigenetic mechanisms for suppression of the expression of genes in this pathway are being uncovered (Tiainen et al., 1999). Here we address a post-translational mechanism for suppression of the LKB1-AMPK pathway in tumor cells. We find that in melanoma cells transformed by mutations in oncogenic B-RAF kinase, LKB1 becomes phosphorylated at two sites that compromise the ability of this enzyme to bind to and phosphorylate AMPK. More importantly, we show that this suppression of LKB1 function in B-RAF-transformed melanoma cells plays an important role in mediating the oncogenic activity of B-RAF.
Results
Melanoma cells with the B-RAF V600E oncogenic mutation have impaired AMPK activation
AICAR (5-aminoinidazole-4-carboxamideribonucleoside), an AMP mimetic, has been shown to activate AMPK and inhibit cell proliferation in several different human tumor cell lines, including MCF-7 (breast cancer), C6 (glioma), PC3 and LNCaP (prostate cancer) cells (Rattan et al., 2005; Xiang et al., 2004). During the investigation of the potential effects of AICAR on melanoma cells, we observed that AICAR stimulated phosphorylation of AMPK at Thr-172 in the human melanoma cell lines MeWo and SK-MEL-31, as expected, but failed to cause phosphorylation of AMPK in several other melanoma cell lines, SK-MEL-28, UACC62 and UACC257 (Figure 1A). Consistent with this observation, AICAR stimulated phosphorylation of the AMPK substrate, Acetyl CoA Carboxylate (ACC) at Ser-79 in the MeWo and SK-Mel-31 cells but not in the other three cell lines (Figure 1A). Total AMPK levels were similar in all five cell lines (Figure 1A). The failure to phosphorylate AMPK in the SK-Mel-28, UACC62 and UACC257-4 cells was not due to lack of LKB1 as judged by western blots with an LKB1 antibody (Figure 1A).
Figure 1.
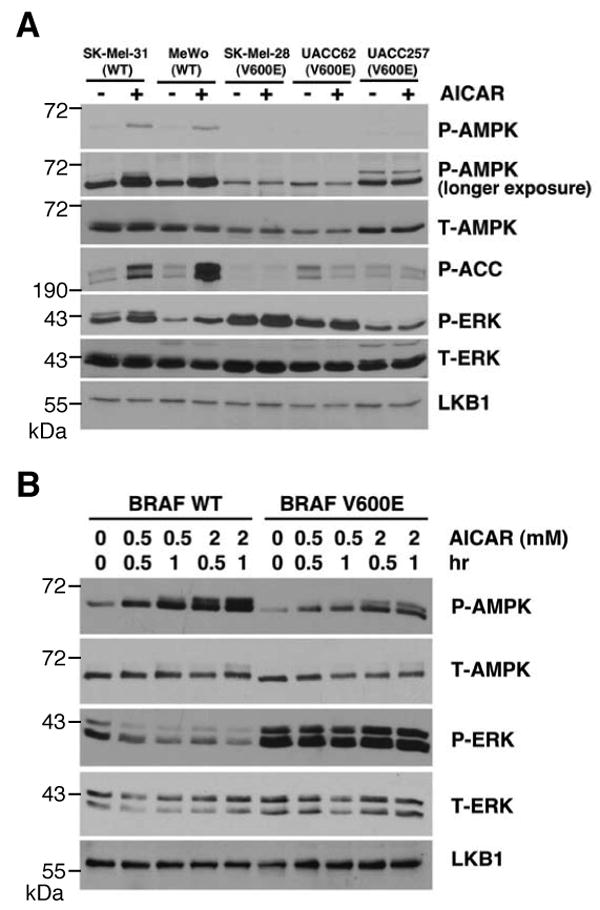
B-RAF V600E suppresses AMPK activity. (A) Phosphorylation of AMPK and ACC in human melanoma cells containing WT B-RAF or V600E mutant. Cells were treated with or without 1 mM AICAR for 1 hr. Cell lysates were used for western blotting with indicated antibodies. (B) Expression of B-RAF V600E attenuates AMPK activation in C140 melanocytes. C140 stably expressed B-RAF WT or V600E mutant were treated with indicated concentration of AICAR.
While MeWo and SK-Mel-31 cells express wild-type B-RAF, SK-MEL-28, UACC62 and UACC 257 cells contain the B-RAF V600E mutation, raising the possibility that activating mutations of B-RAF may suppress LKB1-AMPK signaling. To test this hypothesis, we generated mouse Ink4a/Arf null melanocyte C140 cells stably transfected with WT B-RAF or V600E mutant and found that cells transfected with the B-RAF V600E showed a reduction in AICAR-induced phosphorylation of AMPK (Figure 1B). Transfection with B-RAF V600E enhanced ERK1/2 phosphorylation as expected but did not affect the total level of endogenous AMPK or LKB1 compared to WT B-RAF (Figure 1B). Expression of the B-RAF V600E mutant but not WT B-RAF in Cos-7 cells also suppressed activation of AMPK in response to AICAR (Supplemental Figure S1). These results together indicate that expression of B-RAF V600E inhibits the activity of AMPK. It's noteworthy that suppression of AMPK activation correlated with the B-RAF mutational status of the melanoma cell lines better than with the level of ERK1/2 phosphorylation. For example AICAR activates AMPK in SK-Mel-31 cells, which have elevated ERK1/2 phosphorylation but lack B-RAF mutations (Figure 1A). This result, discussed in more detail below, suggests that B-RAF activation may channel downstream signaling to the suppression of AMPK more than other ERK1/2 activation mechanisms.
Down-Regulation of oncogenic B-RAF signaling stimulates AMPK Activation
To further examine the role of B-RAF in the regulation of AMPK activation, we used RNA interference to knock down the expression of B-RAF (Hingorani et al., 2003) in a melanoma cell line containing the B-RAF V600E mutation. As shown in Figure 2A, down-regulation of B-RAF expression by using two different shRNA constructs in SK-MEL-28 cells led to a decrease in ERK1/2 phosphorylation, as expected, and also resulted in increased phosphorylation of AMPK at Thr-172, further supporting the inhibitory effect of oncogenic B-RAF on AMPK activation.
Figure 2.
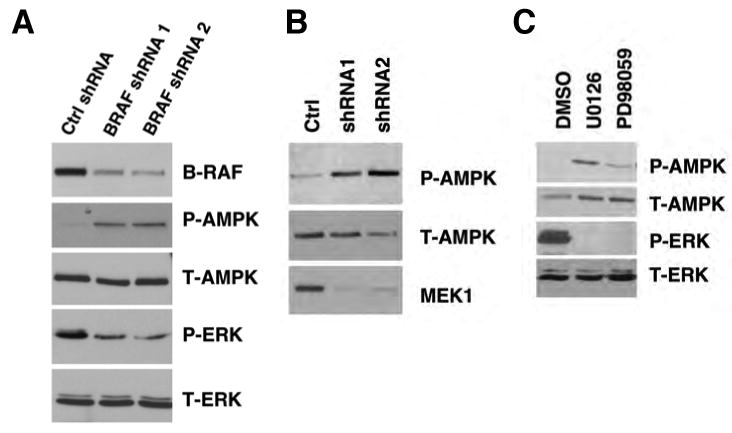
Down-regulation of B-RAF signaling activates AMPK. (A) Knockdown of B-RAF expression by RNA interference activates AMPK. SK-Mel-28 cells were infected with retrovirus containing two different shRNA constructs in pSUPER-retro against B-RAF or pSUPER-retro empty vector.
(B) Knockdown of MEK1 expression by RNA interference activates AMPK. SK-Mel-28 cells were infected with retrovirus containing two different shRNA constructs in pSM2C against MEK1 or control empty vector.
(C) Induction of AMPK phosphorylation by various inhibitors against the RAF-MEK-ERK signaling cascade. SK-MEL-28 cells were treated with DMSO, 20 μM U0126, or 50 μM PD98059 for 1 hr.
To evaluate the role of B-RAF downstream effectors (MEK and ERK) in the regulation of AMPK, we used retroviral shRNA constructs specifically targeting MEK1 or ERK2 inSK-MEL-28 cells, and found that down-regulation of either MEK1 (Figure 2B) or ERK2 (Supplemental Figure S5) led to activation of AMPK. Similar activation of AMPK was also observed when SK-MEL-28 (Figure 2C) and UACC62 cells were treated with the MEK inhibitors U0126, PD98059 or CI-1040 (also known as PD184352) (Supplemental Figures S3 and S4). Taken together, our results suggest that the activity of AMPK is negatively regulated by the oncogenic B-RAF V600E mutant, probably through its downstream MEK-ERK kinase signaling cascade.
Phosphorylation of LKB1 by ERK and p90Rsk, two kinases downstream of B-RAF
The negative regulation of AMPK by B-RAF signaling suggests that AMPK itself or its upstream kinases may be targets of the RAF-MEK-ERK protein kinase signaling cascade. Because LKB1, an upstream activator of AMPK, is known to be phosphorylated on multiple sites in vivo (Sapkota et al., 2002a; Sapkota et al., 2002b; Sapkota et al., 2001), we first examined whether the effect of U0126 treatment on AMPK activity is dependent on the presence of LKB1. As shown in Figure 3A, unlike the immortalized mouse embryonic fibroblast (MEF) cells originated from Lkb1+/+ mouse, U0126 treatment did not cause activation of AMPK in Lkb1-/- MEFs. Similarly, knockdown of LKB1 in SK-Mel-28 cells by using two different shRNA constructs impaired the activation of AMPK in response to U0126 in these cells (Figure 3B), suggesting the presence of LKB1 is critical for the effect of the MEK inhibitor on AMPK activity.
Figure 3.
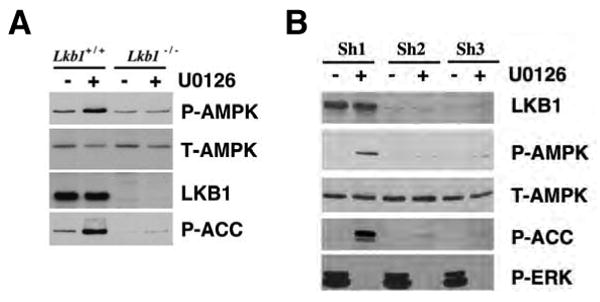
Activation of AMPK by U0126 is dependent on the presence of LKB1.
(A) U0126-induced activation of AMPK is dependent on LKB1 in MEFs. Immortalized Lkb1+/+ and Lkb1-/- MEFs were serum-starved and treated with 20 μM of U0126 for 2 hr.
(B) U0126-induced activation of AMPK is dependent on LKB1 in SK-Mel-28 cells. SK-Mel-28 cells were infected with lentivirus encoding two different shRNA against LKB1 (sh2 and sh3) or control shRNA (sh1), serum-starved and treated with 20 μM of U0126 for 2 hr.
Next, we examined the degree of LKB1 phosphorylation upon modulation of the RAF-MEK-ERK pathway. HEK293 cells were transfected with FLAG-tagged LKB1 and treated with the phorbol ester PMA, a known activator of the RAF-MEK-ERK cascade, in the presence or absence of U0126. LKB1 was immunopurified using anti-FLAG M2 agarose beads (Figure 4A), digested with trypsin or chymotrypsin and subjected to LC-MS/MS analysis to assess phosphorylation status. Two peptides containing phosphorylated Ser325 and Ser428 were consistently found in the PMA treated samples but rarely detected in LKB1 from the untreated cells or cells treated with U0126. The relative quantities of the peptides containing phospho-Ser325 and phospho-Ser428 to the same peptides in their unphosphorylated state were analyzed using rations of the total ion current (TIC) over the LC elution peaks (Asara et al., 2008; Tsay et al., 2000). This analysis revealed that treatment with U0126 dramatically reduced the fraction of LKB1 phosphorylated at Ser325 and Ser428 by approximately 80% and 65%, respectively (Supplemental Figure S6). A previous study demonstrated that Ser428 could be phosphorylated by PKA or by p90Rsk in vitro and in vivo (Sapkota et al., 2001), and the results presented here indicate that Ser428 phosphorylation occurs downstream of MEK signaling, consistent with p90Rsk being the kinase involved in this case. Although the S428 phospho-specific antibody is not sufficiently sensitive to detect endogenous LKB1 in SK-Mel-28 cells or MEFs by western blotting (Supplemental Figure S7), this antibody detected phospho-S428 in SK-MEL-28 cells stably expressing FLAG-LKB1 and the phosphorylation was reduced by treatment with the MEK inhibitors U0126 and PD98059, or by knockdown of B-RAF expression by RNAi (Figure 4B and 4C). These results further support that Ser428 is a target of the RAF-MEK-ERK signaling cascade.
Figure 4.
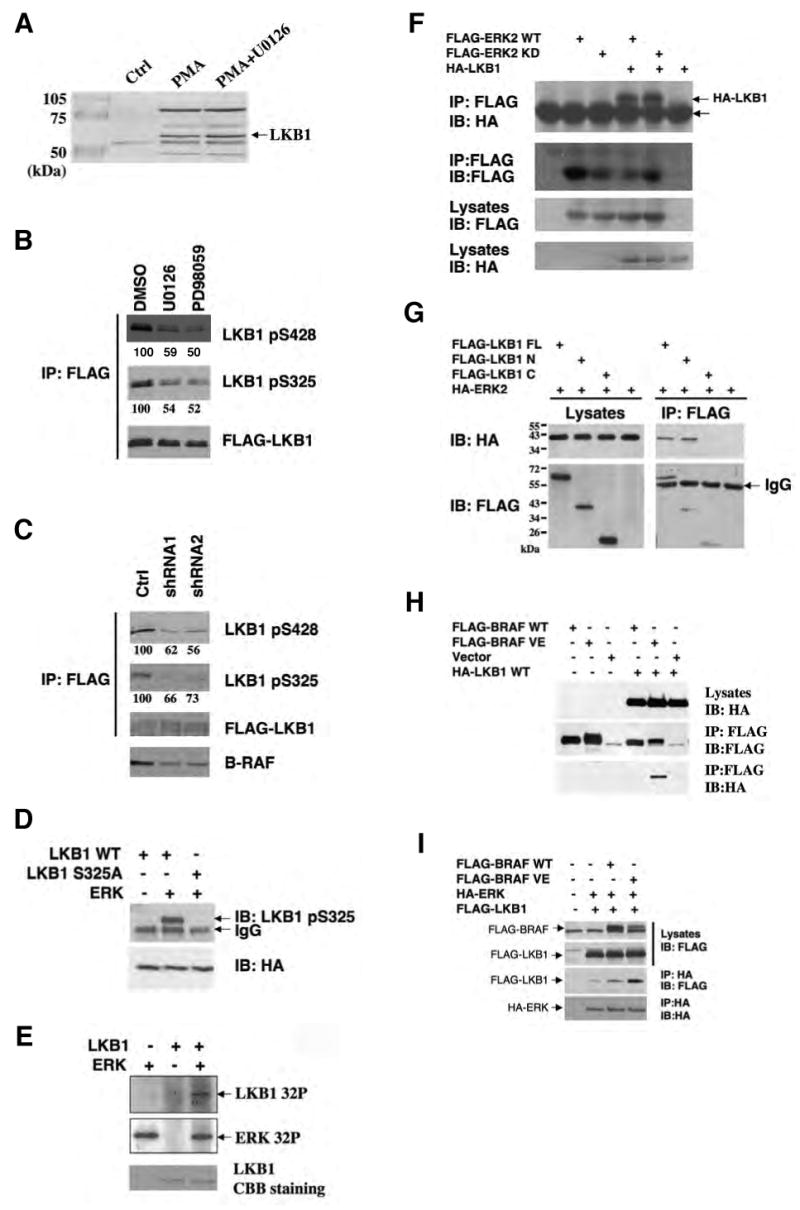
Phosphorylation of Ser325 and Ser428 of LKB1 by two downstream kinases of B-RAF, ERK and p90Rsk, respectively.
(A) Identification of phosophorylated LKB1 peptides containing Ser325 and Ser428 by LC-MS/MS analysis. HEK293 cells transfected with FLAG-LKB1 were serum-starved and pretreated with or without 20 μM of U0126 for 2 hr before the addition of 200 nM PMA for 20 min. FLAG-LKB1 proteins were immunoprecipitated using anti-FLAG M2 agarose beads and subjected to trypsin or chymotrypsin digestion followed by the LC-MS/MS analysis.
(B) Inhibition of LKB1 Ser325 and Ser428 phosphorylation by MEK inhibitors U0126 and PD98059. Cell lysates from SK-Mel-28 stably expressing FLAG-LKB1 were immunoprecipitated with anti-FLAG M2 agarose beads followed by western blotting using indicated antibodies. Numbers indicate relative intensity as quantified by image J analysis.
(C) Attenuation of LKB1 Ser325 and Ser428 phosphorylation upon knockdown of B-RAF expression. SK-Mel-28 cells stably expressing FLAG-LKB1 were infected with retrovirus containing two different shRNA constructs in pSUPER-retro against B-RAF or pSUPER-retro empty vector. Cell lysates were immunoprecipitated with anti-FLAG M2 agarose beads followed by western blotting using indicated antibodies. Numbers indicate relative intensity as quantified by image J analysis.
(D) ERK directly phosphoryates LKB1 in vitro. GST-LKB1 (D194A) proteins were expressed in E. coli, purified and incubated with active recombinant ERK proteins in the presence of γ-32P-ATP. Autoradiography was performed.
(E) Ser325 is critical for phosphorylation of LKB1 by ERK in vitro. HA-LKB1 WT and S325A mutant were immunoprecipitated from HEK293 cells and incubated with recombinant ERK proteins. Protein from the assays and HEK293 cell lysates were used for western blotting analysis with phospho-S325 LKB1 antibody and HA antibody, respectively.
(F) HA-LKB1 coimmunoprecipitates with FLAG-ERK2. HEK293 cells were transfected with HA-LKB1 together with FLAG-ERK2 wildtype or kinase dead mutant. Cell lysates were immunoprecipitated with anti-FLAG M2 agarose beads followed by western blotting with HA antibody.
(G) HA-ERK2 coimmunoprecipitates with FLAG-LKB1-N, but no FLAG-LKB1-C. Cos-7 cells were transfected with HA-ERK2 together with FLAG-LKB1 full-length (FL), N (a.a 1-309), C (a.a.310-433) or control vector. Cell lysates were immunoprecipitated with anti-FLAG M2 agarose beads followed by western blotting with HA antibody.
(H) HA-LKB1 coimmunoprecipitates with B-RAF V600E, but not WT B-RAF. HEK293 cells were transfected with FLAG-BRAF, HA-LKB1 or empty vectors as indicated. Cell lysates were immunoprecipitated with anti-FLAG M2 agarose beads followed by immunoblotting with indicated antibodies.
(I) Expression of BRAF V600E enhances the association between LKB1 and ERK. HEK293 cells were transfected with FLAG-LKB1, HA-ERK together with control vector, FLAG-B-RAF WT or B-RAF V600E constructs as indicated. Cell lysates were immunoprecipitated with anti-HA antibodies followed by immunoblotting with indicated antibodies.
Ser325 in mouse LKB1 was also previously found to be phosphorylated (Sapkota et al., 2002a), but the responsible kinase was not identified. Sequence analysis by Scansite (Yaffe et al., 2001) indicated that Ser325 is a candidate phosphorylation site by proline-dependent kinases such as cyclin-dependent kinases and MAPKs including ERK. Moreover, Scansite predicts that LKB1 contains two putative ERK-docking D domains and one ERK1 binding domain. These predictions together with the sensitivity of Ser325 phosphorylation to MEK inhibitors imply that LKB1 could be directly phosphorylated by ERK on Ser325. To test whether ERK can directly phosphorylate LKB1 in vitro, GST-tagged LKB1 was expressed in E. coli, purified and incubated with active recombinant ERK protein in the presence of γ-32P-ATP. To avoid background 32P incorporation due to autophosphorylation of LKB1, a kinase-dead mutant of LKB1 (D194A) was used. As shown in Figure 4D, ERK proteins were found to phosphorylate recombinant LKB1 protein in vitro. In addition, ERK was found to phosphorylate HA-tagged LKB1 immunoprecipitated from HEK293 cells, and mutation of Ser325 to Ala abolished the phosphorylation (Figure 4E).
To further characterize the phosphorylation of LKB1 Ser325 in vivo, an antibody against phospho-Ser325 of LKB1 was generated. This antibody recognizes over-expressed HA-LKB1 WT, but not the HA-LKB1 S325A mutant, and the reactivity towards HA-LKB1 was greatly reduced upon U0126 treatment (Supplemental Figure S8). Similarly, Ser325 phosphorylation of FLAG-LKB1 in SK-MEL-28 stable cell lines was also sensitive to the treatment of U0126 and PD98059, or knockdown of BRAF expression by RNA interference (Figure 4B and 4C). Moreover, HA-LKB1 co-immunoprecipitated with FLAG-ERK2 in HEK293 cells (Figure 4F). Further mapping experiments showed that the kinase domain of LKB1, but not the C-terminal region of LKB1, mediates its association with ERK2 (Figure 4G). Collectively, these results suggest that LKB1 Ser325 is a direct phosphorylation target of ERK.
The inhibitory effect of RAF-MEK-ERK signaling on AMPK activation was only observed in melanoma cell lines with B-RAF V600E mutant, but not in those with WT B-RAF (Figure 1A). To investigate potential mechanisms underlying this specific effect of mutant B-RAF, we co-expressed FLAG-tagged WT B-RAF or V600E mutant together with HA-LKB1 in HEK293 cells, and found that HA-LKB1 co-immunoprecipitated with FLAG-B-RAF V600E, but not FLAG-BRAF WT (Figure 4H), suggesting that LKB1 preferentially associates with B-RAF V600E. To further examine whether this B-RAF V600E can channel ERK activity to LKB1, we tested the effects of B-RAF V600E expression on the association between LKB1 and ERK. As shown in Figure 4I, there was a dramatic increase of FLAG-LKB1 co-immunoprecipitated with HA-ERK2 in cells expressing B-RAF V600E compared to cells expressing either WT B-RAF or control pCDNA3 vector. These results further support the idea that the BRAF V600E mutant facilitates targeting of ERK to LKB1.
Phosphorylation of Ser325 and Ser428 is critical for the regulation of AMPK activation by LKB1
To address the functional consequence of LKB1 phosphorylation at Ser325 and Ser428, we assessed the ability of LKB1 WT and phosphorylation-deficient mutants to activate endogenous AMPK. As shown in Figure 5A, expression of LKB1 S325A, S428A, or S325A/S428A (AA) double mutants in Lkb1-/- MEFs resulted in enhanced AMPK phosphorylation compared to expression of WT LKB1. The difference was particularly apparent in the absence of AICAR. Enhanced phosphorylation of ACC was also observed in cells expressing phosphorylation-defective mutants of LKB1. These results suggest that phosphorylation at either Ser325 or Ser428 suppresses the ability of LKB1 to activate AMPK. Similar results were obtained when these various constructs were expressed in HeLa cells (Supplemental Figure S10), which lack LKB1 (Tiainen et al., 1999).
Figure 5.

Phosphorylation of LKB1 on Ser325 and Ser428 is involved in the regulation of AMPK activation by LKB1.
(A) Mutation of Ser325 or Ser428 of LKB1 to Ala enhances its activity on AMPK activation. Lkb1-/- MEFs were infected with retrovirus containing the vector control, WT LKB1, S325A, S428A or S325A/S428A LKB1, and treated with 1 mM AICAR for 1 hr. Cell lysates were used for western blotting with indicated antibodies.
(B) Expression of LKB1 S325A/S428A mutant stimulates AMPK in SK-MEL-28 cells. SK-MEL-28 cells were infected with retrovirus containing WT LKB1, S428A, S325A or S325A/S428A (AA) LKB1 mutants. Cell lysates were used for western blotting analysis with indicated antibodies.
(C) Mutation of Ser325 and Ser428 to Ala enhances the ability of LKB1 to associate with AMPK, but not STRAD and MO25. HEK293 cells were transfected with GST-AMPKα1, Omni-STRAD, FLAG-MO25 together with WT or S325A/S428A (AA) mutant of HA-LKB1. Cell lysates were immunoprecipitated with indicated antibodies or incubated with GSH agarose beads for 2hr followed by western blotting with indicated antibodies.
(D) Mutation of Ser325 and Ser428 to alanines or U0126 treatment enhances the ability of LKB1 to associate with endogenous AMPK in SK-Mel-28 cells. SK-Mel-28 cells stably expressing FLAG-LKB1 WT or AA mutant were treated with DMSO or 20 μM of U0126 for 2 hr. Cell lysates were incubated with M2 anti-FLAG agarose beads for 2 hr followed by Western blotting with indicated antibodies.
(E) Knockdown of B-RAF expression enhances the ability of LKB1 to associate with endogenous AMPK in SK-Mel-28 cells. Sk-Mel-28 cells stably expressing FLAG-LKB1 WT were infected with retrovirus containing two different shRNA constructs in pSUPER-retro against B-RAF or pSUPER-retro empty vector. Cell lysates were immunoprecipitated with anti-FLAG M2 agarose beads followed by western blotting using indicated antibodies.
To confirm that phosphorylation of LKB1 on Ser325 and Ser428 mediates the inhibition of AMPK activity in human melanoma cells containing the oncogenic B-RAF V600E mutation, we generated SK-MEL-28 cells stably expressing LKB1 wild-type, S325A, S428A or the AA double mutants of LKB1. As shown in Figure 5B, SK-Mel-28 cells expressing the LKB1 AA mutant showed increased phospho-AMPK compared to cells expressing wild-type LKB1. In these cells, expression of LKB1 with single point mutations had only moderate effects on AMPK phosphorylation, suggesting that phosphorylation of LKB1 at either site may be insufficient to suppress activity. Similar results were obtained in UACC67 cells, another B-RAF V600E containing melanoma cell line (Supplemental Figure S11).
The full activity of LKB1 requires its interaction with two regulatory subunits, STRAD and MO25 (Hawley et al., 2003). In addition, LKB1 has been shown to associate with AMPK (Shaw et al., 2004b). To gain insight into the mechanism underlying the effect of LKB1 phosphorylation on AMPK activation, we compared the ability of LKB1 WT and the phosphorylation-deficient AA mutant to associate with AMPK, STRAD and MO25. As shown in Fig 5C, both HA-tagged WT LKB1 and the AA mutant bind to Omni- STRAD and FLAG-MO25 in HEK293 cells. However, interestingly, the LKB1 AA mutant showed stronger association with GST-AMPK than with WT LKB1 (Figure 5C). Similarly, more endogenous AMPK coimmunoprecipitated with FLAG-tagged LKB1 AA mutant than with WT LKB1 in SK-Mel-28 stable cell lines (Figure 5D). Moreover, U0126 treatment enhanced the interaction between AMPK and WT LKB1, but did not have an additional effect on the AA mutant (Figure 5D). Similarly, knocking down the expression of B-RAF with two different shRNA constructs also enhanced the interaction between AMPK and LKB1 in SK-Mel-28 cells stably expressing LKB1(Figure 5E). These results suggest that phosphorylation of LKB1 on Ser325 and Ser428 suppress its ability to bind to AMPK, thus explaining the decreased phosphorylation of AMPK.
Expression of the LKB1 phosphorylation-deficient mutant suppresses proliferation and anchorage-independent growth of melanoma cells
To examine the biological effects of LKB1 phosphorylation at Ser325 and Ser428, we performed cell proliferation assays on SK-MEL-28 cells stably expressing LKB1 WT or the phosphorylation-deficient AA mutant. We found that cells expressing the AA mutant of LKB1 had significantly reduced proliferation rates compared to cells expressing WT LKB1, suggesting that phosphorylation of LKB1 at Ser325 and Ser428 is important for cell proliferation in BRAF V600E expressing melanoma cells (Figure 6A).
Figure 6.
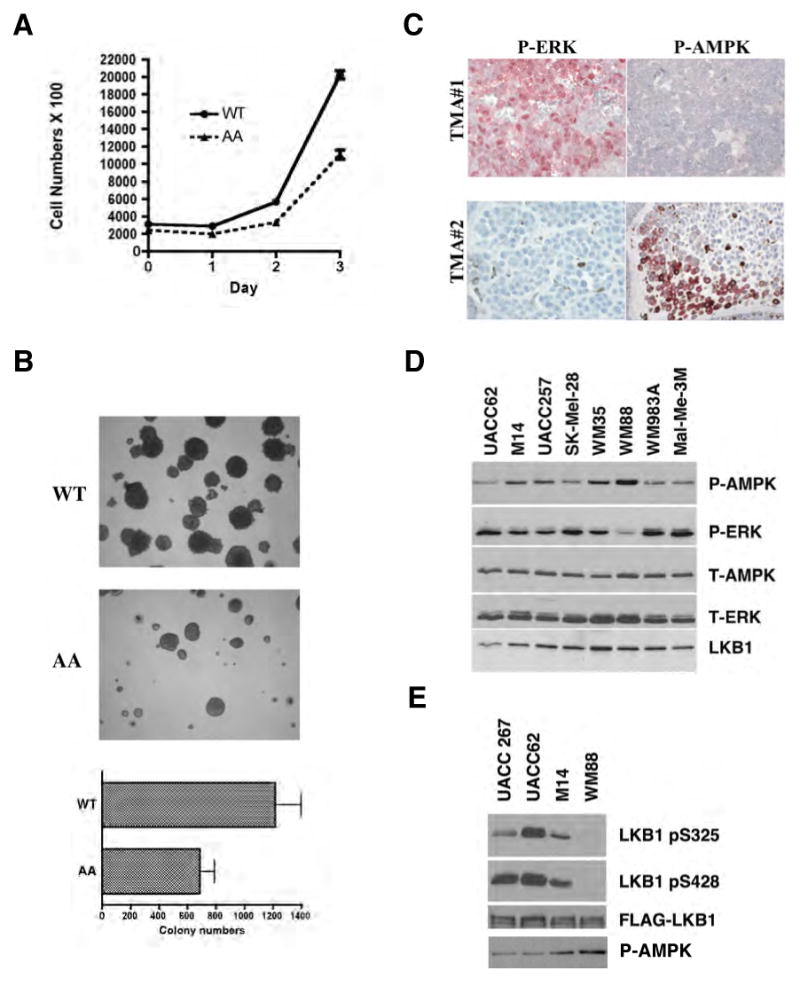
Phosphorylation of LKB1 on Ser325 and Ser428 are critical for cell proliferation and anchorage-independent growth.
(A) Expression of LKB1 S325A/S428A (AA) mutant inhibits cell proliferation. Cell proliferation curves of SK-MEL-28 cells stably expressing WT LKB1 or AA mutant were measured. One representative from three independent experiments is shown.
(B) Expression of LKB1 S325A/S428A (AA) mutant inhibits cell transformation. SK-MEL-28 cells stably expressing LKB1 WT LKB1 or AA mutant were used in sofa agar assay. Colonies were photographed after 28 days of growth. Error bars indicate SD. One representative from three independent experiments is shown.
(C) Inverse correlation between phospho-ERK and phospho-AMPK activities in human melanoma tumor samples. Representative images of human melanoma tumor samples stained with phospho-AMPK or phospho-ERK antibodies in immunohistochemical analysis.
(D) Inverse correlation between phospho-ERK and phospho-AMPK activities in human melanoma cells containing B-RAF V600E mutation. Total cell lysates from several human melanoma cell lines were used in Western blotting analysis with indicated antibodies.
(E) Phosphorylation levels of LKB1 Ser325 and Ser428 in human melanoma cells containing B-RAF V600E mutation. Various melanoma cell lines were stably transfected with pBabe-FLAG-LKB1 WT, and cell lysates were immunoprecipitated with anti-FLAG M2 agarose beads followed by western blotting using indicated antibodies.
To determine the role of phosphorylation in anchorage-independent cell growth, we performed soft agar assays on SK-MEL-28 cells expressing either WT or the AA mutant of LKB1. As shown in Figure 6B, cells expressing the LKB1 AA mutant formed fewer colonies than those expressing WT LKB1, suggesting that phosphorylation at Ser325 and Ser428 are important for cell transformation in B-RAF V600E expressing melanoma cells.
Relationship between P-AMPK activity and P-ERK activity in primary human melanoma specimens and melanoma cell lines
To evaluate the relevance of the inhibition of AMPK activity by RAF-MEK-ERK signaling in human melanoma samples, we examined the levels of phospho-ERK and phospho-AMPK in immunohistochemical studies on human melanoma tumors. We found that among five primary tumor samples showing strong phospho-ERK staining, one of them showed moderate, three showed weak and one showed negative phospho-AMPK staining (Figure 6C). Conversely, among the four tumors that showed strong phospho-AMPK staining, one of them showed moderate and three showed negative phospho-ERK staining, suggesting an inverse correlation between phospho-AMPK and phospho-ERK activities in human melanoma (Chi square test, p = 0.028). Moreover, a similar tendency of inverse correlation between phospho-AMPK and phospho-ERK levels was also observed in an immunoblotting analysis of several human melanoma cell lines containing the BRAF V600E mutation (Figure 6D). We stably expressed FLAG-tagged wild-type LKB1 in a subset of these cell lines, immunoprecipitated LKB1 and examined phosphorylation at Ser325 and Ser428. The cell line with the highest level of phospho-AMPK and lowest level of phospho-ERK1/2 (WM88) exhibited no detectable phosphorylation of LKB1 at either site while the cell line with the lowest level of phospho-AMPK and a high level of phospho ERK1/2 (UACC62) exhibited high phosphorylation at both sites. UACC267 and M14 cells showed intermediate levels of LKB1 phosphorylation and AMPK phosphorylation.
Discussion
The LKB1-AMPK signaling axis plays a central role in bridging cellular metabolic sensing and regulation of cell growth, proliferation and survival (Hardie, 2005; Luo et al., 2005; Motoshima et al., 2006; Shaw, 2006). The activity of AMPK is regulated by the ratio of cytosolic AMP to ATP, which affects the phosphorylation of Thr172. The steady state level of phospho-Thr172 is determined by the relative rate of phosphorylation by kinases and dephosphorylation by phosphatases. Recent studies suggest that AMP binding to the gamma subunit of AMPK decreases the rate of dephosphorylation at Thr172, resulting in increased AMPK activation (Steinberg et al., 2006). The results we present here indicate that phosphorylation of LKB1 by kinases downstream of B-RAF impairs the ability of LKB1 to associate with and phosphorylate AMPK at Thr172, thereby blocking the activation of AMPK even under conditions of elevated AMP. The phosphorylation of LKB1 by ERK and p90RSK, and subsequent inhibition of its ability to phosphorylate and activate AMPK may partially mediate the oncogenic effects of BRAF V600E, as expression of a phosphorylation-deficient mutant of LKB1 (S325A/S428A) inhibited melanoma cell proliferation (Figure 7). This model provides a mechanism for down-regulation of the cellular activity of the tumor suppressor LKB1 in cancer cells through post-translational modification.
Figure 7.

A model for negative regulation of LKB1/AMPK activity by BRAF V600E signaling.
LKB1 contains a central serine-threonine kinase domain, an N-terminal region with a nuclear localization signal and a C-terminal regulatory region (Alessi et al., 2006). Both Ser325 and Ser428 of LKB1 are located in the C-terminal region. While Ser428 was shown previously to be phosphorylated by p90Rsk and PKA in response to different stimuli, there was no evidence that this phosphorylation affected the in vivo or in vitro function of LKB1 (Sapkota et al., 2001). Phosphorylation at Ser325 was also previously detected, but the kinase responsible for this phosphorylation was not determined (Sapkota et al., 2001). In this report, we show that mutation of Ser325 and Ser428 to Ala enhanced the association of LKB1 with AMPK, but did not alter the association with STRAD or MO25. This observation is consistent with previous findings that the C-terminal region of LKB1 is involved in AMPK interaction (Forcet et al., 2005) while the kinase domain mediates the interaction with STRAD and MO25 (Boudeau et al., 2004). It remains to be seen whether phosphorylation at these residues would regulate the interaction between LKB1 and other LKB1 downstream kinases.
Intriguingly, a loss-of-function missense mutation of LKB1 has been identified at codon 324 (Pro to Leu) in the germline of a PJS patient and in a sporadic carcinoma (Alessi et al., 2006). Biochemical studies showed that this mutation impairs LKB1's ability to activate AMPK and its downstream signaling (Forcet et al., 2005), supporting the idea that regulation of AMPK signaling is critical to the activity of this LKB1 mutant. Pro324 is adjacent to the Ser325 ERK phosphorylation site, and the change of Pro to Leu is predicted to make LKB1 a better substrate for ERK2, based on the phosphorylation motif of ERK2 (Yaffe, et al., 2001), which is consistent with the lower activity of this LKB1 mutant on AMPK activation (Forcet et al., 2005). In addition, it is likely that a specific local conformation in this region is required for binding to AMPK and that phosphorylation of Ser325 or mutation of Pro324 to Leu disrupts the local structure needed for this interaction.
In this study, we observed an inverse correlation between the activities of ERK and AMPK in melanoma cells harboring the B-RAF V600E mutation (Figure 1A and Figure 6). However, some melanoma cell lines that lack B-RAF mutations have elevated phospho-ERK without a major suppression of AMPK activation, i.e. SK-Mel-31 cells (Figure 1A). Hence, suppression of AMPK activation correlates better with B-RAF mutations than with total ERK activation in the melanoma cells. Intriguingly, we found that LKB1 coimmunoprecipitates with B-RAF V600E mutant, but not with WT B-RAF, and that expression of B-RAF V600E dramatically enhanced the association between LKB1 and ERK (Figures 4H and 4I). These results strongly suggest that in melanoma cells that harbor B-RAF mutations activated ERK is more efficiently channeled to the substrate, LKB1. Consistent with this concept, previous studies have shown that melanoma with B-RAF mutation and those without B-RAF mutations are wired differently in terms of the regulation of RAS-RAF-MEK-ERK signaling network. For example, melanomas with Ras mutations have been shown to utilize C-RAF rather than B-RAF to activate ERK and the consequence is a disruption of cAMP signaling specifically in the Ras mutant melanomas (Dumaz et al., 2006). In addition, although both Ras mutations and B-RAF mutations activate ERK and cause cell transformation, MEK inhibitors only inhibit growth of B-RAF mutant cell lines (Solit et al., 2006). Both LKB1 and AMPK have been shown to suppress cell growth and proliferation under conditions of energy stress. It's conceivable that tumor cells must turn off this signaling pathway to gain a growth advantage. Mechanisms to suppress LKB1-AMPK signaling include maintaining high levels of cellular ATP through up-regulation of HIF-1 and subsequent transcriptional activation of genes involved in glucose-uptake and glycolysis-dependent ATP synthesis (Ashrafian, 2006; Gatenby and Gillies, 2004; Shaw, 2006). A second mechanism involves deletion or loss-of-function mutations of the LKB1 gene, as occurs in PJS and in non-small cell lung carcinomas (Sanchez-Cespedes et al., 2002), and less frequently in malignant melanomas, colon, breast, ovarian and brain cancers (Alessi et al., 2006; Guldberg et al., 1999; Katajisto et al., 2007; Rowan et al., 1999). Our findings here suggest that phosphorylation of LKB1 represents a new way to “over-ride” the energy brake set by LKB1 and AMPK. We show that AMPK is consistently suppressed in B-RAF V600E transformed cells through phosphorylation and inactivation of LKB1, and that down-regulation of B-RAF signaling by pharmacological inhibitors or RNA interference relieved this inhibition. It would be interesting to study whether similar mechanisms of suppressing LKB1-AMPK signaling through post-translational modifications on LKB1 or AMPK occur in other cancer cells driven by other mechanisms.
Drugs targeting the RAF-MEK-ERK pathway, such as MEK and RAF inhibitors are intensively being developed and under clinical trails for various human cancers, such as melanoma, renal cell carcinoma and colorectal cancer (Beeram et al., 2005; Gray-Schopfer et al., 2007; Schreck and Rapp, 2006; Thompson and Lyons, 2005). Meanwhile, several current prescribed drugs used for metabolic disorders, such as metformin and thiazolidinediones, have been found to activate the LKB1-AMPK pathway (Hardie, 2007; Shaw, 2006; Shaw et al., 2005). The molecular linkage between the RAF-MEK-ERK and LKB1-AMPK signaling pathways reported here has several implications for the potential uses of these drugs in treating cancer and metabolic disorders. First, AMPK activators have been proposed for the use of cancer therapy (Hardie, 2007; Motoshima et al., 2006). Our findings here suggest that AMPK activators and MEK/RAF inhibitors may have synergistic effects on inhibiting proliferation of B-RAF V600E transformed tumor cells. Secondly, our findings suggest that MEK and RAF inhibitors may be useful in the treatment of PJS by activating LKB1 when an intact LKB1 allele is present. Recent studies have shown that haploinsufficiency of LKB1 is sufficient for the formation of hamartomatous polyps in mouse models, and there is evidence that a wild-type allele of LKB1 can be found in the harmatomatous polyps from PJS patients (Hernan et al., 2004). Lastly, in addition to direct AMPK activators, strategies based on modulating post-translational modifications of LKB1, such as MEK and RAF inhibitors, that activate AMPK indirectly, could also be considered for the treatment of metabolic disorders.
In summary, our findings reveal an intriguing molecular linkage between LKB1-AMPK and RAF-MEK-ERK, two important protein kinase signaling pathways involved in cancer. In a recent survey on patterns of somatic mutations in human cancer genomes (Greenman et al., 2007), BRAF and LKB1/STK11 were listed as the second and third of all the protein kinases mutated in cancers (in terms of gene-specific selection pressures). Further understanding of the role of this molecular linkage in tumorigenesis could potentially provide great therapeutic opportunities for cancer treatment.
Experimental Procedures
Materials
Anti-phospho-AMPK (Thr172), anti-AMPK, anti-phospho-ERK1/2 (Thr202/Tyr204), anti-ERK, anti-phospho-ACC and anti-phospho-LKB1 (Ser428) antibodies were purchased from Cell Signaling Technology. Anti-phosopho-LKB1 (Ser325) antibodies were generated at Cell Signaling Technology. Anti-FLAG M2 affinity gel, anti-FLAG M2 monoclonal antibodies and U0126 were purchased from Sigma. Anti-Omni, anti-B-RAF and anti-CRAF antibodies were obtained from Santa Cruz Biotechnology. Anti-HA (11) and anti-LKB1 (Ley37D/G6) monoclonal antibodies were purchased from Covance and Abcam, respectively. AICAR was obtained from Toronto Research Chemicals (Downsview, ON, Canada). PD98059 was from obtained from Cell Signal Technology. LKB1 and AMPK cDNA constructs have been described previously (Shaw et al., 2004b; Zheng and Cantley, 2007). LKB1 S325A, S428A and S325/S428A mutants were generated using PCR mutagenesis and verified by sequencing. FLAG-ERK2 construct was kindly provided by Dr. Melanie Cobb.
Cell culture, transfection and retroviral infection
SK-Mel-28, UACC62, UACC257, SK-Mel-31 and MeWo Cells were cultured in RPMI medium (MediaTech) containing 10% fetal bovine serum (FBS) (Gemini Bio-products). C140 cells stably expressing B-RAF WT or V600E mutant (Kim et al., 2006) were cultured in RPMI medium containing 10% FBS and 2μg/ml doxycycline (Clontech). Immortalized wild type and LKB1-deficient MEFs were gifts from Dr. Nabeel Bardeesy (Massachusetts General Hospital) and cultured in DMEM medium containing 10% FBS. Cos-7 and HeLa cells were obtained from ATCC and cultured in DMEM medium containing 10% FBS. For retroviral transfection, amphotropic retrovirus were generated as described previously (Zheng and Cantley, 2007). When indicated, stable populations were obtained and maintained by selection with 2 μg/ml puromycin (Sigma). For B-RAF RNA interference, pSuper-retro containing short hairpin RNAs against B-RAF (Mu-A and com-4, sh1 and sh2) was obtained from Dr. David Tuveson and described previously (Hingorani et al., 2003). For MEK1 and ERK2 knockdown, pSM2C containing shRNAs were gifts from Dr. Stephen Elledge. For LKB1 interference, pLKO constructs containing shRNAs against human LKB1 were purchased from Sigma. For drug treatment, cells were replaced with fresh media before addition of various drugs as indicated.
Western blotting and immunoprecipitation
Cell lysates were prepared using lysis buffer containing HEPES pH 7.0, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 50 mM NaF, 10 mM β-glycero-phosphate, 10 nM calyculin A, 1 mM Na3VO4 and protease inhibitors, and normalized by protein concentrations using the Bradford method (Bio-Rad). For western blotting, protein samples were separated on 8% - 12% SDS-PAGE and transferred to PVDF membrane (Millipore). Membranes were blocked in TBST containing 5% non-fat milk, incubated with primary antibodies according to antibody manufacturer's instructions, followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Chemicon) and enhanced chemiluminescence detection (Perkin Elmer). For immunoprecipitation, cell lysates were incubated with primary antibodies or anti-FLAG M2 affinity gel overnight at 4°C, followed by incubation with protein A/G sephorase for an additional 1 hr at 4°C, when applicable. Beads were washed three times with lysis buffer and boiled in Laemmli sample buffer, and immune complexes were analyzed by SDS-PAGE and Western blotting. For GST pulldown, cell lysates were incubated with GSH agarose beads (Pharmacia) overnight.
Mass Spectrometry
Coomassie stained SDS-PAGE gel bands containing FLAG-LKB1 isolated from HEK293 cells with different treatments were excised and subjected to in-gel digestion and reversed-phase microcapillary LC/MS/MS using a LTQ 2D linear ion trap mass spectrometer (ThermoScientific) in data-dependent acquisition and positive ion mode. MS/MS spectra were searched against the concatenated target and decoy (reversed) Swiss-Prot protein database using Sequest (Proteomics Browser, ThermoScientific) with differential modifications for Ser/Thr/Tyr phosphorylation(+79.97) and the sample processing artifacts Met oxidation (+15.99) and Cys alkylation (+57.02). Phosphorylated and unphosphorylated peptide sequences were identified if they initially passed the following Sequest scoring thresholds: 1+ ions, Xcorr ≥ 2.0 Sf ≥ 0.4, P ≥ 5; 2+ ions, Xcorr ≥ 2.0, Sf ≥ 0.4, P ≥ 5; 3+ ions, Xcorr ≥ 2.60, Sf ≥ 0.4, P ≥ 5 against the target protein database. Passing MS/MS spectra were manually inspected to be sure that all b- and y-fragment ions aligned with the assigned sequence and modification sites. Determination of the exact sites of phosphorylation were aided using GraphMod software (Proteomics Browser). For relative quantification of phosphorylation peptide signal levels, an isotope-free (label-free) method was used by first integrating the total ion counts (TIC) for each MS/MS sequencing event during a targeted ion MS/MS (TIMM) experiment or a data-dependant acquisition. For each phosphorylation site (Ser325 and Ser428), a ratio of phosphorylated peptide signal (TIC of phosphorylated form) to the total peptide signal (TIC of phosphorylated form + TIC of non-phosphorylated form) from both the PMA and U0126 treated samples were calculated according to the following equation:
The ratios of the Ser325 and Ser428 sites from the PMA treated samples were then compared to the same phosphopeptide ratios from both the untreated and U0126 treated samples according to the following equation:
While a direct comparison of phosphopeptide signals between different experiments is not accurate due to different total protein levels and sample environments, a comparison the ratio of the phosphorylated to non-phosphorylated peptide forms is an accurate measure of signal -level change since the total peptide signal (modified and unmodified) is measured. The above calculations were performed manually using Microsoft Excel and with automated in-house developed software named Protein Modification Quantifier v1.0 (Beth Israel Deaconess Medical Center, Boston, MA).
In vitro MAP kinase assay
Recombinant GST-LKB1 (D194A) proteins were expressed, purified from E.coli, and incubated with recombinant active ERK protein (Stratagene) in the presence of γ-32P-ATP at 37°C for 30 min. For kinase assays using HA-LKB1, HEK293 cells were transfected with HA-LKB1 WT or S325A constructs, and immunoprecipitated HA-LKB1 proteins were used in the kinase assays.
Cell proliferation and soft agar assays
For cell proliferation assays, 2 × 105 cells were seeded in triplicate on a 6-well plate and grown in full medium. At 24 hr intervals for 4 days, cells were trypsinized and the numbers of cells in each well were counted by a Coulter counter. For soft agar assays, cells were suspended in 0.3% agarose in complete medium and plated on a layer of 0.6% agarose in complete medium in 6-well culture plates (3 × 104 cells/well). After 3 weeks, the colonies were stained with iodonitrotetrazolium chloride (Sigma) and photographed. The numbers of colonies were counted using NIH image software.
Immunohistochemical Analysis
Human melanoma tissue microarray was obtained from US Biomax (Rockville MD). Formalin fixed paraffin-embedded tissue microarray was deparaffinized in xylenes and hydrated in a graded series of alcohols. Heat induced epitope antigen retrieval using citrate buffer in a pressure cooker was used. Staining was performed using DAKO LSAB+ Alkaline Phosphatase detection system and Permanent Red as the chromogen. The results were evaluated by pathologist Dr. Scott Granter. The immunoreactivity of phospho-ERK and phospho-AMPK was scored on a scale from 0 to 3+ for negative, weak, moderate and strong staining.
Supplementary Material
Acknowledgments
We thank Drs. Reuben Shaw, Nabeel Bardeesy, David Tuveson and Melanie Cobb for reagents, Lisa Freimark for the assistance with Mass spectrometry analysis, Li Zhang and Szexian Lee for technical assistance, Dr. Yang-Qing Xu and members of the Cantley Lab for helpful discussion. We also would like to thank Shailender Nagpal for helping to create software for quantitative mass spectrometry analysis. B.Z. is supported by a Charles A. King Trust postdoctoral fellowship from the Charles A. King Trust, Bank of America, Co-Trustee. This work is supported by National Institutes of Health Grant GM56203 and CA102694 to L.C.C, K99CA133245 to B.Z., and UO1 CA84313 and RO1 CA93947 to L.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- Asara JM, Christofk HR, Freimark LM, Cantley LC. A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics. 2008;8:994–999. doi: 10.1002/pmic.200700426. [DOI] [PubMed] [Google Scholar]
- Ashrafian H. Cancer's sweet tooth: the Janus effect of glucose metabolism in tumorigenesis. Lancet. 2006;367:618–621. doi: 10.1016/S0140-6736(06)68228-7. [DOI] [PubMed] [Google Scholar]
- Baas AF, Kuipers J, van der Wel NN, Batlle E, Koerten HK, Peters PJ, Clevers HC. Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell. 2004;116:457–466. doi: 10.1016/s0092-8674(04)00114-x. [DOI] [PubMed] [Google Scholar]
- Bardeesy N, Sinha M, Hezel AF, Signoretti S, Hathaway NA, Sharpless NE, Loda M, Carrasco DR, DePinho RA. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419:162–167. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- Beeram M, Patnaik A, Rowinsky EK. Raf: a strategic target for therapeutic development against cancer. J Clin Oncol. 2005;23:6771–6790. doi: 10.1200/JCO.2005.08.036. [DOI] [PubMed] [Google Scholar]
- Boudeau J, Scott JW, Resta N, Deak M, Kieloch A, Komander D, Hardie DG, Prescott AR, van Aalten DM, Alessi DR. Analysis of the LKB1-STRAD-MO25 complex. J Cell Sci. 2004;117:6365–6375. doi: 10.1242/jcs.01571. [DOI] [PubMed] [Google Scholar]
- Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Chong H, Vikis HG, Guan KL. Mechanisms of regulating the Raf kinase family. Cell Signal. 2003;15:463–469. doi: 10.1016/s0898-6568(02)00139-0. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Dhomen N, Marais R. New insight into BRAF mutations in cancer. Curr Opin Genet Dev. 2007;17:31–39. doi: 10.1016/j.gde.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Dumaz N, Hayward R, Martin J, Ogilvie L, Hedley D, Curtin JA, Bastian BC, Springer C, Marais R. In Melanoma, RAS Mutations Are Accompanied by Switching Signaling from BRAF to CRAF and Disrupted Cyclic AMP Signaling. Cancer Res. 2006;66:9483–9491. doi: 10.1158/0008-5472.CAN-05-4227. [DOI] [PubMed] [Google Scholar]
- Forcet C, Etienne-Manneville S, Gaude H, Fournier L, Debilly S, Salmi M, Baas A, Olschwang S, Clevers H, Billaud M. Functional analysis of Peutz-Jeghers mutations reveals that the LKB1 C-terminal region exerts a crucial role in regulating both the AMPK pathway and the cell polarity. Hum Mol Genet. 2005;14:1283–1292. doi: 10.1093/hmg/ddi139. [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. 2004;6:313–319. doi: 10.1016/j.ccr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–857. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- Gray-Schopfer VC, da Rocha Dias S, Marais R. The role of B-RAF in melanoma. Cancer Metastasis Rev. 2005;24:165–183. doi: 10.1007/s10555-005-5865-1. [DOI] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldberg P, thor Straten P, Ahrenkiel V, Seremet T, Kirkin AF, Zeuthen J. Somatic mutation of the Peutz-Jeghers syndrome gene, LKB1/STK11, in malignant melanoma. Oncogene. 1999;18:1777–1780. doi: 10.1038/sj.onc.1202486. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. New roles for the LKB1-->AMPK pathway. Curr Opin Cell Biol. 2005;17:167–173. doi: 10.1016/j.ceb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernan I, Roig I, Martin B, Gamundi MJ, Martinez-Gimeno M, Carballo M. De novo germline mutation in the serine-threonine kinase STK11/LKB1 gene associated with Peutz-Jeghers syndrome. Clin Genet. 2004;66:58–62. doi: 10.1111/j.0009-9163.2004.00266.x. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003;63:5198–5202. [PubMed] [Google Scholar]
- Ikenoue T, Hikiba Y, Kanai F, Tanaka Y, Imamura J, Imamura T, Ohta M, Ijichi H, Tateishi K, Kawakami T, et al. Functional analysis of mutations within the kinase activation segment of B-Raf in human colorectal tumors. Cancer Res. 2003;63:8132–8137. [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Molecular cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Katajisto P, Vallenius T, Vaahtomeri K, Ekman N, Udd L, Tiainen M, Makela TP. The LKB1 tumor suppressor kinase in human disease. Biochim Biophys Acta. 2007;1775:63–75. doi: 10.1016/j.bbcan.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Kim M, Gans JD, Nogueira C, Wang A, Paik JH, Feng B, Brennan C, Hahn WC, Cordon-Cardo C, Wagner SN, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125:1269–1281. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Saha AK, Xiang X, Ruderman NB. AMPK, the metabolic syndrome and cancer. Trends Pharmacol Sci. 2005;26:69–76. doi: 10.1016/j.tips.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Martin SG, St Johnston D. A role for Drosophila LKB1 in anterior-posterior axis formation and epithelial polarity. Nature. 2003;421:379–384. doi: 10.1038/nature01296. [DOI] [PubMed] [Google Scholar]
- Motoshima H, Goldstein BJ, Igata M, Araki E. AMPK and cell proliferation--AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol. 2006;574:63–71. doi: 10.1113/jphysiol.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattan R, Giri S, Singh AK, Singh I. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J Biol Chem. 2005;280:39582–39593. doi: 10.1074/jbc.M507443200. [DOI] [PubMed] [Google Scholar]
- Rowan A, Bataille V, MacKie R, Healy E, Bicknell D, Bodmer W, Tomlinson I. Somatic mutations in the Peutz-Jeghers (LKB1/STKII) gene in sporadic malignant melanomas. J Invest Dermatol. 1999;112:509–511. doi: 10.1046/j.1523-1747.1999.00551.x. [DOI] [PubMed] [Google Scholar]
- Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG, Sidransky D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62:3659–3662. [PubMed] [Google Scholar]
- Sapkota GP, Boudeau J, Deak M, Kieloch A, Morrice N, Alessi DR. Identification and characterization of four novel phosphorylation sites (Ser31, Ser325, Thr336 and Thr366) on LKB1/STK11, the protein kinase mutated in Peutz-Jeghers cancer syndrome. Biochem J. 2002a;362:481–490. doi: 10.1042/0264-6021:3620481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota GP, Deak M, Kieloch A, Morrice N, Goodarzi AA, Smythe C, Shiloh Y, Lees-Miller SP, Alessi DR. Ionizing radiation induces ataxia telangiectasia mutated kinase (ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366. Biochem J. 2002b;368:507–516. doi: 10.1042/BJ20021284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota GP, Kieloch A, Lizcano JM, Lain S, Arthur JS, Williams MR, Morrice N, Deak M, Alessi DR. Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) and cAMP-dependent protein kinase, but not its farnesylation at Cys(433), is essential for LKB1 to suppress cell vrowth. J Biol Chem. 2001;276:19469–19482. doi: 10.1074/jbc.M009953200. [DOI] [PubMed] [Google Scholar]
- Schreck R, Rapp UR. Raf kinases: oncogenesis and drug discovery. Int J Cancer. 2006;119:2261–2271. doi: 10.1002/ijc.22144. [DOI] [PubMed] [Google Scholar]
- Schubbert S, Bollag G, Shannon K. Deregulated Ras signaling in developmental disorders: new tricks for an old dog. Curr Opin Genet Dev. 2007;17:15–22. doi: 10.1016/j.gde.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004a;6:91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004b;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GR, Michell BJ, van Denderen BJ, Watt MJ, Carey AL, Fam BC, Andrikopoulos S, Proietto J, Gorgun CZ, Carling D, et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006;4:465–474. doi: 10.1016/j.cmet.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Thompson N, Lyons J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr Opin Pharmacol. 2005;5:350–356. doi: 10.1016/j.coph.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Tiainen M, Ylikorkala A, Makela TP. Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci U S A. 1999;96:9248–9251. doi: 10.1073/pnas.96.16.9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsay YG, Wang YH, Chiu CM, Shen BJ, Lee SC. A strategy for identification and quantitation of phosphopeptides by liquid chromatography/tandem mass spectrometry. Anal Biochem. 2000;287:55–64. doi: 10.1006/abio.2000.4837. [DOI] [PubMed] [Google Scholar]
- Tuveson DA, Weber BL, Herlyn M. BRAF as a potential therapeutic target in melanoma and other malignancies. Cancer Cell. 2003;4:95–98. doi: 10.1016/s1535-6108(03)00189-2. [DOI] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- Watts JL, Morton DG, Bestman J, Kemphues KJ. The C. elegans par-4 gene encodes a putative serine-threonine kinase required for establishing embryonic asymmetry. Development. 2000;127:1467–1475. doi: 10.1242/dev.127.7.1467. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–844. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Xiang X, Saha AK, Wen R, Ruderman NB, Luo Z. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem Biophys Res Commun. 2004;321:161–167. doi: 10.1016/j.bbrc.2004.06.133. [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Leparc GG, Lai J, Obata T, Volinia S, Cantley LC. A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat Biotechnol. 2001;19:348–353. doi: 10.1038/86737. [DOI] [PubMed] [Google Scholar]
- Zheng B, Cantley LC. Regulation of epithelial tight junction assembly and disassembly by AMP-activated protein kinase. Proc Natl Acad Sci U S A. 2007;104:819–822. doi: 10.1073/pnas.0610157104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.