

Abstract
This protocol describes the primary culture of individual chromaffin cells derived by enzymatic digestion from the adrenal medulla of the bovine adrenal gland. Since the late 1970s, such cells have provided a useful model system to study neurotransmitter biosynthesis, storage and release in the catecholaminergic system. The protocol can be divided into three stages: isolation of cells (4–6 h), determination of viable cell numbers (approximately 30 min) and growth in culture (3–7 d). An alternative procedure is to perform studies in a continuous chromaffin (pheochromocytoma) cell line, such as PC12, although such transformed cells are typically less highly differentiated than primary cells. The bovine chromaffin cell procedure should yield approximately 10–20 million cells, suitable for several experiments over the subsequent 3–7 d. Typical experiments involve transmitter biosynthesis, vesicular storage, exocytotic release, stimulus coupling (signal transduction) toward secretion or transcription, or morphology, including ultrastructure. The total time, from adrenal gland harvest until functional experiments, is typically 4–8 d.
INTRODUCTION
The chromaffin cell is widely used as a model for neuroendocrine secretory cells in general and noradrenergic neurons. Indeed, the structure and function of the components of the secretory apparatus appear to be quite analogous in chromaffin cells and neurons. The ability to obtain reasonably homogeneous chromaffin cells in culture and their characteristic secretory responses have led to their widespread use in neurobiology. As a complement to functional studies of the cellular processes underlying secretion, the adrenal medulla is also a rich preparative source of its secretory organelle, the chromaffin granule, which can be obtained in high purity by differential and gradient centrifugation of adrenal medullary homogenates; indeed, much of what we know about the structure of neuroendocrine secretory granules is derived from such chromaffin granule preparations from the adrenal medulla.
Chromaffin cells are typically maintained in culture on solid plastic dishes or coverslips. The cell preparations can be used for studies of catecholamine and peptide secretion, transcription, signal transduction, intracellular trafficking or morphology/ultra-structure. Most experiments on chromaffin cell biology are now conducted with PC12 rat pheochromocytoma cells, because of the ease of culture of a continuous, transformed cell line. However, primary cultures of bovine chromaffin cells offer advantages that can be useful in many experiments. First, these cells retain their highly differentiated chromaffin phenotype, with dense core secretory (chromaffin) granules distributed throughout the cytosol; in contrast, chromaffin granules in the PC12 line are relatively sparse, and largely docked at the inner face of the plasma membrane. Second, bovine chromaffin cells in primary culture are not transformed; indeed, they seem to be terminally differentiated and incapable of division, and thus may be more representative of the behavior of the normal cells in situ within the gland in vivo. The procedure to obtain such cells described here is quite demanding and time consuming, but over the course of 1–2 d it yields enough cells for several experiments.
Primary culture of bovine chromaffin cells was first accomplished in the late 1970s. Earlier descriptions or reviews of the procedure can be found in refs. 1–13. An especially extensive and systematic review of conditions for chromaffin cell culture, including the optimization of several important parameters, is given by Livett4. Later perspectives on chromaffin cell culture can be found in refs. 14–17. This protocol was developed for bovine chromaffin cells because such cells are the usual model system for studies of chromaffin cell biology and because of the proximity of bovine abattoirs to many large cities. If bovine chromaffin cell primary culture is too difficult for a lab, however, because of the distance from the abattoir or lack of equipment, investigators can consider collaborative studies with other laboratories around the world that study bovine cells. Descriptions of recent meetings of chromaffin cell investigators are available on the Internet: http://www.cnv.cl/isccb13/ (2006); http://webpages.ull.es/users/isccb12 (2003); and http://www.medicine.ucsd.edu/hypertension/ISCCB/home.html (2001). Alternatively, different adrenal cell types could be used (as reviewed in ref. 18); for example, many experiments can be conducted in the more convenient system of PC12 cells, a rat pheochromocytoma cell line in use since 1976 (ref. 19). Although other species’ chromaffin cells have been successfully cultured, such cultures are much less routine and are not regularly undertaken by many (if any) laboratories in the world today. The systematic review by Livett4 also covers the culture of other species’ chromaffin cells. For rodent (mouse, rat) chromaffin cells, in particular, cell yields are likely to permit only studies on single cells at a time, but in the age of transgenic mice and targeted gene ablation, such cells are generating heightened interest. An online protocol is available for obtaining rat chromaffin cells in primary culture20.
Once isolated and cultured, chromaffin cells can be the subject of a variety of experimental procedures, biochemical, physiological, pharmacological, radiochemical and morphological, though such further applications of cultured chromaffin cells are not covered in this protocol, which details only methods for their isolation and culture.
It may also be possible to further separate chromaffin cells into different types, such as adrenergic and noradrenergic cells. The majority of adrenomedullary chromaffin cells store and release primarily epinephrine (adrenaline), and a minority of cells release primarily norepinephrine (noradrenaline). Occasionally investigators may wish to study these two cell populations separately, in which case published methods using gradient centrifugation on chromaffin cells before plating can provide some degree (though incomplete) of differential separation and enrichment of the two cell types, on the principle that epinephrine cells tend to be somewhat denser than norepinephrine cells. The gradient types reported include Renografin followed by Percoll (Moro et al.21) or 10–23% BSA (Dumont et al.12).
MATERIALS
REAGENTS
Bovine adrenal glands ! CAUTION In principle, bovine tissues might be contaminated with bovine microbial or viral pathogens, but in practice this is a very-low-risk protocol. The investigator wears gloves during the sterile cell preparation, antibiotics are present at several steps and we have not encountered problems with human infection.
100× penicillin–streptomycin–GIn (Invitrogen, cat. no. 10378-016)
Fungizone (amphotericin B; Invitrogen, cat. no. 15290-018)
DMEM low-glucose medium (Invitrogen, cat. no. 11885-084) (see REAGENT SETUP)
Heat-inactivated FBS (Gemini Bio-Products, cat. no. 100-106)
AraC stock (cytosine arabinoside; 3 mg AraC per 10 ml DMEM; Sigma, cat. no. C1768) ! CAUTION Might be harmful if inadvertently ingested at high dose.
5-FDU stock (5-fluorodeoxyuridine; 24.6 mg FDU per 10 ml Locke’s; Sigma, cat. no. F0503) ! CAUTION Might be harmful if inadvertently ingested at high dose.
Trypan blue stock (Sigma, cat. no. T8154) (see REAGENT SETUP)
0.2% collagenase P (Roche, cat. no. 11213857001) (see REAGENT SETUP)
0.5% BSA (Sigma, cat. no. A9418)
Percoll (sterile solution; GE-Amersham Biosciences, cat. no. 17-0891-01)
Ethanol (95%/190 proof; manufacturer not critical)
KCl
Locke’s buffered Percoll solution (see REAGENT SETUP)
NaHCO3
10× Locke’s buffer (see REAGENT SETUP)
1× Locke’s buffer (see REAGENT SETUP)
Locke’s transportation buffer (supplemented with antifungal agent) (see REAGENT SETUP)
Glucose
HEPES acid
10 M NaOH
Poly-L-Lys
Poly-D-Lys
EQUIPMENT
Spinner flask; either glass (Bellco, cat. no. 1965-00100) or plastic (disposable; Corning, cat. no. 3152)
100-, 250- and 500-ml glass beakers
500-ml bottle
250-μm nylon mesh plus filter container
Glass rod of a Dounce homogenizing vessel ▴ CRITICAL All the above pieces of equipment should be wrapped in aluminum foil and sterilized by autoclaving before their use in sterile cell preparation.
15-cm polystyrene Petri dish
1.5-ml microcentrifuge tubes
50-ml conical tubes (Corning, Cole-Parmer, cat. no. EW-17400-34)
50-ml round-bottom transparent centrifuge tubes (e.g., Nalgene)
24-well tissue-culture-treated polystyrene cell culture plates (e.g., Becton Dickinson-Falcon; Corning-Costar)
10- and 25-ml plastic pipettes ▴ CRITICAL The above plasticware items should be wrapped and sterile.
37 °C, 5% CO2 cell culture incubator
Inverted light microscope
Neubauer hemacytometer (cell counting chamber; e.g., Canemco H4247-1N) with coverglass
Single channel pipetters
Motorized pipetting aid
Cell culture hood (with sterile laminar air flow)
Filtration unit, 0.22-μm pore size
40-μm (BD-Falcon 352340) and 100-μm (BD-Falcon 352360) filters (e.g., single-use BD-Falcon cell strainer; fits into a 50-ml conical tube)
REAGENT SETUP
10× Locke’s buffer
90 g NaCl, 4.17 g KCl, 3.02 g NaHCO3, 10.09 g glucose, 50 ml 1 M HEPES acid. Adjust to pH 7.4 with 10 M NaOH and make up the volume to 1 l with water. Filter-sterilize with 0.22-μm pore-size filter. Store frozen (for months) or refrigerated (for weeks).
1× Locke’s buffer
Dilute 10× Locke’s buffer 10–fold
Locke’s transportation buffer (supplemented with antifungal agent)
1,500 ml 10× Locke’s buffer containing 100 U ml−1 penicillin, 100 μg ml−1 streptomycin (as 100× penicillin–streptomycin–GIn; add 1%, vol/vol) and 0.5 μM fungizone. Filter-sterilize with 0.22-μm pore-size filtration unit and refrigerate. Prepare on the day of the procedure.
DMEM cell culture medium
DMEM low-glucose medium containing 100 U ml−1 penicillin, 100 μg ml−1 streptomycin (as 100× penicillin–streptomycin–GIn; add 1%, vol/vol), 10% heat-inactivated FBS, 1% AraC stock (10 μM final concentration) and 0.1% 5-FDU stock (10 μM final concentration). Filter-sterilize with 0.22-μm pore-size filtration unit. Store frozen (for months) or refrigerated (for weeks).
0.1% trypan blue stock solution in 1× Locke’s solution
Store refrigerated (for weeks).
0.2% collagenase P solution
50 ml 0.2% collagenase P in 1× Locke’s buffer, supplemented with 0.5% BSA. Prepare and filter-sterilize on day of procedure.
Locke’s buffered-Percoll solution
For 20 ml, mix 2 ml sterile 10× Locke’s buffer with 18 ml Percoll.
PROCEDURE
Isolation of bovine chromaffin cells ● TIMING 4–6 h
1| Collect bovine adrenal glands from vendor or bovine abattoir (we usually obtain ten at once). Transport the glands in a sterile 500-ml capped cell culture bottle containing 300-ml 1× Locke’s buffer with fungizone (1:1,000 of 1,000× stock, where stock is 250 μg ml−1 amphotericin B). Keep the bottle on ice.
▴ CRITICAL STEP This protocol is given for three glands; when preparing more glands, adjust volumes accordingly. From Step 2, a sterile technique should be followed: the investigator should wear gloves and work in a cell culture hood. Sterilized equipment should be used, and surgical instruments can be re-sterilized during the procedure by dipping into 100% ethanol in a 500-ml beaker and then flaming.
2| Remove surrounding fat and connective tissue from glands using scissors and wash the glands by perfusing 1 ml 1× Locke’s buffer through the adrenal vein with a pipetter, until the buffer coming out of the gland is clear and free of blood (approximately 10 ml per gland); extensive washing of the gland in this manner helps to diminish red blood cells in the final preparation. Keep the washed glands in 1× Locke’s buffer in a sterile beaker.
3| Inflate each gland three to four times through the adrenal vein with 1 ml 0.2% collagenase P solution (discarding the effluent that leaks out of the gland), and incubate the gland (without additional buffer) for 15 min at 37 °C in a 250–500-ml sterile glass beaker covered with sterile aluminum foil. Repeat this step.
4| To isolate the medullae, snip off lower pole/tip of gland (furthest away from the vein) and cut longitudinally (along the long axis of the gland). On the convex (rounded-out) outer surface incise the gland once longitudinally from top to bottom with scissors, down to the pink medulla, approximately 5 mm or so. Peel away the cortexes gently and place the medullae into a sterile 15-cm diameter Petri dish.
5| Mince the medullae finely with small scissors.
▴ CRITICAL STEP The medullae must be minced finely to obtain a good yield of cells. Mincing should take 8–10 min.
6| Pour the minced medullae into the remaining sterile-filtered 0.2% collagenase P solution in a sterile spinner flask. Rinse the Petri dish a few times with 0.2% collagenase P solution and add to the spinner flask.
7| Incubate the collagenase/medulla mixture with gentle stirring (at a few revolutions per second) for 30 min at 37 °C.
8| Filter the collagenase/medulla mixture through a 250-μm sterile nylon mesh into a sterile glass beaker; if necessary use a sterile glass rod and facilitate filtration by steering the rod while rinsing with 1× Locke’s buffer.
9| Transfer filtrate into a 50-ml conical tube, adjust the volume to 50 ml with 1× Locke’s buffer and gently pellet the cells by centrifuging for 10 min, at room temperature (20–25 °C), at approximately 400g.
10| Carefully remove the supernatant with a pipette and discard. For three glands, re-suspend the cell pellet in 20-ml 1× Locke’s buffer.
11| Filter the cell suspension into a 50-ml sterile conical tube using a 100-μm single-use cell strainer filter.
12| To separate cells using a Percoll gradient, add 20 ml Locke’s-buffered Percoll to 20 ml cell suspension and mix gently (by upending the capped round-bottom centrifuge tube).
13| Centrifuge at 20,000g at room temperature for 20 min. Red blood cells collect near the bottom of the tube. Chromaffin cells band in the middle of the tube (though you may not actually see a discrete chromaffin cell band; rather, a hazy layer), and cellular debris accumulates in the upper portion of the gradient.
14| Remove the debris (approximately one-tenth of the gradient) at the top with a pipette and discard.
15| Collect the middle chromaffin cell layer (approximately 50–60% of the gradient volume, 10–15 ml) with a 10-ml pipette.
▴CRITICAL STEP Some laboratories avoid the Percoll gradient step by repeated (×4) gentle centrifugation steps, during which the chromaffin cells collect at the bottom and the debris and red blood cells can be carefully removed from the side of the tubes and the top of the pellet, respectively.
16| Filter the middle layer of the gradient containing chromaffin cells through a 40-μm single-use cell strainer filter.
17| Put the filtrate (10–15 ml, which contains the cells) into a 500-ml sterile bottle and add 200 ml sterile DMEM low-glucose medium.
▴ CRITICAL STEP No antibiotic or serum is needed at this stage.
18| Aliquot 200 ml into four 50-ml conical tubes and pellet the cells by centrifuging at approximately 400g for 10 min at room temperature.
19| Remove the supernatant carefully and discard. Re-suspend the cell pellet gently in 20 ml DMEM cell culture medium using a motorized pipetter.
Determination of the number of viable cells ● TIMING Approximately 30 min
20| Clean the hemacytometer with ethanol and mount the coverglass over the ruled areas of the two chambers.
21| Prepare an aliquot of the cell sample for counting by adding 100 μl 0.1% trypan blue stock solution (final concentration of 0.05% trypan blue) and 80 μl 1× Locke’s buffer to 20 μl cell suspension in a 1.5-ml microcentrifuge tube.
22| Mix the contents of the tube by gentle agitation by hand. Allow to stand for a few minutes but not longer than 10 min.
23| Fill the counting chambers with the cell solution.
24| Using the microscope, count all unstained cells (viable cells) in the four large corner squares in both counting chambers.
▴ CRITICAL STEP Very small round cells correspond to contaminating red blood cells (though red cells are usually effectively removed by the Percoll gradient); a high percentage of trypan blue–stained cells is indicative of cell death.
25| Calculate number of viable cells as follows: determine average number (x) of viable (unstained) cells per large square (count four large squares and divide by 4). Then use the following formula to calculate total number of cells: total cells = [x] [10,000 (=hemacytometer factor)] [10 (=dilution factor)] [20 ml (see Step 19)].
▴ CRITICAL STEP Expected range is variable, though approximately 10–20 million cells per adrenal gland is a decent yield.
Cell culture ● TIMING 3–7 d
26| Dilute cells to the appropriate density in DMEM cell culture medium.
27| Pipette the cell suspension into the polystyrene plate or dish, with the desired well size. The cells will settle and attach over time in the incubator.
▴ CRITICAL STEP For secretion studies, for example22–25, cells are plated at approximately 250,000 cells per ml per well in 24-well culture plates (each well is 1.5 cm in diameter).
▴ CRITICAL STEP Plating can be scaled up or down to other vessel sizes by adjusting volumes in proportion to the surface area.
28| The cultures are typically maintained under sterile conditions in a 37 °C per 5% CO2 incubator.
▴ CRITICAL STEP Experiments are typically performed 3–7 d after plating, so there is typically no need to change the medium. However, if the medium turns yellow, there may be contamination (usually fungal).
? TROUBLESHOOTING
● TIMING
Steps 1–19, isolation of bovine chromaffin cells: approximately 4–6 h
Steps 20–25, determination of the number of viable cells: approximately 30 min
Steps 26–28, cell culture: 3–7 d
? TROUBLESHOOTING
Sterility, infection and antibiotics
As with any primary culture, the risk of bacterial infection of the cells is high, though to some extent it is mitigated by the antibiotics in culture medium. Bacterial infection is usually manifested early (within 1 d) by fall in medium pH (yellow color), and microscopic appearance of numerous rods in the medium. In our experience, treatment by supplemental antibiotics cannot ‘rescue’ microbial infections in cultured cells. Fungal infection may occur during summer months, has its onset more slowly and is typically associated microscopically with the appearance of mycelia. Amphotericin B (fungizone) may be helpful in preventing fungal infection.
pH/acidity of the culture medium
The pH indicator (phenol red) in buffered DMEM turns yellow as pH falls; such extracellular acidosis typically indicates either exhaustion of nutrients or, more likely, cell death. During the 3–7-d course of cell culture, a change to fresh medium can be attempted once if medium pH falls.
Cell death and viability
Frequent nuclear staining by trypan blue (just before cell plating) indicates that viable cells did not result from the preparation. Possible causes include excessive postmortem delay and contaminants in the collagenase preparation. Excessive postmortem delay can be obviated by prompt transport of the glands to the laboratory; alternatively, transportation for a few hours on ice is usually successful. Suboptimal collagenase preparations can be identified by systematically testing several lots of the enzyme and reserving sufficient quantities of a successful lot for future studies; the enzyme can be stored frozen. If the collagenase is suboptimal and the cells are not substantially dissociated before mincing, this might also lead to mechanical damage; in this context, increasing the collagenase incubation time within the gland before mincing may be helpful.
Collagenase variability
For more than two decades, investigators have commented on the variability of collagenase preparations from source to source, and even from lot to lot4. We have not encountered such difficulties, but a logical approach may be to test multiple lots of collagenase and then purchase a successful lot in bulk for future cell preparations. Some investigators include DNAseI in collagenase digestion solutions; the rationale is that genomic DNA released by dead cells may form aggregates, trapping cells. We have achieved acceptable results with and without using DNAseI, but have not systematically tested its value.
Other cell types in these cultures
Although the Percoll gradient step enriches the presence of chromaffin cells in these cultures, the resulting cell types are still somewhat heterogeneous; other cell types identified in the cultures include endothelial cells26 and adrenal cortical cells27, although specific identification of other cells in a given culture is typically not performed, and such cells are often referred to non-specifically as ‘fibroblasts’. As (unlike chromaffin cells) these other adrenal cells are not terminally differentiated and continue to divide, antimetabolites (AraC and 5-FDU) are usually included to limit growth of non-chromaffin cells. Chromaffin cells can be enriched by differential plating before final plating4 and may be identified4 from the heterogeneous cell mixture using such stains as formaldehyde-induced fluorescence (in which oxidized catecholamines become fluorescent) or neutral red (which visualizes acidic compartments, such as chromaffin granules); however, we do not routinely subject our cultures to such microanatomic investigation. In any event, the investigator should be aware that the cell types are not homogeneous, and events occurring in the culture might not necessarily be attributable to chromaffin cells. If such uncertainty occurs, experiments with clonal cells (such as PC12) may be useful.
Cell growth substrate
We typically plate cells onto tissue-culture-treated polystyrene plates or glass coverslips; if cell attachment is not firm during growth, change of medium, buffer washing or secretion, attachment can be augmented by pretreatment of the polystyrene with sterile poly-Lys solution, by incubating plates with 100 μg ml−1 poly-L-Lys or poly-D-Lys (Sigma) in sterile PBS for 1 h at 37 °C, followed by plate rinse with sterile water. Cells may be plated on glass coverslips coated with a solution of collagen, by incubating coverslips with 100 μg ml−1 rat tail collagen in sterile PBS, for 120 min at room temperature.
Navigating the bovine abattoir
Obtaining access to adrenal glands at the local bovine abattoir may not be entirely straightforward. First, your laboratory may be at a considerable distance from the facility. In our case, the closest facility is now more than 100 miles from our laboratory; hence we employ a commercial courier to retrieve the glands by prearrangement with the facility manager. Even if the abattoir is proximate, it may be difficult, for reasons of liability or insurance, for the investigator to retrieve glands directly from the animals. If this is not possible, a U.S. Department of Agriculture veterinarian is usually stationed as an inspector at each abattoir, who is usually well acquainted with both bovine anatomy and the customs of the work floor of the particular facility and may be able to assist the investigator in obtaining the required material.
ANTICIPATED RESULTS
Within approximately 4–8 d from commencing the study, the investigator should have enough cells for several experiments.
Number of experiments
At a typical yield of approximately 10–20 million cells per gland, starting with three glands and a requirement for approximately 250,000 cells per well, then approximately 120–240 wells will be available for experiments.
Cell performance
Typically cells are best studied within 3–7 d of plating. After that, a variety of factors may hamper the continued functioning of these cells, including microbial infection (despite antibiotics), toxicity of antimetabolites (AraC and 5-FDU), overgrowth by non-chromaffin cells in the culture (despite antimetabolites) or exhaustion of the nutrients within the medium. Exhaustion of the nutrients within the medium can be circumvented by replacement of the medium.
Cell appearance
After cell attachment and growth for 3–7 d, chromaffin cells yield a somewhat heterogeneous appearance on phase-contrast (Fig. 1) and immunofluorescence (Fig. 2a and b) microscopy. The classical appearance of a chromaffin cell in culture is usually described as rounded (see Figure 1); however, specific immunostaining of chromaffin cells in culture results in a sail-like appearance (Fig. 2a and b).
Figure 1.
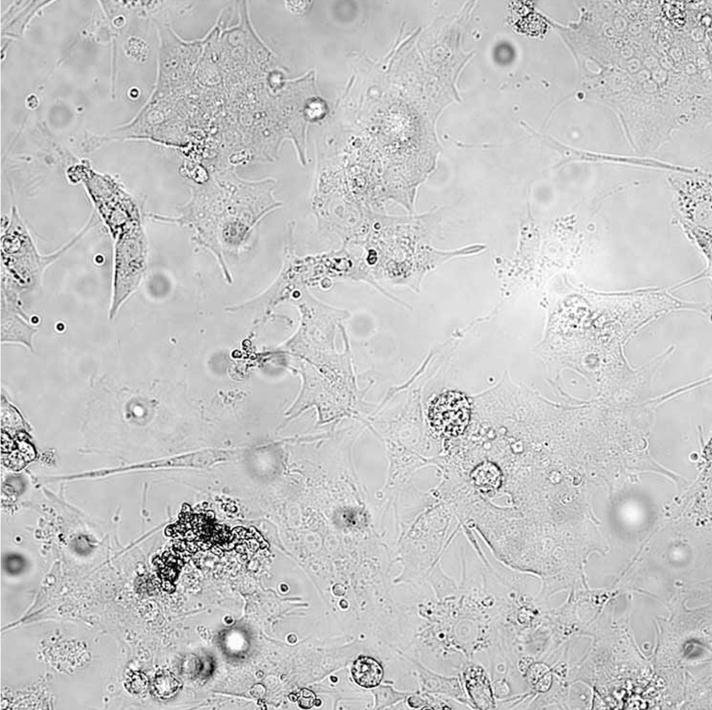
Phase-contrast micrograph of a primary culture of bovine chromaffin cells after cell attachment to the substratum. Note the classical appearance of rounded chromaffin cells. The average diameter of rounded chromaffin cells is ~20 μm.
Figure 2.

Immunofluorescence micrograph of a primary culture of bovine chromaffin cells after cell attachment to the substratum. Dual staining was achieved for the nucleus (with Hoechst dye 33342; in blue) and chromaffin granules (for chromogranin A). Chromogranin A was visualized with rabbit anti-chromogranin A, followed by a secondary antibody (anti-rabbit) coupled to the red dye AlexaFluor 594 (Molecular Probes/Invitrogen). (a) Lower-power field. (b) Higher-power field. The nuclear diameter is approximately 10 μm.
Example application of cultured chromaffin cells: use of cells in catecholamine release studies
Within 3–7 d, cells should be able to take up and store [3H]-L-norepinephrine and then release the catecholamine in response to typical secretagogue stimulation, such as membrane depolarization with 55 mM K+ in the extracellular medium, or 10–20 μM nicotine (acting as a nicotinic cholinergic agonist), over a time course of 10–30 min. In a healthy cell preparation, basal (unstimulated) release of [3H]-L-norepinephrine should be well under 5% of cell total stores, and secretagogue stimulation should yield at least three to four times basal (unstimulated) release. For our results on studies of secretion using bovine chromaffin cells, see refs. 22–25. Further information on studies of catecholamine release, including catecholamine labeling and release protocols is available at http://medicine.ucsd.edu/hypertension.
Footnotes
COMPETING INTERESTS STATEMENT The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Fenwick EM, Fajdiga PB, Howe NB, Livett BG. Functional and morphological characterization of isolated bovine adrenal medullary cells. J Cell Biol. 1978;76:12–30. doi: 10.1083/jcb.76.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hochman J, Perlman RL. Catecholamine secretion by isolated adrenal cells. Biochim Biophys Acta. 1976;421:168–175. doi: 10.1016/0304-4165(76)90180-x. [DOI] [PubMed] [Google Scholar]
- 3.Schneider AS, Herz R, Rosenheck K. Stimulus-secretion coupling in chromaffin cells isolated from bovine adrenal medulla. Proc Natl Acad Sci USA. 1977;74:5036–5040. doi: 10.1073/pnas.74.11.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Livett BG. Adrenal medullary chromaffin cells in vitro. Physiol Rev. 1984;64:1103–1161. doi: 10.1152/physrev.1984.64.4.1103. [DOI] [PubMed] [Google Scholar]
- 5.Livett BG, Boksa P, Dean DM, Mizobe F, Lindenbaum MH. Use of isolated chromaffin cells to study basic release mechanisms. J Auton Nerv Syst. 1983;7:59–86. doi: 10.1016/0165-1838(83)90069-3. [DOI] [PubMed] [Google Scholar]
- 6.Unsicker K, Griesser GH, Lindmar R, Loffelholz K, Wolf U. Establishment, characterization and fibre outgrowth of isolated bovine adrenal medullary cells in long-term cultures. Neuroscience. 1980;5:1445–1460. doi: 10.1016/0306-4522(80)90006-8. [DOI] [PubMed] [Google Scholar]
- 7.Trifaro JM, Lee RW. Morphological characteristics and stimulus-secretion coupling in bovine adrenal chromaffin cell cultures. Neuroscience. 1980;5:1533–1546. doi: 10.1016/0306-4522(80)90018-4. [DOI] [PubMed] [Google Scholar]
- 8.Greenberg A, Zinder O. Alpha- and beta-receptor control of catecholamine secretion from isolated adrenal medulla cells. Cell Tissue Res. 1982;226:655–665. doi: 10.1007/BF00214792. [DOI] [PubMed] [Google Scholar]
- 9.Hesketh JE, Ciesielski-Treska J, Aunis D. A phase-contrast and immunofluorescence study of adrenal medullary chromaffin cells in culture: neurite formation, actin and chromaffin granule distribution. Cell Tissue Res. 1981;218:331–343. doi: 10.1007/BF00210348. [DOI] [PubMed] [Google Scholar]
- 10.Wilson SP, Kirshner N. Preparation and maintenance of adrenal medullary chromaffin cell cultures. Methods Enzymol. 1983;103:305–312. doi: 10.1016/s0076-6879(83)03020-7. [DOI] [PubMed] [Google Scholar]
- 11.Waymire JC, et al. Bovine adrenal chromaffin cells: high-yield purification and viability in suspension culture. J Neurosci Methods. 1983;7:329–351. doi: 10.1016/0165-0270(83)90026-2. [DOI] [PubMed] [Google Scholar]
- 12.Dumont M, Day R, Lemaire S. Distinct distribution of immunoreactive dynorphin and leucine enkephalin in various populations of isolated adrenal chromaffin cells. Life Sci. 1983;32:287–294. doi: 10.1016/0024-3205(83)90042-5. [DOI] [PubMed] [Google Scholar]
- 13.Burgoyne RD. Mechanisms of secretion from adrenal chromaffin cells. Biochim Biophys Acta. 1984;779:201–216. doi: 10.1016/0304-4157(84)90009-1. [DOI] [PubMed] [Google Scholar]
- 14.Livett BG, Borges R. Advances in cell culture for chromaffin cells and related cell types. In: Borges R, Gandia L, editors. Cell Biology of the Chromaffin Cell 2004 Workshop. Laguna; Spain: 2004. pp. 261–267. [Google Scholar]
- 15.Livett BG, Mitchelhill KI, Dean DM. In vitro methods for studying secretion. In: Poisner AM, Trifaro JM, editors. The Secretory Process. Vol. 3. Elsevier; Amsterdam: 1987. pp. 171–204. [Google Scholar]
- 16.Marley PD, Livett BG. Chromaffin cells: model cells for neuronal cell biology. In: Adelman G, Smith BH, editors. Encyclopedia of Neuroscience. Elsevier Science Publishers; Amsterdam: 2004. [Google Scholar]
- 17.Goncalves CA, Gottfried C, Dunkley PR. The use of permeabilized cells to assay protein phosphorylation and catecholamine release. Neurochem Res. 2000;25:885–894. doi: 10.1023/a:1007533927813. [DOI] [PubMed] [Google Scholar]
- 18.Eaton MJ, Duplan H. Useful cell lines derived from the adrenal medulla. Mol Cell Endocrinol. 2004;228:39–52. doi: 10.1016/j.mce.2003.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilabert JA, Montalvo GB, Artalejo AR. Rat chromaffin cells primary cultures: standardization and quality assessment for single-cell assays. Nat Protoc. 2006 doi: 10.1038/nprot.2006.294. http://www.natureprotocols.com/2006/09/29/rat_chromaffin_cells_primary_c.php. [DOI]
- 21.Moro MA, Lopez MG, Gandia L, Michelena P, Garcia AG. Separation and culture of living adrenaline- and noradrenaline-containing cells from bovine adrenal medullae. Anal Biochem. 1990;185:243–248. doi: 10.1016/0003-2697(90)90287-j. [DOI] [PubMed] [Google Scholar]
- 22.Lee JC, et al. Primary sequence characterization of catestatin intermediates and peptides defines proteolytic cleavage sites utilized for converting chromogranin A into active catestatin secreted from neuroendocrine chromaffin cells. Biochemistry. 2003;42:6938–6946. doi: 10.1021/bi0300433. [DOI] [PubMed] [Google Scholar]
- 23.Mahata SK, et al. Novel autocrine feedback control of catecholamine release. A discrete chromogranin a fragment is a noncompetitive nicotinic cholinergic antagonist. J Clin Invest. 1997;100:1623–1633. doi: 10.1172/JCI119686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barbosa JA, Gill BM, Takiyyuddin MA, O’Connor DT. Chromogranin A: posttranslational modifications in secretory granules. Endocrinology. 1991;128:174–190. doi: 10.1210/endo-128-1-174. [DOI] [PubMed] [Google Scholar]
- 25.Takiyyuddin MA, et al. Is physiologic sympathoadrenal catecholamine release exocytotic in humans? Circulation. 1990;81:185–195. doi: 10.1161/01.cir.81.1.185. [DOI] [PubMed] [Google Scholar]
- 26.Banerjee DK, Ornberg RL, Youdim MB, Heldman E, Pollard HB. Endothelial cells from bovine adrenal medulla develop capillary-like growth patterns in culture. Proc Natl Acad Sci USA. 1985;82:4702–4706. doi: 10.1073/pnas.82.14.4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirshner N, Corcoran JJ, Erickson HP. Synthesis of alpha 2-macroglobulin by bovine adrenal cortical cell cultures. Am J Physiol. 1989;256:C779–C785. doi: 10.1152/ajpcell.1989.256.4.C779. [DOI] [PubMed] [Google Scholar]