

Abstract
Background:
Pathogenic mutations in rapsyn result in endplate acetylcholine receptor (AChR) deficiency and are a common cause of postsynaptic congenital myasthenic syndromes.
Methods:
Clinical, electrophysiologic, pathologic, and molecular studies were done in 39 patients.
Results:
In all but one patient, the disease presented in the first 2 years of life. In 9 patients, the myasthenic symptoms included constant or episodic ophthalmoparesis, and 1 patient had a pure limb-girdle phenotype. More than one-half of the patients experienced intermittent exacerbations. Long-term follow-up was available in 25 patients after start of cholinergic therapy: 21 became stable or were improved and 2 of these became asymptomatic; 3 had a progressive course; and 1 died in infancy. In 7 patients who had endplate studies, the average counts of AChR per endplate and the synaptic response to ACh were less reduced than in patients harboring low AChR expressor mutations. Eight patients were homozygous and 23 heterozygous for the common p.N88K mutation. Six mutations, comprising 3 missense mutations, an in-frame deletion, a splice-site mutation, and a nonsense mutation, are novel. Homozygosity for p.N88K was associated with varying grades of severity. No genotype-phenotype correlations were observed except in 8 Near-Eastern patients homozygous for the promoter mutation (c.-38A>G), who had a mild course.
Conclusions:
All but 1 patient presented early in life and most responded to cholinergic agonists. With early diagnosis and therapy, rapsyn deficiency has a benign course in most patients. There was no consistent phenotype-genotype correlation except for an E-box mutation associated with jaw deformities.
GLOSSARY
- 3,4-DAP
= 3,4-diaminopyridine;
- α-bgt
= α-bungarotoxin;
- AChR
= acetylcholine receptor;
- CMAP
= compound muscle action potential;
- CMS
= congenital myasthenic syndrome;
- EP
= endplate;
- MEPC
= miniature endplate currents;
- MEPP
= miniature endplate potentials;
- TPR
= tetratricopeptide.
The congenital myasthenic syndromes (CMS) are heterogenous disorders of neuromuscular transmission caused by defects in presynaptic, synaptic, and postsynaptic proteins of the neuromuscular junction.1 Among the 295 patients with CMS investigated at the Mayo Clinic, 15% carry mutations in rapsyn, a postsynaptic protein.1
Rapsyn binds to the long cytoplasmic loop of the AChR subunits2 and is essential for clustering and anchoring AChR in the postsynaptic membrane. Rapsyn is composed of several functionally distinct regions (figure 1A): a myristoylated N-terminal is required for membrane interaction3; 7 tetratricopeptide repeats (TPR) are important for rapsyn self-aggregation4 and binding to the cytoplasmic portion of the muscle-specific kinase MuSK5; the coiled-coil domain interacts with the cytoplasmic loops of AChR subunits6; and the C-terminal domain binds to β dystroglycan and thereby links the rapsyn-AChR complex to the cytoskeleton.7 Mutations in rapsyn compromise the safety margin of neuromuscular transmission by causing endplate (EP) AChR deficiency.8

Figure 1 Genetic analysis of rapsyn
(A) Rapsyn domains and interactions. (B) Schematic representations of the RAPSN mutations identified in the 39 patients investigated at the Mayo Clinic. The novel mutations are indicated in red. (C) The mutated residues in the coding region are conserved across species.
The clinical spectrum of rapsyn-CMS varies from severe hypotonia and arthrogryposis at birth to mild limb muscle weakness. Early and late-onset phenotypes have been described.9 Episodic respiratory crises and lack of ophthalmoparesis have been reported as hallmarks of rapsyn-CMS that differentiate it from the CMS caused by low expressor mutations in the AChR ɛ subunit.10 Here we review our findings in 39 rapsyn-CMS patients investigated at the Mayo Clinic.
METHODS
This study includes 21 male subjects and 18 female subjects with rapsyn-CMS. All human studies were in accord with the guidelines of the Institutional Review Board of the Mayo Clinic. Written consent was obtained from all patients participating in the study. Nineteen of the patients were seen at the Mayo Clinic. Additional information came from follow-up letters from referring physicians or correspondence from patients regarding their management. In 20 patients, the clinical and EMG data were obtained from other medical centers.
All patients underwent neurologic examination and 32 had EMG studies using 2-3 Hz repetitive nerve stimulations of selected proximal, distal, or facial muscles, or single fiber EMG. Seven patients underwent intercostal or anconeus muscle biopsies for EP studies by in vitro microelectrode and electron microscopy studies, as described.11,12 The number of AChRs per EP was estimated with 125I-labeled-α-bungarotoxin11 and acetylcholinesterase was visualized on glutaraldehyde-fixed teased single muscle fibers by Gautron’s method.13 All 8 exons and flanking untranslated regions of RAPSN including 800 bp upstream of the translational start site were sequenced as previously described.14 We used allele-specific PCR to screen for new mutations in the proband’s relatives and in 100 unrelated control subjects. PolyPhen (http://genetics.bwh.harvard.edu/pph/index.html) software was used to predict the possible effect of the missense mutations.
RESULTS
Phenotype.
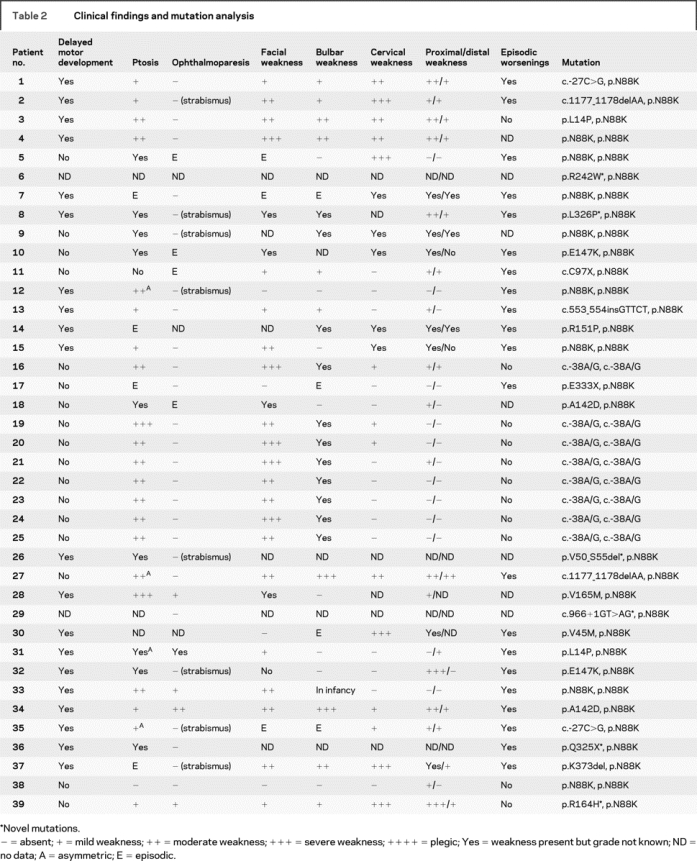
Table 1 shows the presenting symptoms as well as age at onset, age at diagnosis, and age at the last follow-up. Table 2 indicates other clinical features of the disease. Thirteen patients (patients 3, 4, 5, 13,8 1, 19, 20, 21, 22, 23, 24, 25,14 and 215) had been previously reported. Seven of the 20 patients whose disease presented at birth had decreased fetal movements. Eight patients with symptoms at birth were born by caesarian section due to breech presentation and 4 of these had decreased fetal movements. Only 6 patients (patients 10, 11, 18, 31, 32, and 39) reported intermittent diplopia. Three patients (patients 3, 37, and 39) had mild and one (patient 5) had severe torso muscle weakness. Twenty patients had episodic exacerbations precipitated by infection, fever, or no obvious trigger, and 12 of these required ventilatory support during one or more exacerbations. Four patients had dyspnea at rest. The tendon reflexes were normally active or mildly decreased. Eight patients with onset of symptoms at birth had arthrogryposis, 10 had a high arched palate, 3 developed scoliosis, and 2 had lordosis. Eight Near-Eastern patients (patients 16 and 19-25), 3 from consanguineous marriages, and patient 27 had facial asymmetry and mandibular prognathism. One non-Near-Eastern patient (patient 2) also had mandibular prognathism, elongated face, and low-set malformed ears. Other dysmorphic features were small jaw (patient 28) and small chin (patient 29). Eleven patients (patients 3, 4, 6, 9, 10, 18, 19, 20, 21, 30, and 31) had a similarly affected sibling.
Table 1 Presenting symptoms, age (in years) at onset, at diagnosis, and at last follow-up

Table 2 Clinical findings and mutation analysis
Thirty-six patients were treated with pyridostigmine (Mestinon; Valeant Pharmaceuticals International, Aliso Viejo, CA) and all responded positively. In 11 patients, addition of 3,4-diaminopyridine (3.4-DAP; Jacobus Pharmaceutical Co., Princeton, NJ) resulted in further clinical improvement. Patient 2 responded incompletely to pyridostigmine and 3,4-DAP, but improved further on receiving ephedrine.15
Follow-up information was available in 25 patients after start of therapy with cholinergic agents. The mean follow-up time after start of therapy was 6.6 years (range 0.5-18 years). In this subset of patients, 21 became stable or were improved except for intermittent worsening, but the exacerbations decreased in frequency and severity and 2 became asymptomatic. Three patients had a progressive worsening: patient 1 experienced increased weakness after pregnancy associated with much weight gain; she improved with weight loss but still remained weaker than previously. Patient 2 experienced progressive worsening of dysphagia and fatigability between 6 and 11 years of age, and then became stable. Patient 39 became progressively weaker between the first and fifth decade of life and was wheelchair-dependent when last examined at the age of 45 years. Despite combined therapy with pyridostigmine (360 mg/day), 3,4-DAP (1 mg/kg/day), and albuterol extended release (Vospire ER; DAVA Pharmaceuticals, Fort Lee, NJ) (4 mg/day), he can take only a few steps independently. Patient 29 died at age 1 of respiratory failure.
EMG studies.
These were done in 32 patients. A decremental response on 2-3 Hz stimulation was detected in 23 but in patient 28 this appeared only after exercise. In 3 patients (patients 13, 15, and 30), a decremental response at 2 Hz appeared only after 10-Hz stimulation for 5 minutes. Five patients (patients 12, 24, 25, 30, and 38) with no EMG decrement at rest had abnormal jitter on single-fiber EMG. IV edrophonium (Tensilon; Baxter Healthcare, Deerfield, IL) partially or fully repaired the EMG decrement in 5 patients (patients 3, 10, 17, 27, and 34). In 6 patients (patients 1, 4, 13, 17, 27, and 39), the decrement was also improved by an oral dose of 3,4-DAP. The compound muscle action potentials were of normal amplitude in all. Needle EMG examination, performed in 11, showed rapid recruitment of short duration and unstable motor unit potentials in 9. Spontaneous muscle activity was not observed in any patient.
Endplate studies.
Between 1992 and 2008, 7 patients underwent intercostal or anconeus muscle biopsies for in vitro electrophysiologic and morphologic studies. The number of AChRs per EP was between 19% and 55% of normal; mean 39% of normal. All had decreased MEPP amplitude (12% to 47% of normal; mean 36%) and a decreased MEPC amplitude (20% to 80% of normal; mean 38%). The mean number of AChRs per EP varied from 16% to 80% of normal. Quantal release by nerve impulse at 1 Hz fell in the normal range, except in 2 patients in whom it was increased to 60 and 66 (normal mean ± SE: 31 ± 1; range: 17-46; n = 11). Single-channel studies of the EP in 7 patients revealed no kinetic abnormality of the AChR channel.
All muscle specimens showed type 1 fiber predominance without myopathic features. Teased single muscle fibers displayed multiple small EP regions distributed along an elongated length of the fibers (figure 2A). In each patient, electron microscopy studies revealed simplified postsynaptic regions with no or only few junctional folds (figure 2, B and C), yet the structural integrity of the postsynaptic region was preserved. The distribution of AChR on the simplified postsynaptic membrane was patchy and discontinuous (figure 2B).

Figure 2 Structural features of rapsyn-deficient endplates (EPs)
(A) Small cholinesterase reactive EP regions are dispersed over an extended length of the muscle fiber. These synaptic contacts differ from the compact pretzel-shaped contacts at normal EPs. (B, C) Multiple small nerve terminals are apposed against highly simplified postsynaptic regions with no (B) or few (C) junctional folds. In (B), the distribution of AChR on the postsynaptic membrane, visualized with peroxidase-labeled α-bungarotoxin, is patchy. Bars 50 μm in (A) and 1 μm in (B) and (C).
Mutation analysis.
Results of the mutation analysis are summarized in table 2 and figure 1B. Six patients (patients 7, 9, 27, 30, 37, and 40) carried p.N88K and a second novel mutation. None of the novel mutations was detected in 100 unrelated control subjects. Analysis by the PolyPhen software predicted R242W and L326P probably deleterious, and R164H possibly deleterious. Each unaffected parent carries a RAPSN mutation on one allele, and the unaffected siblings carry one or no mutation. Affected siblings of 11 patients (patients 3, 4, 6, 9, 10, 18, 19, 20, 21, 30, and 31) carry the same mutations as the proband. The 8 patients (patients 16 and 19-25) homozygous for the c.-38A>G E-box mutation in the rapsyn promoter region were all from the Near-East and had mandibular prognathism and a mild clinical course.
DISCUSSION
In 38 patients and in their affected siblings, the disease manifested at birth or in the first 2 years of life. Only one patient presented at 5 years of age. This contrasts with previous reports of onset as late as the fifth decade of life.9,16 Most patients had ptosis of varying severity; in 5 it was asymmetric, a finding thought to be uncommon in congenital myasthenias.17 As previously observed,9,18 strabismus was relatively common, but 9 patients in our series have had constant or episodic ophthalmoparesis. Therefore, ophthalmoparesis is not a reliable negative criterion for distinguishing rapsyn-CMS from CMS caused by low expressor mutations in the AChR ɛ subunit as previously suggested.9 Still, except for the CMS caused by mutations in DOK7, ophthalmoparesis is less common than in other forms of CMS. Facial and bulbar weakness were common, often associated with neck muscle weakness. Proximal muscle weakness was as or more severe than distal weakness. One patient had only mild distal but no proximal weakness, but his proximal muscles fatigued abnormally on exertion and he also had ptosis, ophthalmoparesis, and facial weakness. Out-of-proportion weakness of the foot dorsiflexors was reported a feature of the late-onset phenotype,9 but it was not detected in our series of early onset patients. Intermittent exacerbations occurred in more than half of the patients. Arthrogryposis manifesting at birth occurred in less than a third of the patients.
Patient 38 with a homozygous N88K mutation had limb-girdle weakness and abnormal fatigability present at least since the age of 5 years. He had no ocular, facial, or bulbar symptoms, intermittent exacerbations, and his clinical course was stable. His muscle biopsy showed no tubular aggregates. This patient’s case extends the genetic causes of the congenital limb-girdle myasthenias beyond mutations in Dok-7, or with tubular aggregates in muscle with no mutations in Dok-7.19–21
Not all patients had a decremental EMG response at rest. Six with mild weakness of muscles accessible for repetitive nerve stimulations had none, but in 5 of these single-fiber EMG demonstrated abnormal jitter and blocking. Three patients with mild weakness had a transient decremental EMG response at 2 Hz immediately after a conditioning train at 10 Hz for 5 minutes. This is a nonspecific finding in different types of CMS; it is specific only if it persists for 5-10 minutes after stimulation, in which case it would point to a defect in choline acetyltransferase.22
The in vitro electrophysiologic and electron microscopy studies of intercostal or anconeus muscle EPs in 7 patients showed multiple small synaptic contacts dispersed over an extended length of the muscle fiber. Electron microscopy revealed EPs with few or no junctional folds, and a patchy distribution of AChR on the highly simplified postsynaptic membrane. The safety margin of neuromuscular transmission in rapsyn-CMS is compromised by the decreased amplitude of the MEPP which is due to the decreased number of AChRs per EP, and by the paucity of junctional folds which decreases the input resistance of the postsynaptic membrane and hence the amplitude of the MEPP.23 Interestingly, the decrease in the number of AChRs per EP (mean decrease 32% of normal) is less marked than in patients with low-expressor mutations in AChR subunits in whom it is typically less than 10% of normal. This likely explains why the amplitude of the MEPP and MEPC is not as markedly reduced, and why in some patients the EMG decrement is more difficult to detect, than in patients with low-expressor mutations of the AChR.
Since the discovery that mutations in rapsyn cause a CMS,8 a total of 45 rapsyn mutations have been identified, 15 by us14,15,24 and 30 by other investigators.9,10,16,18,25–31 The mutations are dispersed through the entire RAPSN gene. N88K occurred with high frequency, as observed in other series.9,18,26,32 Six mutations reported here are novel and family analysis indicates they are recessive.
The mutated arginine at codon 164 (p.R164H) and 242 (p.R242W), and the mutated leucine at codon 326 (p.L326P), are highly conserved among species (figure 1C).
p.R164H is located in TPR5. Another mutation at the same location (p.R164C) was shown to reduce co-clustering of AChR with rapsyn by about 50%.29 Although arginine and histidine are both positively charged amino acids, histidine has an imidazole group which likely affects the secondary structure of rapsyn.
p.R242W is located in the linker region between TPR6 and TPR7. Replacing the positively charged arginine with the larger nonpolar tryptophan may distort the structure of the close TPR7, interfering with its role in co-clustering of AChR with rapsyn. A new mutation was previously reported in rapsyn-CMS 4 amino acids downstream (p.A246V).18
p.L326P is the first mutation observed in the coiled-coil domain (298-331) of rapsyn. This domain interacts with the long cytoplasmic loop of each AChR subunit and is essential for clustering AChRs.4 Leucine at position 326 contributes to the continuous hydrophobic surface of the coiled-coil domain. The integrity of this hydrophobic surface has been shown to be essential for AChR clustering by site-directed mutagenesis studies.4
Regarding the novel p.V50_S55del, 3 of the 5 deleted amino acids are conserved across species (figure 1C). The deletion is located in the TPR2, one of the most important domains for rapsyn self-clustering. The deletion may also affect the folding of the protein. Rapsyn mutant lacking TPR2 does not form clusters efficiently and exerts a dominant negative effect when expressed with wild type rapsyn by disrupting rapsyn self-association.33
The nonsense mutation p.Q325X results in truncated rapsyn lacking part of the coiled coil domain and the C-terminal Ring-H2 that mediates association of rapsyn with AChR subunits and dystroglycan.4,7 The mutant rapsyn also lacks the extreme C-terminus essential for MuSK-induced tyrosine phosphorylation of the AChR β subunit.34
The splice-site mutation c.966 + 1GT>AG may result in exon skipping, intron retention, creation of an intronic pseudo-exon, or activation of cryptic splice site.35 Point mutations at position IVS + 1G>A more likely lead to cryptic 5′ splice sites.36 Therefore, the splice site likely affects the coiled-coil and the C-terminal RING-H2 domains of rapsyn.
We found no genotype-phenotype correlation except the association of the homozygous c.-38A>G rapsyn E-box mutation in Near Eastern patients with mandibular prognathism and a mild clinical course.14,37 In particular, heterozygous truncation mutations are not associated with a more severe phenotype and the phenotypic spectrum of p.N88K homozygosity in our series encompasses respiratory failure at birth (patient 4), mild ptosis with intermittent exacerbations (patient 12), and pure limb-girdle weakness (patient 38). Arthrogryposis occurred in patients with missense, frameshift, in-frame, and promoter region mutations. The type of mutation did not correlate with the clinical course, which included stability, improvement, or progressive worsening. The type of mutation also did not correlate with the extent of the EMG decrement or with the decreased amplitude of the MEPP and MEPC.
ACKNOWLEDGMENT
The authors thank Drs. B. Banwell, L. Bird, P. Bourque, D. Buckley, I. Butler, F. Cornelio, D. Gendron, D. Gill, E. Goldstein, P. Jacob, N. Kuntz, V. Lewis, W. Marks, M. Sadeh, D. Morris, H. Mussell, J. Nemeth, K. North, R. Ouvrier, M. Pollack, S. Robb, M. Ryan, R. Stone, and T. Williams for patient referral and follow-up.
DISCLOSURE
Dr. Milone reports no disclosures. Dr. Shen reports no disclosures. D. Selcen receives research support from NIH/NINDS [K08, NS50106]. Dr. Ohno reports no disclosures. Mrs. Brengman reports no disclosures. Dr. Iannaccone served on the Muscular Dystrophy Advisory Board of NicOx; serves on the Editorial Boards of Neuromuscular Disorders, Journal of Clinical Neuromuscular Disease, and Journal of Pediatric Rehabilitation Medicine; receives research support from PTC Therapeutics for clinical trial of PTC124 in Duchenne muscular dystrophy [Site PI], from NIH for clinical trial of phenylbutyrate in spinal muscular atrophy [Site PI], and from the SMA Foundation, North American Charcot-Marie Tooth Network, and the Muscular Dystrophy Association. Dr. Harper reports no disclosures. Dr. Engel serves as Associate Editor for Neurology®; receives royalties from McGraw-Hill for editing the 3rd edition of Myology; received honorarium for editing volume 91 of the Handbook of Clinical Neurology, 3rd series; and receives research support from NIH [NS6277] and the Muscular Dystrophy Association.
Address correspondence and reprint requests to Dr. Margherita Milone, Department of Neurology, Mayo Clinic, 200 First Street SW, Rochester, MN 55905 Milone.Margherita@mayo.edu
Supported by NIH grant 6277 and by a research grant from the Muscular Dystrophy Association to Dr. Engel.
Disclosure: Author disclosures are provided at the end of the article.
Received February 16, 2009. Accepted in final form April 13, 2009.
REFERENCES
- 1.Engel AG, Shen XM, Selcen D, Sine SM. Further observations in congenital myasthenic syndromes. Ann NY Acad Sci 2008;1132:104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maimone MM, Enigk RE. The intracellular domain of the nicotinic acetylcholine receptor alpha subunit mediates its coclustering with rapsyn. Mol Cell Neurosci 1999;14:340–354. [DOI] [PubMed] [Google Scholar]
- 3.Ramarao MK, Cohen JB. Mechanism of nicotinic acetylcholine receptor cluster formation by rapsyn. Proc Natl Acad Sci USA 1998;95:4007–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramarao MK, Bianchetta MJ, Lanken J, Cohen JB. Role of rapsyn tetratricopeptide repeat and coiled-coil domains in self-association and nicotinic acetylcholine receptor clustering. J Biol Chem 2001;276:7475–7483. [DOI] [PubMed] [Google Scholar]
- 5.Antolik C, Catino DH, Resneck WG, Bloch RJ. The tetratricopeptide repeat domains of rapsyn bind directly to cytoplasmic sequences of the muscle-specific kinase. Neuroscience 2006;141:87–100. [DOI] [PubMed] [Google Scholar]
- 6.Maimone MM, Merlie JP. Interaction of the 43 kd postsynaptic protein with all subunits of the muscle nicotinic acetylcholine receptor. Neuron 1993;11:53–66. [DOI] [PubMed] [Google Scholar]
- 7.Bartoli M, Ramarao MK, Cohen JB. Interactions of the rapsyn RING-H2 domain with dystroglycan. J Biol Chem 2001;276:24911–24917. [DOI] [PubMed] [Google Scholar]
- 8.Ohno K, Engel AG, Shen XM, et al. Rapsyn mutations in humans cause endplate acetylcholine-receptor deficiency and myasthenic syndrome. Am J Hum Genet 2002;70:875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burke G, Cossins J, Maxwell S, et al. Rapsyn mutations in hereditary myasthenia: distinct early- and late-onset phenotypes. Neurology 2003;61:826–828. [DOI] [PubMed] [Google Scholar]
- 10.Burke G, Cossins J, Maxwell S, et al. Distinct phenotypes of congenital acetylcholine receptor deficiency. Neuromuscul Disord 2004;14:356–364. [DOI] [PubMed] [Google Scholar]
- 11.Engel AG, Nagel A, Walls TJ, Harper CM, Waisburg HA. Congenital myasthenic syndromes: I. Deficiency and short open-time of the acetylcholine receptor. Muscle Nerve 1993;16:1284–1292. [DOI] [PubMed] [Google Scholar]
- 12.Milone M, Hutchinson DO, Engel AG. Patch-clamp analysis of the properties of acetylcholine receptor channels at the normal human endplate. Muscle Nerve 1994;17:1364–1369. [DOI] [PubMed] [Google Scholar]
- 13.Gautron J. [Localization of cholinesterases at the level of the nerve-electroplate junction in mottled torpedo.] C R Acad Sci Hebd Seances Acad Sci D 1970;271:714–717. [PubMed] [Google Scholar]
- 14.Ohno K, Sadeh M, Blatt I, Brengman JM, Engel AG. E-box mutations in the RAPSN promoter region in eight cases with congenital myasthenic syndrome. Hum Mol Genet 2003;12:739–748. [DOI] [PubMed] [Google Scholar]
- 15.Banwell BL, Ohno K, Sieb JP, Engel AG. Novel truncating RAPSN mutations causing congenital myasthenic syndrome responsive to 3,4-diaminopyridine. Neuromuscul Disord 2004;14:202–207. [DOI] [PubMed] [Google Scholar]
- 16.Cossins J, Burke G, Maxwell S, et al. Diverse molecular mechanisms involved in AChR deficiency due to rapsyn mutations. Brain 2006;129:2773–2783. [DOI] [PubMed] [Google Scholar]
- 17.Beeson D, Hantai D, Lochmuller H, Engel AG. 126th International Workshop: congenital myasthenic syndromes, 24-26 September 2004, Naarden, the Netherlands. Neuromuscul Disord 2005;15:498–512. [DOI] [PubMed] [Google Scholar]
- 18.Muller JS, Mildner G, Muller-Felber W, et al. Rapsyn N88K is a frequent cause of congenital myasthenic syndromes in European patients. Neurology 2003;60:1805–1810. [DOI] [PubMed] [Google Scholar]
- 19.Beeson D, Higuchi O, Palace J, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science 2006;313:1975–1978. [DOI] [PubMed] [Google Scholar]
- 20.Selcen D, Milone M, Shen XM, et al. Dok-7 myasthenia: phenotypic and molecular genetic studies in 16 patients. Ann Neurol 2008;64:71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodolico C, Toscano A, Autunno M, et al. Limb-girdle myasthenia: clinical, electrophysiological and morphological features in familial and autoimmune cases. Neuromuscul Disord 2002;12:964–969. [DOI] [PubMed] [Google Scholar]
- 22.Ohno K, Tsujino A, Brengman JM, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci USA 2001;98:2017–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wood SJ, Slater CR. The contribution of postsynaptic folds to the safety factor for neuromuscular transmission in rat fast- and slow-twitch muscles. J Physiol 1997;500:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohno K, Engel AG. Lack of founder haplotype for the rapsyn N88K mutation: N88K is an ancient founder mutation or arises from multiple founders. J Med Genet 2004;41:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunne V, Maselli RA. Identification of pathogenic mutations in the human rapsyn gene. J Hum Genet 2003;48:204–207. [DOI] [PubMed] [Google Scholar]
- 26.Richard P, Gaudon K, Andreux F, et al. Possible founder effect of rapsyn N88K mutation and identification of novel rapsyn mutations in congenital myasthenic syndromes. J Med Genet 2003;40:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ioos C, Barois A, Richard P, Eymard B, Hantai D, Estournet-Mathiaud B. Congenital myasthenic syndrome due to rapsyn deficiency: three cases with arthrogryposis and bulbar symptoms. Neuropediatrics 2004;35:246–249. [DOI] [PubMed] [Google Scholar]
- 28.Muller JS, Abicht A, Christen HJ, et al. A newly identified chromosomal microdeletion of the rapsyn gene causes a congenital myasthenic syndrome. Neuromuscul Disord 2004;14:744–749. [DOI] [PubMed] [Google Scholar]
- 29.Muller JS, Baumeister SK, Rasic VM, et al. Impaired receptor clustering in congenital myasthenic syndrome with novel RAPSN mutations. Neurology 2006;67:1159–1164. [DOI] [PubMed] [Google Scholar]
- 30.Maselli RA, Dris H, Schnier J, Cockrell JL, Wollmann RL. Congenital myasthenic syndrome caused by two non-N88K rapsyn mutations. Clin Genet 2007;72:63–65. [DOI] [PubMed] [Google Scholar]
- 31.Michalk A, Stricker S, Becker J, et al. Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am J Hum Genet 2008;82:464–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maselli RA, Dunne V, Pascual-Pascual SI, et al. Rapsyn mutations in myasthenic syndrome due to impaired receptor clustering. Muscle Nerve 2003;28:293–301. [DOI] [PubMed] [Google Scholar]
- 33.Eckler SA, Kuehn R, Gautam M. Deletion of N-terminal rapsyn domains disrupts clustering and has dominant negative effects on clustering of full-length rapsyn. Neuroscience 2005;131:661–670. [DOI] [PubMed] [Google Scholar]
- 34.Lee Y, Rudell J, Yechikhov S, Taylor R, Swope S, Ferns M. Rapsyn carboxyl terminal domains mediate muscle specific kinase-induced phosphorylation of the muscle acetylcholine receptor. Neuroscience 2008;153:997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rogan PK, Faux BM, Schneider TD. Information analysis of human splice site mutations. Hum Mutat 1998;12:153–171. [DOI] [PubMed] [Google Scholar]
- 36.Buratti E, Chivers M, Kralovicova J, et al. Aberrant 5′ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res 2007;35:4250–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldhammer Y, Blatt I, Sadeh M, Goodman RM. Congenital myasthenia associated with facial malformations in Iraqi and Iranian Jews. A new genetic syndrome. Brain 1990;113:1291–1306. [DOI] [PubMed] [Google Scholar]