

Abstract
Ceftobiprole is a cephalosporin with potent activity against methicillin (meticillin)-resistant Staphylococcus aureus (MRSA). In order to treat patients with severe staphylococcal pneumonia, it is important to understand the drug exposure required to mediate the killing of multiple log10 cells in a preclinical-infection model. We measured drug exposure in terms of the percentage of penetration of the drug into epithelial lining fluid (ELF) and in terms of the time for which the drug concentration was above the MIC (time>MIC) in plasma and ELF. In a murine model of staphylococcal pneumonia, we demonstrated that ceftobiprole penetrated into ELF from the plasma at a median level of nearly 69% (25th to 75th percentile range, 25 to 187%), as indexed to the ratio of values for the area under the concentration-time curve in ELF and plasma. The total-drug times>MIC in ELF that were required to kill 1 log10 and 2 log10 CFU/g of lung tissue were 15% and 25% of the dosing interval. We also examined the penetration of ELF by ceftobiprole in volunteers, demonstrating mean and median penetration percentages of 25.5% and 15.3%, respectively (25th to 75th percentile range, 8 to 30%). Attainment rates were calculated for kill targets of 1 log10 and 2 log10 CFU/g, taken from the murine model, but using the volunteer ceftobiprole ELF penetration data. The standard dose for ceftobiprole is 0.5 g every 8 h as a 2-h infusion. The attainment rates remained above 90% for 1-log10 and 2-log10 CFU/g kill targets at MICs of 1 and 0.5 mg/liter, respectively. Taking the expectation over the distribution of ceftobiprole MICs for 4,958 MRSA isolates showed an overall target attainment of 85.6% for a 1-log10 CFU/g kill and 79.7% for a 2-log10 CFU/g kill. It is important to derive exposure targets in preclinical-infection models of the infection site so that these targets can be explored in clinical trials in order to optimize the probability of a good clinical outcome.
A critical but little-studied issue is the exposure targets required at different infection sites. As an example, because of penetration issues due to the blood-brain barrier, one would not expect a priori that the dose choice that would be successful for a skin or soft-tissue infection would be adequate for meningitis therapy. Similarly, identifying the minimal amount of drug exposure necessary for therapy in a mouse thigh infection model may not provide good guidance for the therapy of pneumonia. It is critical to identify the concentration-time profile of a drug at the primary infection site of interest and to link this to the microbiological effect in our preclinical models.
For β-lactam antibiotics, the time for which the concentration of free drug is above the MIC (free-drug time>MIC) is most closely linked to outcome (2, 8). Surprisingly, β-lactam antibiotics show a broad range of abilities to penetrate into the lung, as measured by their concentrations in epithelial lining fluid (ELF). For instance, ceftazidime has 21% penetrance (4), while cefepime's penetration exceeds 100% (3). Likewise, there may well be differences across species. It is well known that macrolides have differential penetration into ELF, depending on the species studied (17, 20). It may also be that the half-time of concentration at the infection site is also important to the outcome.
Ceftobiprole is a new cephalosporin with good activity against methicillin (meticillin)-resistant Staphylococcus aureus (MRSA) (1). Because this pathogen is a major problem in hospital- and ventilator-associated pneumonia and is an emerging problem in community-acquired pneumonia, we believed that it was important to study the penetration and microbiological activity of ceftobiprole in a murine pneumonia model, to define the exposure targets associated with good microbiological activity in this model, to determine the penetration of this drug into ELF in humans, and thereby to evaluate the utility of standard ceftobiprole drug doses for MRSA pneumonia in humans.
MATERIALS AND METHODS
For the murine pneumonia model employed, the methods have been fully defined previously (7). All animal experimentation was approved by the Hartford Hospital IACUC.
Antimicrobials.
Ceftobiprole (BAL9141) and ceftobiprole medocaril (BAL5788; the prodrug of ceftobiprole) were supplied by Johnson & Johnson Pharmaceutical Research & Development (Raritan, NJ) for in vitro and in vivo experiments, respectively.
Bacteria.
As previously reported (14), eight strains of S. aureus were employed in this evaluation for ceftobiprole. There were two methicillin-susceptible S. aureus isolates, three community-acquired MRSA isolates, and three hospital-acquired MRSA isolates. Details of handling have been provided previously (14). MICs were determined by CLSI methodology (7).
Animals.
BALB/cAnNCr (14) mice (age, 7 to 9 weeks; weight, 15 to 22 g; female) were studied. They were obtained from the National Cancer Institute, Frederick, MD. IACUC approval was obtained for all experiments. Mice were acclimated for 7 to 14 days and were rendered neutropenic with cyclophosphamide as previously described (14).
Induction of experimental pneumonia.
A bacterial inoculum containing 107 CFU/ml of S. aureus was prepared in a 3% suspension of mucin from porcine stomach, type II (Sigma Chemical Co.), and normal saline (14). The neutropenic mice were anesthetized with vaporized isoflurane. An inoculum of 0.05 ml was administered orally, with nasal blockage until the challenge inoculum was aspirated. An oxygen-rich environment was used for recovery and the subsequent randomization of the mice into groups.
Pharmacokinetic studies.
Ceftobiprole medocaril was prepared in sterile water. All injection volumes were 0.2 ml. Single doses (subcutaneous) of 1.0, 2.5, 10.0, and 25.0 mg/kg of body weight were studied. Injections were given to infected animals 6 h post-bacterial challenge. Blood was obtained terminally by cardiac puncture and was collected in EDTA-containing tubes (six per time point) at 0.25, 0.5, 1, 1.5, 2, 2.5, 3, and 4 h after administration. Plasma was collected by centrifugation and stabilized by addition of 3 μl of 2 M citric acid. Bronchoalveolar lavage (BAL) was performed on each animal, at 0.25, 1, 2, and 4 h, in order to obtain ELF. The methodology has been described previously (11, 14). A 10-μl volume of 2 M citric acid was added for the stabilization of ceftobiprole medocaril. Concentrations of ceftobiprole in plasma and BAL fluid were determined by a validated high-performance liquid chromatography assay at Johnson & Johnson. The limit of quantification was 0.01 mg/liter in all matrices, and the between-day coefficient of variation was <12%. The assay distinguishes between the prodrug and the active drug. Plasma and BAL fluid samples were tested for the urea concentration with a commercially available urea kit (Teco Diagnostics, Anaheim, CA). The performance of the kit has been reported previously, as have the calculation of the concentration of ceftobiprole in ELF from that in BAL fluid and the calculation of the concentrations of urea in plasma and BAL fluid (14).
Pharmacodynamic studies.
Multiple dosing regimens were administered subcutaneously to study mice in order to provide different exposures. Five mice were used in each cohort (regimen, infecting organism). At zero hour (approximately 6 h after inoculation), lungs were collected from a group of untreated controls. This provided a baseline measurement of the bacterial density in the lung. Ceftobiprole and sterile water (the latter was used as a no-treatment control) were initiated 6 h after inoculation and continued for 24 h. Dosages of 1 to 25 mg/kg administered once to five times daily were used in order to provide a wide range of ceftobiprole exposures. Lungs were harvested aseptically 24 h after the beginning of drug administration. They were then homogenized in 1.0 ml of normal saline. Homogenate dilutions (100 to 105 in saline) were plated onto 5% sheep blood agar and Columbia nutrient agar (Remel Inc., Lenexa, KS) for the prevention of contamination with gram-negative bacteria. The cultures were incubated at 35°C for as long as 48 h. The limit of detection in lung tissue was 2 × 102 CFU/ml. Protein binding data for ceftobiprole in mice (19%) were provided by the study sponsor (Johnson & Johnson Pharmaceutical Research & Development).
Healthy human subjects for determination of the penetration of ELF by ceftobiprole.
Twenty-five healthy, nonsmoking subjects, >18 years of age, were studied at a presumed steady state (around the 4th dose of ceftobiprole [a 500-mg dose was given every 8 h as a 2-h constant-rate intravenous infusion]) for ceftobiprole pharmacokinetics. For all subjects, a history was taken, a physical examination was performed, and clinical laboratory parameters were assessed as normal for study inclusion. The study was approved by the institutional review board. All subjects gave written informed consent.
Bronchoscopy, BAL, and blood sampling.
Twenty-four subjects each underwent one standardized bronchoscopy and BAL in an outpatient surgical facility at 2.5, 4, 6, or 8 h following the start of the last intravenous infusion of antibiotic. One subject who had blood obtained for determination of ceftobiprole plasma concentrations declined to have a bronchoscopy. A blood sample to determine the concentrations of the drug and urea was obtained just prior to the scheduled bronchoscopy and was kept on ice until it was centrifuged. Blood samples to determine ceftobiprole concentrations were obtained at hours 8, 16, and 24 prior to the last dose and at hours 24.5, 25.25, 26, 26.5, 27, 28, 30, and 32 during the last dosing interval, where a 2-h intravenous infusion was employed.
Population modeling approach and Monte Carlo simulation.
The population modeling approach and Monte Carlo simulation are described in the Appendix.
Drug assay.
Ceftobiprole concentrations in plasma and ELF were quantitated at SFBC Analytical Laboratories by a validated liquid chromatography-tandem mass spectrometry assay. The standard curves for ceftobiprole in plasma and BAL fluid were linear (r2 ≥ 0.99; ranges of concentrations, 50 to 25,000 ng/ml and 20 to 10,000 ng/ml, respectively). The precision of quality control samples for plasma ranged from 4.98% to 6.40%. Accuracy ranged from 3.74% to 7.24%. For BAL fluid, precision and accuracy ranged from 1.31% to 11.53% and −1.47% to −0.07%, respectively. The lower limits of quantitation in plasma and BAL fluid were 50 and 20 ng/ml, respectively. The assay distinguishes between the prodrug and the active drug.
Urea assay.
Concentrations of urea in plasma were analyzed spectrophotometrically by Quest Diagnostics using an Olympus AU5431 Chemistry Immuno Analyzer with a commercially available assay kit (OSR6636). The lower limit of quantitation for urea in plasma was 2 mg/dl. Concentrations of urea in ELF were measured at SFBC Analytical Laboratories using a liquid chromatography-tandem mass spectrometry assay. The standard curves for urea in BAL fluid were linear (r2 = 0.999; range of concentrations, 0.02 to 1.0 mg/dl). The intraday precision and accuracy for quality control samples ranged from 1.33% to 5.34% and −3.82% to 4.15%, respectively. The interday precision and accuracy ranged from 1.53% to 5.73% and 1.37% to 3.00%, respectively. The lower limit of quantitation for urea in BAL fluid was 0.02 mg/dl.
Concentration of the drug in ELF.
The concentration of ceftobiprole in ELF was calculated as ceftobiproleBAL × VBAL/VELF, where ceftobiproleBAL is the measured concentration of the antimicrobial agent in BAL fluid, VBAL is the volume of aspirated BAL fluid, and VELF is the volume of ELF sampled by BAL. VELF is calculated as VBAL × ureaBAL/ureaserum, where ureaBAL is the concentration of urea in BAL fluid and ureaserum is the concentration of urea in serum.
Inhibitory sigmoid Emax effect modeling.
The decline in the measured CFU per gram of lung tissue served as the dependent variable. The free-drug time>MIC in plasma and the total-drug time>MIC in ELF served as the independent variables. The equation linking them is as follows: log10 CFU/g = Econ − {Emax × [time>MICH/(time>MICH + EC50H)]}, where Econ is the number of organisms at 24 h in the mouse lung in the absence of treatment; Emax is the maximal decline in the number of organisms achievable by drug administration at 24 h; EC50 is the time>MIC associated with 50% of maximal effect; and H is Hill's constant. All data for all eight isolates of Staphylococcus aureus were considered together for the purposes of generating time>MIC targets.
RESULTS
MICs for the challenge organism.
Of the eight isolates employed, two were methicillin-susceptible S. aureus isolates (ceftobiprole MIC, 0.25 mg/liter) and six were MRSA isolates (three community-acquired MRSA isolates with MICs of 1, 1, and 2 mg/liter; three hospital-acquired MRSA isolates, with MICs of 0.5, 1, and 1 mg/liter).
Murine ceftobiprole pharmacokinetics.
The model fit the data well. After the Bayesian step, the regression equation for plasma data was as follows: observed = 1.037 × predicted − 0.00365 (r2 = 0.977; P ≪ 0.001; n = 157). For ELF data, the regression equation was as follows: observed = 0.945 × predicted + 0.0482 (r2 = 0.972; P ≪ 0.001; n = 93).
After the Bayesian step, the pharmacokinetic parameter values were grouped by dose. Extensive simulation demonstrated that using modal values as the measure of central tendency produced the best fit of the model to the data. After breakout by dose, the median of the values derived from the modes gave the most faithful simulation, especially for ELF values.
The area under the concentration-time curve (AUC) in plasma (AUCplasma), the AUCELF, and the penetration of ELF by the drug were determined by a 9,999-subject Monte Carlo simulation. For AUCplasma, the median was 68.8 mg·h/liter and the interquartile range was 37.2 to 122.6 mg·h/liter. For AUCELF, the median was 46.0 mg·h/liter and the interquartile range was 14.5 to 153.8 mg·h/liter. The median penetration percentage was 68.8%, and the interquartile range was 25.1 to 187.3%.
Ceftobiprole pharmacodynamics in the murine pneumonia model.
All of the cell kill data for all the strains were comodeled simultaneously after doses and schedules were transformed into free-drug time>MIC, with the assumption of a mouse plasma protein-binding level of 19% (free fraction, 81%), and were also transformed into total-drug time>MIC in ELF.
The results are displayed in Fig. 1A for plasma and in Fig. 1B for ELF. The overall r2 for plasma was quite acceptable, at 0.817. For stasis, ceftobiprole as free drug must be present in plasma at a concentration in excess of the MIC for 8.8% of a 24-h period. For cell kills of 1 and 2 log10 CFU/g, these percentages are 13.5% and 23%, respectively. The 95% confidence intervals (Fig. 1A) around the point estimates of the mean parameters are quite tight.
FIG. 1.

Relationship between ceftobiprole exposure, expressed as free-drug time>MIC in murine plasma (A) or total-drug time>MIC in murine ELF (B), and cell kill for eight strains of Staphylococcus aureus.
Examination of Fig. 1B demonstrates that the overall r2 for ELF was, again, quite acceptable, at 0.801. For stasis, ceftobiprole as total drug must be present in ELF at a concentration in excess of the MIC for 7.7% of a 24-h interval. For cell kills of 1 and 2 log10 CFU/g, these percentages are 12.9% and 24%, respectively. As in Fig. 1A, the 95% confidence intervals are tight.
For extrapolation to humans, we decided that since bacterial pneumonia is a very serious infection, we would employ only the 1- and 2-log10 CFU/g cell kill exposure targets. Further, for ELF, we rounded these numbers to 15% and 25% of a 24-h interval.
Ceftobiprole pharmacokinetics and ELF penetration in healthy volunteers.
Plasma ceftobiprole concentrations (means ± standard deviations [SD]) before the second, third, and fourth infusions were 3.06 ± 0.73, 3.60 ± 0.77, and 3.92 ± 1.07 mg/liter, respectively. These trough concentrations were consistent within and between subjects. Ceftobiprole concentrations in ELF (means ± SD) at 2.5, 4, 6, and 8 h were 2.55 + 0.99, 2.00 ± 1.07, 4.58 ± 5.82, and 1.51 ± 0.39 mg/liter, respectively; the concurrent concentrations in plasma were 17.68 ± 4.48, 12.77 ± 2.26, 6.91 ± 4.58, and 3.65 ± 1.05 mg/liter, respectively. Eight subjects had ELF drug concentrations below the limit of detection.
Ceftobiprole concentrations from 268 plasma samples and 16 BAL fluid samples were modeled using the population pharmacokinetic analysis program BigNPAG. The fit of the model to the data was quite acceptable. In Fig. 2A, we show the predicted-observed plot after the Bayesian step for the plasma concentrations. In Fig. 2B, the predicted-observed plot for ELF data is displayed.
FIG. 2.

(A) Predicted-observed plot of plasma ceftobiprole concentrations in healthy volunteers after the Bayesian estimation step for 268 samples. (B) Predicted-observed plot of ELF ceftobiprole concentrations in healthy volunteers after the Bayesian estimation step for 16 samples.
As a further check on the modeling process, we performed a 9,999-subject simulation and simulated drug concentrations in plasma and ELF hourly for 32 h. We used this to generate the 95% confidence interval for these values (2.5% to 97.5%). The observed plasma and ELF data are scattered around these intervals for plasma (Fig. 3A) and ELF (Fig. 3B). The appropriate number of observations fell within the 95% confidence intervals, adding certitude to the appropriateness of the modeling process.
FIG. 3.

Plasma (A) and ELF (B) concentration-time profiles simulated from the mean parameter vectors employing a 9,999-subject Monte Carlo simulation. Black circles represent the means; blue upright and inverted triangles represent the upper and lower 95% confidence intervals, respectively. Red squares represent actual observations and demonstrate the appropriate fit of the model to the data.
The estimates are presented in Table 1. The point estimate of clearance is appropriate and similar to that seen previously for this agent (18).
TABLE 1.
Population parameter values for ceftobiprole from healthy volunteers
| Parametera (unit) | Mean | Median | SD |
|---|---|---|---|
| Vc (liters) | 2.849 | 2.374 | 1.409 |
| K23 (h−1) | 6.171 | 4.744 | 4.504 |
| K32 (h−1) | 5.207 | 2.652 | 5.691 |
| K24 (h−1) | 5.512 | 6.021 | 2.276 |
| K42 (h−1) | 2.962 | 1.957 | 2.422 |
| CL (liters/h) | 5.162 | 5.380 | 0.685 |
| VELF (liters) | 39.10 | 44.79 | 13.43 |
| Khydrolysis (h−1) | 44.74 | 39.46 | 33.22 |
Vc, volume of the central compartment; K23, K32, K24, and K42, first-order intercompartmental transfer rate constants; CL, clearance; VELF, volume of the ELF compartment; Khydrolysis, first-order rate constant of hydrolysis of prodrug to active ceftobiprole.
These data were employed to perform a 9,999-subject simulation to examine the penetration of ceftobiprole into ELF (Table 2). The mean penetration was 25.5% (median, 15.3%; interquartile range, 7.9% to 30.4%). These percentages of penetration differ markedly from those seen in the mouse.
TABLE 2.
Penetration of ELF by ceftobiprole in healthy volunteers from a 9,999-subject simulation
| Parameter (unit) | Mean | Median | SD | Percentile
|
|||
|---|---|---|---|---|---|---|---|
| 10th | 25th | 75th | 90th | ||||
| AUCplasma (mg·h/liter) | 98.7 | 98.1 | 13.1 | 82.4 | 89.3 | 107.1 | 115.6 |
| AUCELF (mg·h/liter) | 25.2 | 15.0 | 35.9 | 4.20 | 7.67 | 30.1 | 55.5 |
| Penetration (%) | 25.5 | 15.3 | 36.6 | 4.32 | 7.89 | 30.4 | 55.7 |
We then used the kill targets of 1 and 2 log10 CFU/g for ELF in the mouse to bridge to humans. For the concentration-time profile of ceftobiprole in the ELF of humans, employing a 9,999-subject Monte Carlo simulation, we found the target attainment as displayed in Fig. 4. Also on this graph is the ceftobiprole MIC distribution for MRSA isolates (n = 4,958). Taking the expectation over the MIC distribution and the target attainments, we found that a 2-log10 CFU/g kill was achieved 79.7% of the time, while a 1-log10 CFU/g kill was achieved 85.6% of the time.
FIG. 4.

Probabilities of target attainment for cell kills of 2 log10 CFU/g (circles) and 1 log10 CFU/g (triangles) by ceftobiprole in ELF, as calculated from the penetration data of healthy volunteers. Targets were derived from a model of Staphylococcus aureus pneumonia in neutropenic mice.
It is critical to remember that mice and humans have very different ELF penetrations. Consequently, one cannot predict the microbiological effect for humans by using the plasma targets in the mouse for pneumonia. If one attempted to do so, a 99.9% target attainment would be expected for the 2-log10 CFU/g kill target (data not shown).
DISCUSSION
The emergence of MRSA in the community and the increasing importance of MRSA as a cause of nosocomial pneumonia make the advent of new agents active against this pathogen important (12, 13, 15, 21). There is currently no class of drug that works as well for MRSA as nafcillin/oxacillin works for methicillin-sensitive Staphylococcus aureus. Consequently, β-lactams, such as ceftobiprole, that have been designed to bind with high affinity to penicillin-binding protein (PBP2a) have attracted considerable interest among clinicians, in hopes of having a nafcillin/oxacillin equivalent in the fight against MRSA.
The mouse thigh model has been used in the past as a surrogate for many types of infections not located in a privileged space and has been used to guide dose choice. One would not expect that privileged spaces (central nervous system, eye, prostate) would be treatable with the same dose and schedule of drug as those used for a skin or skin structure infection, because of penetration. The penetration properties of antibiotics into lungs have been thought to be well represented by the mouse thigh model. This is because much work has been done over the past decade with fluoroquinolone antimicrobials (10, 19). Here, penetration into ELF is known to approximate the total AUC in plasma in animals and in humans. Consequently, the mouse thigh model with fluoroquinolones, where site penetration is excellent and reflects the plasma AUC, is a reasonable surrogate for setting exposure targets for pneumonia.
Until recently, little has been known about the penetration of β-lactam agents into ELF. For hospitalized, infected patients, Boselli et al. (4-6) and Bayat et al. (3) have examined a number of different β-lactams (e.g., ceftazidime, cefepime, piperacillin-tazobactam, and ertapenem) and have shown discordant penetration that seems not to be dependent on protein binding or structure.
Since ceftobiprole is being studied for pneumonia, we believed it was important to examine this agent in a preclinical murine model of pneumonia, including the penetration of ELF, and to determine the exposure targets in ELF necessary to produce a near-maximal microbiological effect in this system (a cell kill of approximately 2 log10 CFU/g). We were surprised to find that the penetration into murine ELF, while variable, was quite reasonable, with a median of almost 69%. We also identified the times>MIC in ELF driving cell kills of 1 log10 and 2 log10 CFU/g as 15% and 25% of a 24-h interval. These results are concordant with the recent report by Andes and Craig (8), whose mean time>MIC for a 2-log10 CFU/g kill was 29.3% (range, 24.4% to 39.1%) for eight strains of Staphylococcus aureus in murine infection models, particularly when one recognizes that the Craig-Andes data are for total drug in plasma. Our 2-log10 CFU/g kill exposure was a free-drug time>MIC in plasma of 23% of the dosing interval.
In order to bridge this information to humans so as to support a rational dose choice, we needed to identify the penetration and concentration-time course of ceftobiprole in the ELF of healthy volunteers. It became apparent that ceftobiprole penetrated into ELF very differently in humans than in mice, with a mean, median, and interquartile range of penetration of 25.5%, 15.3%, and 7.9 to 30.4%, respectively. This sort of species difference has been seen previously with macrolides and macrolide-type antibiotics (17, 20). The basis of such differences is currently unclear, but they are important. If one were to measure the time>MIC target in mouse plasma and use it to bridge to humans, a suboptimal dose would be chosen. This is because the attainment of a specific target in mouse plasma infers the attainment of a specific amount of exposure in the lung (ELF). If one then matches the free-drug time>MIC in the plasma of humans, one would drive a much shorter time>MIC in the lung (ELF) because of the lower penetration into this space seen in humans.
Figure 4 demonstrates that target attainment for a 500-mg dose of ceftobiprole every 8 h as a 2-h infusion falls below 90% for a cell kill of 2 log10 CFU/g at a MIC of 1.0 mg/liter and for a 1-log10 CFU/g cell kill at a MIC of 2.0 mg/liter. When one takes the overall expectation over the distribution of ceftobiprole MICs for MRSA isolates into account, one achieves the 2-log10 CFU/g cell kill target 79.7% of the time, accounting for the variability of pharmacokinetics and the variability in MICs likely to be encountered, and the 1-log10 CFU/g cell kill target 85.6% of the time. It should also be recognized that stasis (time>MIC in ELF, 10%) is achieved 94.1% of the time.
For seriously ill patients, particularly in the intensive-care unit, we believe a 1- or 2-log10 CFU/g cell kill target is most appropriate. Consequently, it may be that somewhat higher ceftobiprole doses or longer infusion times (to prolong the time>MIC), or both, will be required to comfortably ensure a 90% target attainment for seriously ill patients with MRSA pneumonia.
What issues remain for which we have little or no data? The first and foremost is that we are employing ELF concentrations as a surrogate for lung penetration and the ability to control infections in this site. While ELF penetration is certainly theoretically a better measure than concentrations in whole-lung tissue for ascertaining lung penetration, it is not certain that this is the best metric of penetration. However, it is as good as or better than any other currently available competing metric, because it comes from the anatomic site where the causative microorganisms are found.
The second issue is that the preclinical murine model of pneumonia was granulocytopenic. In the murine thigh and lung infection models, Craig and Andes (8) demonstrated that the activity of ceftobiprole against Streptococcus pneumoniae and Klebsiella pneumoniae is markedly enhanced by the presence of neutrophils. The required time>MIC for a static effect against Streptococcus pneumoniae was less than 10% in nonneutropenic mice, compared to more than 20% in neutropenic mice. It is not known what the impact of a full granulocyte response would be in terms of altering the exposure target (time>MIC in ELF). There are currently insufficient data to make clear recommendations for different subpopulations.
Finally, we currently do not have insight into the impact of infection on the penetration of ceftobiprole into ELF in humans. It should be noted that the mice in the preclinical-infection model were infected and had substantially higher penetration of ELF by the drug than that seen in the healthy volunteers. It will be critical to identify the impact of inflammation and infection on the penetration of ELF by ceftobiprole in humans.
In summary, to identify a safe and efficacious drug dose for patients with pneumonia, it is important to delineate the penetration into the affected site and to delineate the between-patient variability in penetration, so that the chosen dose will reach the infection site for a large percentage of patients and in an amount that will enable substantial cell killing for organisms with MICs that are likely to be encountered in the clinic.
Acknowledgments
M. Khashab, G. J. Noel, and S. Nicholson are current employees of Johnson & Johnson. J. B. Kahn is an immediate past employee of Johnson & Johnson. G. L. Drusano, K. A. Rodvold, M. Gotfried, and D. P. Nicolau received grant support from Johnson & Johnson for generating data for portions of this article. G. L. Drusano, K. A. Rodvold, M. Gotfried, and D. P. Nicolau have been consultants to Johnson & Johnson. D. P. Nicolau has spoken for Johnson & Johnson over the past 2 years.
APPENDIX
Population modeling approach. (i) Murine modeling.
The BigNPAG program of Leary, Jelliffe, Schumitzky, and Van Guilder was employed (16). Adaptive γ was not employed because of the single-observation nature of the data. Here, the ELF is its own sampling compartment with its own apparent volume of distribution. It takes eight parameters and four differential equations to define this system. The agent administered was the prodrug. The equations are as follows:
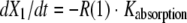 |
(1) |
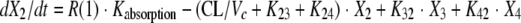 |
(2) |
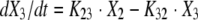 |
(3) |
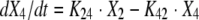 |
(4) |
where R(1) is a piecewise input function in which the drug is administered at a fixed rate for a specified period; X1 is the amount of drug administered into the absorption compartment; X2, X3, and X4 are the amounts of active drug in the central, peripheral, and ELF compartments, respectively; CL is clearance; Vc is the volume of the central compartment; and K23, K32, K24, and K42 are first-order intercompartmental transfer rate constants.
The concentration of the drug in plasma is calculated as X2/Vc, and the concentration in ELF as X4/VELF, where VELF is the volume of the ELF compartment.
Weighting of observations was as the inverse of the between-day assay error variance. Bayesian estimates were obtained for each individual using the “population of one” utility in BigNPAG. The model was evaluated by predicted-observed plots. The mean error served as the measure of bias. The bias-adjusted mean squared error served as the measure of precision. Since the same doses were used for pharmacokinetic study as for the pharmacodynamics study, we employed the measures of central tendency for the pharmacokinetic parameters after the Bayesian step for all animals in a particular dosing group that best described the raw data (the prodrug was administered). These parameter values were then used in simulation studies to calculate the time>MIC (for free drug in plasma and total drug in ELF) for each MIC studied. Simulation studies were performed with the ADAPT II package of programs of D'Argenio and Schumitzky (9).
(ii) Healthy-volunteer modeling.
The same form of the model system was employed for healthy-volunteer modeling as for murine modeling. However, in the murine system, the drug was injected subcutaneously, whereas the volunteers had ceftobiprole medocaril injected intravenously as a 2-h infusion. The prodrug was then rapidly hydrolyzed to the active drug ceftobiprole. Consequently, the “Ka” in this instance represents a first-order hydrolysis rate constant, Khydrolysis or Kabsorption. All other modeling choices were the same as those described for the murine modeling.
Monte Carlo simulation.
Point estimates of the population parameter values and their dispersions were inserted into the simulation module of ADAPT II (9). A 9,999-subject Monte Carlo simulation was performed using the mean parameter values as the measure of central tendency. Both normal and log-normal distributions were evaluated, and the choice of distribution was determined by the fidelity with which the original parameter values and their dispersions were re-created by the simulation (log-normal distribution was employed). Plasma and ELF drug concentrations were simulated hourly for 32 h to provide the 95% confidence intervals (2.5% to 97.5%).
In addition, a single-dose simulation was performed. In this simulation, other differential equations were added to identify the AUC from time zero to 1,000 h (an approximation of zero to infinity) for both plasma and ELF. The penetration was determined as the AUCELF/AUCplasma ratio. The form of the differential equation is as follows: dX5,6/dt = Xn/Vn, where n is 2 for plasma and 4 for ELF. Other system outputs were added to provide the AUCs and penetration: Y3 = X5 (AUCplasma); Y4 = X6 (AUCELF); Y5 = X6/X5 (penetration).
In this single-dose simulation, the time>MIC was also calculated for the first dosing interval. This was done by adding the following differential equations: if {[X(2)·0.82]/Vc}.GE. MIC, then dX7/dt = 1.0; else, dX7/dt = 0.0.
The MIC could be varied over the range from 0.25 mg/liter to 4 μg/ml. This provided the time>MIC for plasma as corrected for protein binding (free fraction, 82% in humans). The time>MIC in ELF (no protein binding correction) was provided similarly, except that [X(2)·0.82]/Vc was replaced by X4/VELF.
Confidence intervals were determined directly from the Monte Carlo simulations.
Footnotes
Published ahead of print on 18 May 2009.
REFERENCES
- 1.Adis International Data Information. 2006. Ceftobiprole medocaril: BAL5788, JNJ 30982081, JNJ30982081, RO 65-5788, RO 655788. Drugs R. D. 7:305-311. [DOI] [PubMed] [Google Scholar]
- 2.Andes, D. R., and W. A. Craig. 2000. In vivo pharmacodynamics of RO 63-9141 against multiple bacterial pathogens, abstr. 1079. Abstr. 40th Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC.
- 3.Bayat, S., K. Louchahi, B. Verdière, D. Anglade, A. Rahoui, P. M. Sorin, M. Tod, O. Petitjean, F. Fraisse, and F. A. Grimbert. 2004. Comparison of 99mTc-DTPA and urea for measuring cefepime concentrations in epithelial lining fluid. Eur. Respir. J. 24:150-156. [DOI] [PubMed] [Google Scholar]
- 4.Boselli, E., D. Breilh, T. Rimmelé, J. C. Poupelin, M. C. Saux, D. Chassard, and B. Allaouchiche. 2004. Plasma and lung concentrations of ceftazidime administered in continuous infusion to critically ill patients with severe nosocomial pneumonia. Intensive Care Med. 30:989-991. [DOI] [PubMed] [Google Scholar]
- 5.Boselli, E., D. Breilh, M. Cannesson, F. Xuereb, T. Rimmelé, C. D. Chassard, M. C. Saux, and B. Allaouchiche. 2004. Steady-state plasma and intrapulmonary concentrations of piperacillin/tazobactam 4 g/0.5 g administered to critically ill patients with severe nosocomial pneumonia. Intensive Care Med. 30:976-979. [DOI] [PubMed] [Google Scholar]
- 6.Boselli, E., D. Breilh, M. C. Saux, J. B. Gordien, and B. Allaouchiche. 2006. Pharmacokinetics and lung concentrations of ertapenem in patients with ventilator-associated pneumonia. Intensive Care Med. 32:2059-2062. [DOI] [PubMed] [Google Scholar]
- 7.Clinical and Laboratory Standards Institute. 2006. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 7th ed. CLSI publication M7-A7. Clinical and Laboratory Standards Institute, Wayne, PA.
- 8.Craig, W. A., and D. R. Andes. 2008. In vivo pharmacodynamics of ceftobiprole against multiple bacterial pathogens in murine thigh and lung infection models. Antimicrob. Agents Chemother. 52:3492-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D'Argenio, D. Z., and A. Schumitzky. 1997. ADAPT II. A program for simulation, identification, and optimal experimental design. User manual. Biomedical Simulations Resource, University of Southern California, Los Angeles. http://bmsr.usc.edu/Software/ADAPT/ADAPTguide.html.
- 10.Drusano, G. L., S. L. Preston, M. H. Gotfried, L. H. Danziger, and K. A. Rodvold. 2002. Levofloxacin penetration into epithelial lining fluid as determined by population pharmacokinetic modeling and Monte Carlo simulation. Antimicrob. Agents Chemother. 46:586-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du, X., C. Li, H. K. Sun, C. H. Nightingale, and D. P. Nicolau. 2005. A sensitive assay of amoxicillin in mouse serum and broncho-alveolar lavage fluid by liquid-liquid extraction and reversed-phase HPLC. J. Pharm. .Biomed. Anal. 39:648-652. [DOI] [PubMed] [Google Scholar]
- 12.Francis, J. S., M. C. Doherty, U. Lopatin, C. P. Johnston, G. Sinha, T. Ross, M. Cai, N. N. Hansel, T. Perl, J. R. Ticehurst, K. Carroll, D. L. Thomas, E. Nuermberger, and J. G. Bartlett. 2005. Severe community-onset pneumonia in healthy adults caused by methicillin-resistant Staphylococcus aureus carrying the Panton-Valentine leukocidin genes. Clin. Infect. Dis. 40:100-107. [DOI] [PubMed] [Google Scholar]
- 13.Kowalski, T. J., E. F. Berbari, and D. R. Osmon. 2005. Epidemiology, treatment, and prevention of community-acquired methicillin-resistant Staphylococcus aureus infections. Mayo Clin. Proc. 80:1201-1207. [DOI] [PubMed] [Google Scholar]
- 14.Laohavaleeson, S., P. R. Tessier, and D. P. Nicolau. 2008. Pharmacodynamic characterization of ceftobiprole in experimental pneumonia caused by phenotypically diverse Staphylococcus aureus strains. Antimicrob. Agents Chemother. 52:2389-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LaPlante, K. L., M. J. Rybak, M. Amjad, and G. W. Kaatz. 2007. Antimicrobial susceptibility and staphylococcal chromosomal cassette mec type in community- and hospital-associated methicillin-resistant Staphylococcus aureus. Pharmacotherapy 27:3-10. [DOI] [PubMed] [Google Scholar]
- 16.Leary, R., R. Jelliffe, A. Schumitzky, and M. Van Guilder. 2001. An adaptive grid non-parametric approach to pharmacokinetic and dynamic (PK/PD) models, p. 389-394. In Proceedings of the 14th IEEE Symposium on Computer-Based Medical Systems. IEEE Computer Society, Bethesda, MD.
- 17.Lodise, T. P., S. L. Preston, V. Barghava, A. Bryskier, R. Nusrat, S. Chapel, M. Rangaraju, and G. L. Drusano. 2005. Pharmacodynamics of an 800 mg dose of telithromycin in patients with community-acquired pneumonia caused by extracellular pathogens. Diagn. Microbiol. Infect. Dis. 52:45-52. [DOI] [PubMed] [Google Scholar]
- 18.Lodise, T. P., R. Pypstra, J. B. Kahn, B. P. Murthy, H. C. Kimko, K. Bush, G. J. Noel, and G. L. Drusano. 2007. Probability of target attainment for ceftobiprole as derived from a population pharmacokinetic analysis of 150 subjects. Antimicrob. Agents Chemother. 51:2378-2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Louie, A., C. Fregeau, W. Liu, R. Kulawy, and G. L. Drusano. 2008. Pharmacodynamics of levofloxacin in a murine pneumonia model with Pseudomonas aeruginosa: determination of epithelial lining fluid targets, abstr. A-043. 48th Intersci. Conf. Antimicrob. Agents Chemother., Washington, DC, 25 to 28 October 2008. [DOI] [PMC free article] [PubMed]
- 20.Ong, C. T., P. K. Dandekar, C. Sutherland, C. H. Nightingale, and D. P. Nicolau. 2005. Intrapulmonary concentrations of telithromycin: clinical implications for respiratory tract infections due to Streptococcus pneumoniae. Chemotherapy 51:339-346. [DOI] [PubMed] [Google Scholar]
- 21.Zetola, N., J. S. Francis, E. L. Nuermberger, and W. R. Bishai. 2005. Community-acquired methicillin-resistant Staphylococcus aureus: an emerging threat. Lancet Infect. Dis. 5:275-286. [DOI] [PubMed] [Google Scholar]