

Abstract
The mouse chlamydial pathogen Chlamydia muridarum has been used as a model organism for the study of human Chlamydia trachomatis urogenital and respiratory tract infections. To date, two commonly used C. muridarum isolates have been used interchangeably and are essentially taken to be identical. Herein, we present data that indicate that this is not the case. The C. muridarum Weiss isolate and C. muridarum Nigg isolate varied significantly in their virulences in vivo and possessed different growth characteristics in vitro. Distinct differences were observed in intravaginal 50% infectious doses and in challenge infections, with the Weiss isolate displaying greater virulence. Respiratory infection by the intranasal route also indicated a greater virulence of the Weiss isolate. In vitro, morphometric analysis revealed that the Weiss isolate produced consistently smaller inclusions in human cervical adenocarcinoma cells (HeLa 229) and smaller plaques in monolayers of mouse fibroblasts (L929) than did the Nigg isolate. In addition, the Weiss isolate possessed significantly higher replicative yields in vitro than did the Nigg isolate. In plaque-purified isolates derived from our stocks of these two strains, total genomic sequencing identified several unique nonsynonymous single nucleotide polymorphisms and insertion/deletion mutations when our Weiss (n = 4) and Nigg (n = 5) isolates were compared with the published Nigg sequence. In addition, the two isolates shared 11 mutations compared to the published Nigg sequence. These results prove that there is genotypic and virulence diversity among C. muridarum isolates. These findings can be exploited to determine factors related to chlamydial virulence and immunity.
Until quite recently, there has been no means to consistently and predictably mutate chlamydiae in order to study virulence and immunity (2). Until such a method is available for routine use, one could potentially study closely related chlamydial strain variants in which one isolate is more virulent than the other and then employ a readily available animal model with which to assess the putative differences, as was done previously by Kari et al. (14). In this report, we will present evidence to demonstrate that the natural mouse chlamydial pathogen Chlamydia muridarum could prove to be such a model but has gone unrecognized for the 60 years that it has been used by chlamydial researchers.
While studying human influenza virus and certain mouse respiratory pathogens, several investigators in the late 1930s and early 1940s identified a separate and distinct pathogen that caused pneumonitis in mice but was native to the mouse colonies of the day (10, 11, 20). Each investigator individually concluded that they had isolated a new “virus” from mice that produced disease similar to, but distinct from, human influenza virus and other viral respiratory tract infections. This “virus” was found to create inclusion bodies in infected host cells and was termed the agent of mouse pneumonitis (MoPn). It was also noted to be serologically related to the causative agent of human lymphogranuloma venereum and avian psittacosis (10, 21). Eventually, MoPn was accepted as a distinct biovar of the bacterium Chlamydia trachomatis (19). Subsequently, it was asserted through phylogenetic analyses of the 16S and 23S rRNA genes that although closely related to C. trachomatis, MoPn actually should be assigned to a separate species (8). As such, it was reclassified as Chlamydia muridarum. It is still referred to by most investigators by the original term, MoPn.
In 1981, Barron et al. first reported a successful MoPn infection in the urogenital tract of mice and proposed this as a model for human urogenital chlamydial infection (1). Since that time, MoPn urogenital and respiratory tract infections in mice have become popular animal models for the study of immune responses and pathological host responses to chlamydiae (18, 26, 27).
To date, two isolates of MoPn have been commonly employed in this model, MoPn Nigg and MoPn Weiss. It has been generally assumed that both MoPn isolates were essentially identical and derived from a single original strain. Each laboratory using these strains has maintained their own lineage from the original stocks, which have been passed from one laboratory to another, or they obtained them from the culture depository at the American Type Culture Collection (ATCC). Initially passed through embryonated hen's homogenized yolk sacs, each line has been variously maintained in mouse fibroblasts (L929 or McCoy cells) or in human adenocarcinoma lines (HeLa 229 cells).
Through some initial coincidental observations, serendipity, and subsequent systematic study, we have concluded that these two isolates, while closely related, possess distinct growth characteristics in vitro and vary significantly in virulence in vivo. We believe that the findings described herein not only will be important to those employing this model but ultimately will contribute significantly to an understanding of the basis of chlamydial pathogenesis and immunity, chlamydia population biology, as well as chlamydia-host cell interactions and biology.
MATERIALS AND METHODS
Origins and maintenance of culture stocks.
C. muridarum isolate MoPn Nigg was obtained and originally maintained by the laboratories of Roger Rank and the late Almen Barron at the University of Arkansas for Medical Science. This isolate was acquired in approximately 1977 from the original deposit of chicken embryo yolk sack-grown MoPn at the ATCC (Rockville, MD). MoPn Nigg was originally deposited as yolk sac homogenate at the ATCC by Francis B. Gordon, who obtained the isolate from Clara Nigg (20, 21). In the early literature, this strain was referred to as Atherton II. It should be noted that MoPn derived from mammalian cell culture has since been redeposited to the ATCC by Julius Schachter. Schachter indicated that this strain is likely the Weiss isolate because all of his MoPn stock was derived from an original seed stock from Emilio Weiss (J. Schachter, personal communication).
The MoPn Weiss isolate used in this study was obtained by the Ramsey laboratory at Midwestern University in 1995 from Todd Cotter during Cotter's postdoctoral fellowship in the laboratory of Gerry Byrne. Cotter had obtained his stocks of MoPn Weiss while working in the laboratory of Harlan Caldwell. Caldwell, in turn, had obtained the Weiss isolate from Julius Schachter, who had obtained the isolate from Emilio Weiss (29; J. Schachter, personal communication). The Weiss strain was originally referred to as the Chicago strain (12; J. Molder, personal communication).
Thus, while we cannot rule out a convergence somewhere in the distant history of the two isolates, it is quite clear that for several years and innumerable in vitro passages, likely under various growth conditions, these two strains have been maintained separately.
Note that great caution was exercised in order to prevent cross-contamination of stocks of the MoPn Weiss and MoPn Nigg strains in the laboratory. Stocks were maintained by culture in HeLa 229 cells using standard protocols except that working with either strain in both the Ramsey and Rank laboratories on the same day was strictly forbidden. Similarly, growth of stock cultures of either strain at the same time in the laboratory was also strictly forbidden. Both strains were periodically checked and verified to be mycoplasma free either by a rapid carbohydrate utilization test (MycoAlert; Cambrex, East Rutherford, NJ) or by culture in PPLO broth with subsequent multiple subordinate cultures in PPLO agar (Difco Laboratories, Detroit, MI).
Mice, infections, and infection assessments.
We selected a susceptible strain of mice for these experiments (7). Five- to six-week-old female C3H/HeN mice were obtained from Harlan Sprague Dawley (Madison, WI) and housed with rodent chow and water ad libitum with a 12-h-12-h light-dark cycle.
For genital tract infections, following a 10-day acclimation period subsequent to arrival from the supplier, mice were pretreated with 2.5 mg of medroxyprogesterone acetate subcutaneously (DepoProvera, P4; Upjohn, Kalamazoo, MI) and were inoculated intravaginally 7 days later with approximately 100 50% infectious doses (ID50) (104 inclusion-forming units [IFU]) of HeLa 229-grown C. muridarum (MoPn) exactly as described elsewhere previously (5). In order to verify and track genital infection, cervical-vaginal swabs were collected from all mice at day 4 postinfection, and the infection course was followed at postinfection days 4, 7, 10, and 14 and every 7 days thereafter through day 49 postinfection in each experiment. C. muridarum was isolated; inclusions were visualized by indirect immunofluorescence and quantified as IFU in HeLa 229 cell cultures as previously described (24). For challenge infections, mice were pretreated again with P4 at day 49 postinfection and inoculated with 105 IFU (approximately 1,000 ID50) of either strain at day 56 as specified previously (5). To confirm previous culture titrations of our stocks on which challenge infections were based, and thus confirm the inoculating dose of MoPn as being accurate, we again determined the titer of the diluted stock preparations used to challenge mice on the same day of the challenge by culture. In each case, our stock titers and day-of-challenge titers were confirmed to be well within a range of ±20% of the original titer and often much less than that.
To compare the ID50 values between the two strains, 40 mice were similarly pretreated with progesterone as described above and inoculated with log10 dilutions of either strain intravaginally. Cervical-vaginal swabs were collected at days 4, 7, 10, and 14 postinfection for detection of MoPn in culture. As an indicator of an active infection below the sensitivity of our culture methods, blood was collected from culture-negative mice at day 35 postinfection, and total plasma anti-MoPn immunoglobulin G (IgG) levels were determined by enzyme-linked immunosorbent assay (ELISA) (see below).
For intranasal infection, 5- to 6-week-old C3H/HeN mice (Harlan Sprague Dawley) were obtained and acclimated to the Midwestern University Animal Resources Facility for 10 days. Mice were anesthetized and placed in dorsal recumbency, and 10 μl of log10-diluted stocks of either the Nigg or Weiss isolate were placed dropwise in an alternating fashion over the nares to effect a dose of 10° to 106 IFU per mouse in groups of 10 per dose. Mice were weighed daily, and weight gain or loss was used as a measure of morbidity. Mice were also monitored daily for other signs of morbidity, including a lack of grooming, suppressed response to tactile stimuli, and social withdraw. Mortalities were noted daily, but when mice became visibly moribund, ethical practices dictated that we euthanize any such mice. These mice were considered in the mortality curves presented herein. As in urogenital infections, seroconversion was used to detect infection below the sensitivity of weight change alone.
In vitro culture comparison of MoPn strains.
HeLa 229 cells were used to compare replication curves of the two strains in cell culture. Initially, the titers of both strains were determined using HeLa 229 cells to determine their multiplicity of infection (MOI). This was performed by adding MOIs of 0.01, 0.05, 0.1, 0.5, 1.0, 5.0, and 10.0 for each strain in triplicate to 13-mm coverslips in a 36-well culture dish. After 32 h of culture, the coverslips were fixed with methanol and stained with immune mouse serum (diluted 1:100 from serum collected 35 days or more after urogenital infection), followed by fluorescein isothiocyanate-labeled rabbit anti-mouse IgG (1:80). The number of inclusions per 20 fields was determined, and the two strains were compared. No significant difference was observed between the MOIs for each strain (data not shown), suggesting that the infectivity of each strain was the same for this host cell type.
To compare the replication curves of each strain, an MOI of 1.0 was chosen. As described above, 13-mm coverslips were added to each well in 36-well culture plates, and HeLa 229 cell cultures were grown to confluence. The cultures were inoculated with both strains of C. muridarum, and at various times after inoculation, triplicate wells were harvested for each strain. The supernatants were removed and frozen individually at −70°C until they were later thawed for determinations of progeny IFU. One milliliter of additional sucrose potassium glutamate was added to each well, and the well was sonicated with a probe to disrupt the cell monolayers. The plate was then centrifuged at 600 × g for 10 min at 4°C to sediment the cell and coverslip debris. The supernatant of this suspension (coverslips) was removed and frozen at −70°C until it was thawed for progeny IFU determinations as described above.
Inclusion size determination.
Stock of the Weiss and Nigg strains of MoPn were inoculated at an MOI of approximately 0.1 (104 IFU per well) in HeLa 229 cell monolayers in a 24-well plate. At 12, 18, and 24 h postinoculation, monolayers well were fixed in methanol and stained by indirect immunofluorescence (6). Several fields were photographed using an AxioCamMRc5 apparatus (Carl Zeiss Microimaging, Inc., Thornwood, NY) attached to an inverted fluorescence microscope (Axiovert 25; Zeiss). Inclusions were measured by highlighting the edge of the inclusion using Axiovision 4.5 software, which was previously calibrated and set by the manufacturer.
Plaque-forming assay and plaque size determination.
Stocks of C. muridarum strains were inoculated onto confluent monolayers of L929 mouse fibroblasts in six-well tissue culture plates (Cellstar; Greiner Bio-One, Monroe, NC) and centrifuged at 3,000 rpm for 1 h at 37°C (HermleZ400K; Labnet). The inoculum was then removed and replaced with overlay medium (1× Dulbecco's modified Eagle's medium, 10% fetal bovine serum, 50 μg/ml gentamicin, 0.2 μg/ml cycloheximide, and a final concentration of 0.2% of agarose). The cells were then incubated at 37°C in an atmosphere of 5% CO2 for 5 days to allow plaques to form. The solid overlay was removed, and the monolayers were fixed in methanol and then stained with 0.25% Neutral Red in phosphate-buffered saline for 10 to 15 min at room temperature. The six-well plate containing the monolayers was scanned using an HP Scanjet 5470C device, and images were saved in tagged image file format using the HP Precision Pro 3.1 program. The area of each plaque was measured by Scion Image free software (http://www.scioncorp.com). Measurements were validated using the total area of a well as provided by the manufacturer compared to the total area measured by the Scion Image software.
ELISA.
To detect antibody responses against MoPn antigen, blood was collected via puncture of the retro-orbital venous plexus of anesthetized mice, and total plasma IgG (heavy- and light-chain-specific) ELISA for MoPn-specific antibodies was performed exactly as described elsewhere previously (25).
Total genome sequencing and identification of SNPs and insertion/deletion mutations.
MoPn Nigg (here Nigg2 to distinguish it from the published reference sequence) and MoPn Weiss were isolated from single plaques as described above and amplified in HeLa 229 cell monolayers. DNA was isolated from renograffin gradient-purified elementary bodies (EBs) (3) and subjected to whole-genome sequencing utilizing the 454 GS-FLX sequencing platform (Roche Applied Sciences, Indianapolis, IN). Both isolates were sequenced as previously described (16). Genome sequencing was conducted in microfabricated high-density picoliter reactors to an average depth of 45× coverage using one-half of a Pico Titer plate per isolate (16). Draft sequences for the Nigg and Weiss isolates were obtained by assembling each isolate data set against the previously published C. muridarum Nigg sequence (28) using GS Reference Mapper software (Roche Applied Sciences), identifying putative high-quality single nucleotide polymorphisms (SNPs) and insertions or deletions (indels). To confirm identified SNPs and indels identified by high-throughput sequencing, targeted regions were PCR amplified in the forward and reverse directions, and purified amplicons were sequenced on contract by the University of Chicago Cancer Research Center DNA Sequencing Facility (Chicago, IL). Low-throughput full service sequencing was done using Applied Biosystems 3730XL 96-well capillary DNA sequencers.
Statistics.
Infection courses as assessed by the shedding of viable organisms from the lower genital tract were compared by two-way analysis of variance (ANOVA) (group and days) with repeated measures (days) with post hoc analysis with a Tukey-Kramer multiple-comparisons test. For intranasal infection, morbidities (measured as weight change) were also compared between groups by two-way repeated-measures ANOVA (group and days) with repeated measures (days) and post hoc analysis as described above. In vitro replication curves were compared between groups by two-way ANOVA (group and time) with repeated measures on IFU counts. Inclusion and plaque size variation were compared by a two-sample unpaired Student's t test. The ID50 (urogenital infection) and 50% lethal dose (LD50) (intranasal infection) were estimated using probit analysis and a log-rank test to determine statistical significance. All statistical tests were accomplished using either the NCSS and PASS (Kaysville, UT) combined software package or SigmaStat (Systat Software, Chicago, IL).
Nucleotide sequence accession numbers.
Data from this whole-genome shotgun project have been deposited in the DDBJ/EMBL/GenBank database under accession number ACOV00000000 for Chlamydia muridarum Nigg. The version described in this paper is the first version, under GenBank accession number ACOV01000000. For Chlamydia muridarum Weiss, data from the whole-genome shotgun project has been deposited in the DDBJ/EMBL/GenBank database under accession number ACOW00000000.
RESULTS
Differences in protective immunity in urogenital infections with C. muridarum strains in vivo.
Our first indication that we were dealing with two distinct strains of C. muridarum was a coincidental observation by our laboratory during the process of studying heterotypic immunity in the mouse genital model (24). We began to observe that following intravaginal challenge at day 56 after primary infection with MoPn, we often achieved a high culture-positive rate albeit at lower IFU counts than those for primary infection. This was not unusual and was reported previously by ourselves and others (as summarized in references 18 and 26). However, in several other experiments using identical challenge doses, we found that animals were solidly immune to challenge infection, and no mice were culture positive following challenge. At first, we attributed this inconsistency to a low sensitivity of our culture method and the corresponding low IFU counts in challenge infections. We also realized that we had introduced another source of MoPn into our laboratory (source, Todd Cotter, during collaborative efforts with the laboratory of Gerry Byrne). Thus, after carefully reviewing laboratory protocols and stock records, we noted that regardless of which strain was used in the primary infection, if the challenge infection was conducted with MoPn Weiss, immunity was not complete, and a second, albeit less intense, infection could be detected upon culture of cervical-vaginal swabs; however, mice challenged with MoPn Nigg were solidly immune. Conversely, if the primary infection had been conducted utilizing MoPn Nigg, mice were routinely culture positive upon challenge, regardless of the challenge strain. Thus, we set out to confirm these observations in head-to-head experiments. Combined primary and challenge infection course IFU kinetics from these controlled replicate experiments are depicted in Fig. 1, and the ratios of culture-positive to culture-negative mice in the challenge infections are summarized in Table 1.
FIG. 1.

Primary and challenge infection courses with C. muridarum strains. (A) Course of primary and challenge infections of mice inoculated intravaginally with 104 IFU of MoPn Weiss (closed circles). The primary infection is shown in the left half of the graph. Each point represents the mean and standard deviation (error bars) for IFU isolated from cervical-vaginal swabs (n = 38; two experiments). Following the resolution of infection, mice were then challenged intravaginally at day 56 after primary infection either homotypically with MoPn Weiss (open circles) or heterotypically with MoPn Nigg (open squares) at 105 IFU. Only mice challenged with MoPn Weiss displayed any susceptibility to challenge infection (P < 0.02 by repeated-measures ANOVA). (B) Course of primary infection of mice inoculated intravaginally with MoPn Nigg (closed squares) (n = 20; one experiment). Mice were then challenged intravaginally at day 56 after primary infection with MoPn Weiss (open circles) (n = 10) or MoPn Nigg (open squares) (n = 10) at 105 IFU. A tendency toward higher rates of culture-positive mice and higher infectious burdens was observed in the mice challenged with MoPn Weiss, but this did not prove to be significant (P = 0.07 by repeated-measures ANOVA). Primary infection courses between MoPn Weiss and MoPn Nigg were not significantly different (P = 0.789 by repeated-measures ANOVA). Comparison between any primary infection and any challenge infection yielded highly significant differences (P < 0.0001).
TABLE 1.
Challenge infection with MoPn strains
| Infection-challenge isolatesa | No. of positive mice/total no. of mice on day postchallengec:
|
||||||
|---|---|---|---|---|---|---|---|
| 0b | 4 | 7 | 10 | 14 | 21 | 35 | |
| Weiss-Weiss | 0/18 | 7/18 | 9/18 | 7/18 | 0/18 | 0/18 | ND |
| Weiss-Nigg | 0/20 | 0/20 | 0/20 | 0/20 | 0/20 | 0/20 | ND |
| Nigg-Nigg | 0/10 | 8/10 | 6/10 | 3/10 | 0/10 | 0/10 | 0/10 |
| Nigg-Weiss | 0/10 | 10/10 | 10/10 | 9/10 | 6/10 | 6/10 | 4/10 |
Primary infection was accomplished with 104 IFU of either MoPn Weiss or MoPn Nigg. Challenge inoculation was accomplished with 105 IFU of either MoPn Weiss or MoPn Nigg. Two experiments were conducted by utilizing primary infection with MoPn Weiss, and one experiment was conducted by utilizing primary infection with MoPn Nigg.
Corresponds to day 56 after primary infection.
Number of culture-positive swabs collected from individual mice of the total number of mice. ND, not done.
The primary infection courses for MoPn Nigg and MoPn Weiss were not different (Fig. 1). However, regardless of the source of the primary or challenge infection, the level of the second infection, when detected, was always significantly lower than that of the primary one. This indicated to us that the strains likely possessed a good deal of antigenic similarity. Thus, as would be expected, the isolates were likely to be far more related than, for example, when either MoPn strain is compared to human C. trachomatis serovars, as was observed in cross-protective immunity experiments reported previously (24).
Another interesting observation was the persisting culture-positive mice with a primary MoPn Nigg infection that had been challenged with MoPn Weiss (Table 1 and Fig. 1B). This group achieved a 100% infection rate upon challenge, and the challenge infection cultures persisted beyond all other challenge infections in this study. Six of ten mice remained culture positive on day 14 through day 28 after challenge infection, and 4 of 10 mice were still positive on day 35 after challenge infection. When the collection of cervical-vaginal swabs ceased on day 42 after challenge infection, one mouse in this group was still culture positive albeit at a low level (1.5 × 102 IFU) (not shown). While the absolute numbers of IFU isolated from these mice were relatively low (Fig. 1B), the chronology of the infection course mirrored that of primary infection.
Cumulatively, these data indicated to us that the two strains are likely to be very closely related but are indeed distinct strains of C. muridarum that behave differently in their natural host, the mouse.
ID50 determinations for MoPn strains in urogenital and respiratory tract infections.
An accepted measure of virulence is the ID50 for a specific host. To this end, we inoculated mice with log10-graded dilutions of MoPn Nigg or MoPn Weiss intravaginally. Two groups of 20 mice were pretreated with P4, and 7 days later, they were inoculated with either 101, 102, 103, or 104 IFU, with five mice per dilution group. Higher doses were not utilized because we routinely achieve 100% infection with a dose of 104 IFU with MoPn Weiss (5). Swabs were collected at days 4, 7, 10, and 14 postinfection. In addition to microbiological detection of infection, we used seroconversion as a supplemental measure in order to detect any infection below the level of sensitivity of our culture method. Thus, on day 35 postinfection, all mice were bled, and the plasma obtained was assessed by ELISA for total anti-MoPn IgG antibodies. The results are shown in Table 2.
TABLE 2.
ID50 determinations for MoPn strains in urogenital tract infection
| Dose (IFU) | No. of positive mice/total no. of micec
|
|||
|---|---|---|---|---|
| Weiss
|
Nigg
|
|||
| Culture positivea | Seroconversionb | Culture positivea | Seroconversionb | |
| 101 | 0/5 | 0/5 | 0/5 | 0/5 |
| 102 | 1/5 | 4/5 | 0/5 | 0/5 |
| 103 | 3/5 | 5/5 | 4/5 | 5/5 |
| 104 | 5/5 | 5/5 | 4/5 | 5/5 |
Number of mice that were culture positive of the total number mice inoculated with either MoPn Weiss or MoPn Nigg at the given dose. Culture detection of infection was attempted using swabs collected at 4, 7, 10, and 14 days postinfection.
Number of mice of the total number of mice tested displaying seroconversion for plasma IgG (day 35 postinfection) against MoPn antigen (titers ranged from 640 to 5,120).
The ID50 values for the culture-positive group and the seroconversion group were 502 and 58 IFU, respectively, for mice infected with MoPn Weiss; those for the culture-positive group and the seroconversion group were 316 and 316 IFU, respectively, for mice infected with MoPn Nigg.
By culture detection of infection, the calculated ID50 values for urogenital inoculation were similar for the two strains, with MoPn Weiss displaying an estimated ID50 of 502 IFU and with MoPn Nigg displaying an estimated ID50 of 316 IFU (probit analysis). However, the seroconversion rates indicated a different pattern, with ID50 values of 58 IFU for MoPn Weiss and 316 IFU for MoPn Nigg. It is particularly interesting that four of five mice inoculated with MoPn Weiss at a dose of 102 IFU seroconverted with high antibody titers despite no microbiological evidence of infection and despite repeated attempts at culture, whereas mice inoculated with an identical dose of MoPn Nigg showed neither microbiological nor serological evidence of infection. It should also be noted that we previously observed that mice inoculated intravaginally with high titers of UV light-inactivated MoPn (108 IFU equivalents) do not seroconvert, as determined by ELISA for plasma IgG or vaginal IgA (K. H. Ramsey and R. G. Rank, unpublished data). Thus, any immunogenic stimulus from nonviable organisms in our stocks of these strains is not likely to account for seroconversion to the degree displayed in Table 2 (titers ranging from 640 to 5,120 in culture-negative but seropositive mice). Lastly, no differences were observed in the mean IFU isolated from any culture-positive mice in either the MoPn Weiss- or MoPn Nigg-infected group, regardless of initial inoculating dose (data not shown). Essentially, once microbiologically evident infection was detected, the infection courses were quite similar among mice regardless of the dose or strain.
Because MoPn urogenital tract infections in mice cause neither overt morbidity nor mortality, we decided to employ a more sensitive measure to determine virulence differences between the subject strains. To this end, mice were inoculated intranasally with log10 dilutions of the two MoPn strains to establish a respiratory tract infection and weighed daily, and the percent weight change was recorded as a measure of morbidity. Lethality or signs of becoming moribund were also noted daily and recorded, and mice that were obviously moribund were euthanized. Blood was collected from surviving animals on day 0 and day 35 postinfection for total IgG antibody against MoPn antigen as described above. The results of these experiments are summarized in Table 3 (ID50 and LD50 determinations), Fig. 2 (weight change), and Fig. 3 (mortality).
TABLE 3.
ID50 and LD50 determinations for MoPn strains in respiratory tract infection
| Dose (IFU) | No. of positive mice/total no. of micec
|
|||
|---|---|---|---|---|
| Weiss
|
Nigg
|
|||
| Mortalitya | Seroconversionb | Mortalitya | Seroconversionb | |
| 100 | 0/10 | 2/10 | 0/10 | 0/10 |
| 101 | 0/10 | 10/10 | 0/10 | 2/10 |
| 102 | 0/10 | 10/10 | 0/10 | 7/10 |
| 103 | 0/10 | 10/10 | 0/10 | 10/10 |
| 104 | 1/10 | ND | 0/10 | 10/10 |
| 105 | 9/10 | ND | 2/10 | ND |
| 106 | ND | ND | 4/10 | ND |
Number of moribund mice of the total number of mice inoculated at the given dose with either MoPn Weiss or MoPn Nigg through an 18- to 24-day monitoring period.
Number of mice of the total number of mice tested displaying seroconversion for plasma IgG (day 35 postinfection) against MoPn antigen (titers ranged from 40 to 5,120). All mice were seronegative at day 0.
The LD50 values for the mortality group were 3.7 × 104 IFU for mice infected with MoPn Weiss and >106 IFU for mice infected with MoPn Nigg. The ID50 values for the seroconversion group were 3 IFU for mice infected with MoPn Weiss and 58 IFU for mice infected with MoPn Nigg. ND, not done.
FIG. 2.
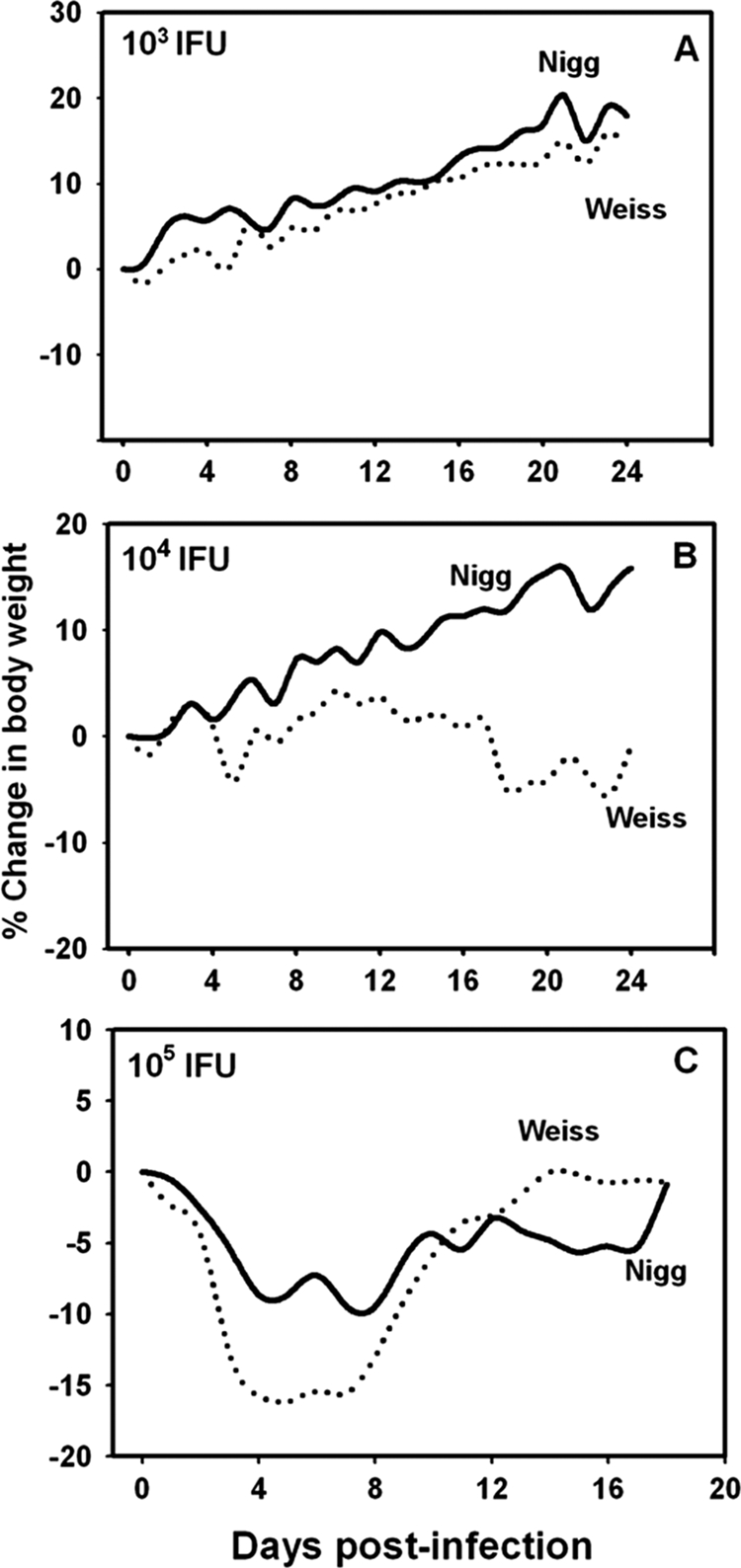
Morbidity in mice with MoPn respiratory infection. The solid line in each panel represents the mean daily percent change in body weight of 10 mice inoculated intranasally with MoPn Nigg. The dashed line represents the mean daily percent change in body weight of 10 mice inoculated with MoPn Weiss. (A) Mice were inoculated with 103 IFU of either strain. MoPn Weiss-infected mice gained less weight over time than did the MoPn Nigg-infected mice (P < 0.02 by repeated-measures ANOVA). (B) Mice were inoculated with 104 IFU of either strain. MoPn Weiss-infected mice had a significant weight loss compared to the weight gain experienced by mice infected with MoPn Nigg (P < 0.005). One mouse died in the MoPn Weiss-infected group on day 17. (C) Mice were inoculated with 105 IFU of either strain. All but two mice died by day 8 postinfection in the MoPn Weiss-infected group, and two mice died in the MoPn Nigg-infected group on days 9 and 17 (Fig. 3). The mean weight losses of the groups over time were significantly different (P < 0.02).
FIG. 3.
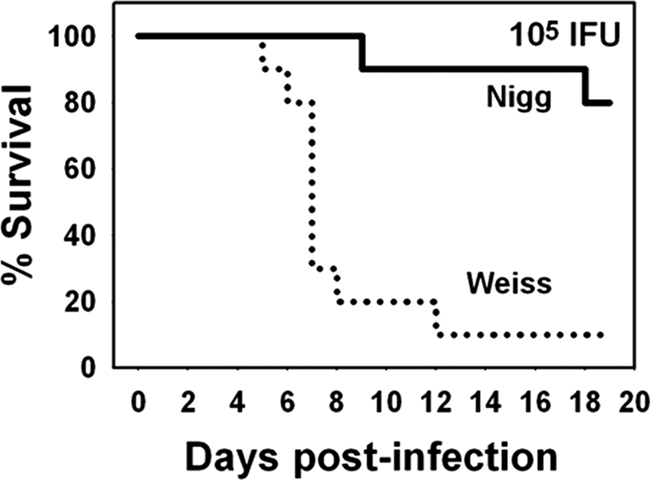
Kaplan-Meier survival curve. Mice were infected with 105 IFU of either MoPn Weiss or MoPn Nigg intranasally. Nine of ten mice infected with MoPn Weiss died through day 12 postinfection, whereas 2 of 10 mice infected with MoPn Nigg died on days 9 and 17 postinfection. The survival curves are significantly different (P < 0.004 by log-rank test).
No discernible differences in morbidity as measured by weight loss were noted between the strains at intranasal inoculating doses of 10°, 101, or 102 IFU per mouse (not shown) despite seroconversion in many mice in these groups, which would indicate that an infection had been achieved (Table 3). Of particular note was the seroconversion in 2 of 10 mice at low titers (titer of 40) and 100% of mice at high titers (titer of 2,560 to 10,240) at doses of 10° and 101 IFU, respectively, for the MoPn Weiss-infected group, yielding a surprisingly small estimated ID50 of 3 IFU (probit analysis). This is in contrast to 2 of 10 seroconversions at a dose of 101 IFU and 7 of 10 seroconversions at a dose of 102 IFU in the MoPn Nigg-inoculated mice. By serology, a 100% infection was not achieved unless 103 IFU of MoPn Nigg were used. Thus, the ID50 for MoPn Nigg as measured by seroconversion was estimated to be 58 IFU.
Note that inactivated MoPn antigen administered intranasally without adjuvant can cause a seroconversion of mice with low total IgG antibody titers (titers of 40 to 160) if high IFU equivalents are used (108 to 109 IFU [20 μg]) in at least two separate doses (C. J. Denman and K. H. Ramsey, unpublished data). Thus, we censored the lower seroconversions at the 10° dose for our calculations. The high-titer seroconversion that we observed in all of the mice inoculated at a dose of 101 IFU of MoPn Weiss was not likely to be due to the presence of large numbers of dead organisms in this single inoculum. In this sense, the results are quite similar to what we observed with MoPn Weiss urogenital seroconversion as described above.
Some interesting patterns began to emerge at a dose of 103 IFU (Fig. 2A). While both groups of mice displayed weight gain over time and, thus, no morbidity from infection with either strain, those inoculated with MoPn Weiss gained significantly less weight during the monitoring period (P < 0.02 by repeated-measures ANOVA). Additionally, when mice were infected with 104 IFU of MoPn Weiss, an overall mean weight loss throughout the monitoring period was noted, and one death was observed. This is in contrast to the mean group weight gain for the MoPn Nigg-infected group of approximately 15%, with no mortalities (P < 0.005). At a dose of 105 IFU, although both groups displayed a mean weight loss, MoPn Weiss-infected mice lost more weight (Fig. 2C) (P < 0.02) and had a higher mortality rate than did the MoPn Nigg-infected mice (P < 0.004 by log-rank test) (Fig. 3). In Fig. 2C, the data displayed beyond day 8 for the MoPn Weiss-infected mice represent those from only two remaining mice, and beyond day 12, the data are for the sole surviving mouse. Based on these observations, the estimated LD50 for MoPn Weiss was 3.7 × 104 IFU. Even at a dose of 106 IFU, the MoPn Nigg strain did not achieve a sufficient lethality to accurately calculate the LD50. We conclude from these experiments that the two strains display different pathogenic properties in the mouse host and that the MoPn Weiss strain is more virulent.
In vitro characteristics of C. muridarum strains.
To determine if the disparate pathogenic properties that the strains displayed in vivo translated to different properties in vitro, we used three different approaches. First of all, a chlamydial replication curve was established by using a host cell line that is known to be highly susceptible to chlamydial infection. HeLa 229 cells were infected at an MOI of 1 for both strains, and progeny viable EBs were determined for both strains over a 72-h period, likely representing two rounds of replication (Fig. 4). Progeny IFU were measured both in supernatant (taken to be extracellular EBs from chlamydia-lysed cells) and from sonicated coverslips (considered to be intracellular or cell-associated EBs) from which the monolayer was grown. In both samples, we observed a higher rate of production of progeny IFU for MoPn Weiss than for MoPn Nigg (P < 0.001 by two-factor repeated-measures ANOVA).
FIG. 4.
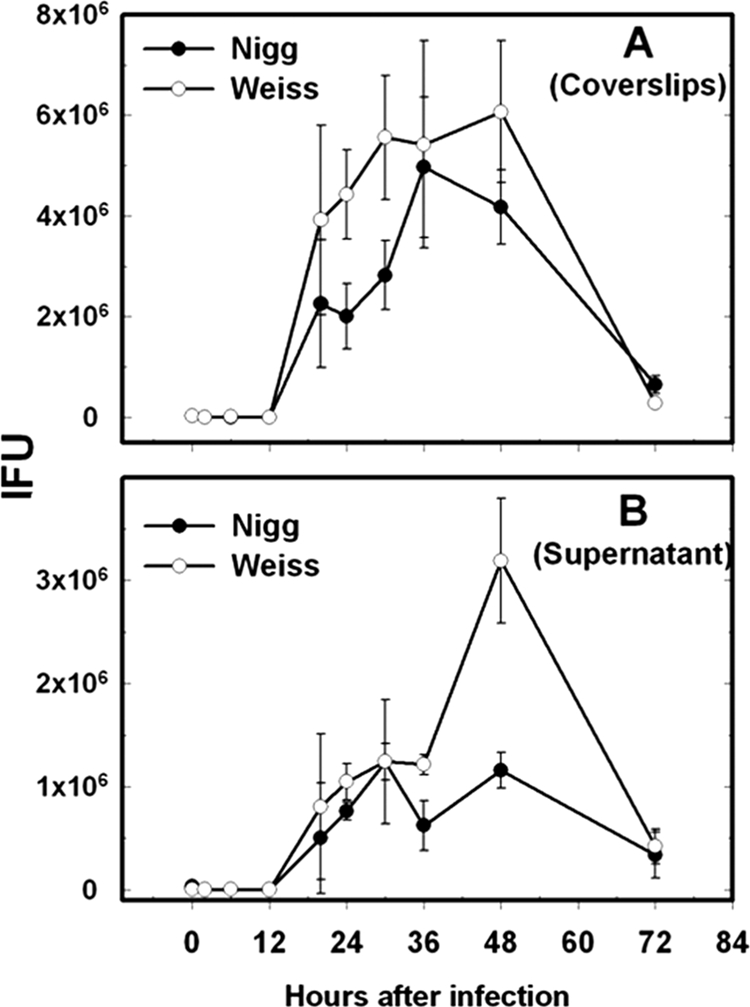
Replication rate of MoPn isolates in vitro. Each point represents the mean IFU at the specified time postinfection. Error bars are standard errors of the means of triplicate cultures (20 fields per replicate). Open circles represent MoPn Weiss; closed circles represent MoPn Nigg. (A) Results from sonicated coverslips representing cell-associated EBs. (B) Results from supernatants representing extracellular EBs. For both panels, the results represent a single experiment. A separate experiment yielding essentially identical results was also conducted (not shown). In both cases, MoPn Weiss displayed a higher yield of progeny than did MoPn Nigg (P < 0.001 by repeated-measures ANOVA).
Casual observations when comparing these strains also led us to believe that there were significant variations in the sizes of the inclusions formed in host cells between the two strains. Thus, we used morphometric analysis to assess inclusions size (area) at 12, 18, and 24 h postinfection (Fig. 5). At each time point, we observed that inclusions produced by MoPn Nigg infection displayed a larger mean area than those produced by MoPn Weiss infection. At first, this seemed somewhat counterintuitive in light of the higher yield of EBs in the MoPn Weiss strain, but this assumes that larger inclusions yields more infectious EBs, which apparently is not true.
FIG. 5.

Inclusion size variation in MoPn isolates. Inclusion size was measured as the area of each inclusion at 12, 18, and 24 h postinfection for the two strains. Each point represents the morphometric analysis of a single inclusion (n = 150 to 220 per time point). Closed circles are values for MoPn Nigg; open circles are values for MoPn Weiss. Horizontal bars are the means of each group at each time. P values provided at each time point represent those derived by comparing the two isolates using an unpaired, two-tailed t test.
Lastly, like other chlamydiae, C. muridarum is known to form plaques in monolayers of appropriate susceptible host cells, and the size of these plaques is known to vary considerably (14, 23). Again, using morphometric analysis, we assessed the mean plaque sizes of the two strains following identical incubation periods (Fig. 6). While variable from plaque to plaque, the MoPn Nigg strain of C. muridarum produced larger mean plaque size than did the MoPn Weiss strain.
FIG. 6.
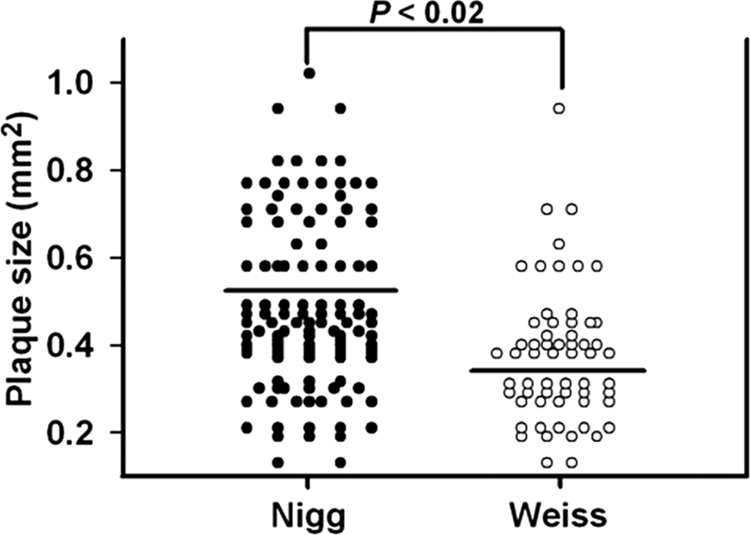
Plaque size variation in MoPn isolates. Plaque sizes in cultures of MoPn isolates grown for 5 days were measured. Each point represents the morphometric analysis of a single plaque of either strain (n = 118 for Nigg and n = 69 for Weiss). Closed circles are values for MoPn Nigg; open circles are values for MoPn Weiss. The means (horizontal bars) of the MoPn Nigg plaque sizes were larger than those of MoPn Weiss. The P value shown was derived by comparing the two isolates using an unpaired, two-tailed t test.
Comparative genomic sequencing and analysis of MoPn Nigg and MoPn Weiss.
Total genomic DNA was submitted for sequencing at the University of Maryland Institute for Genome Studies. Identified SNPs and indels were assessed as synonymous versus nonsynonymous mutations using LaserGene (v.8; DNAStar, Inc., Madison, WI). The results of the comparative sequence analysis are summarized in Table 4.
TABLE 4.
Nonsynonymous SNPs and insertion or deletion mutations identified in C. muridarum strains
| Open reading frame | Description | Position | Mutation (no. of mutations present/total no. sequences [%])d
|
Resulte | ||
|---|---|---|---|---|---|---|
| Nigga | Nigg2b | Weissc | ||||
| TC0052 | Major outer membrane protein, porin (ompA) | 59062 | GTT | — (18/18 [100]) | GTT | del→Cys |
| TC0052 | Major outer membrane protein, porin (ompA) | 59065 | C | C | T (26/27 [96]) | SNP, Ser→Phe |
| TC0107 | Lipoprotein, putative (intragenic) | 126406 | G | — (18/18 [100]) | — (20/20 [100]) | del→ |
| TC0107 | Lipoprotein, putative (intragenic) | 126416 | — | A (18/18 [100]) | A (20/20 [100]) | ins→ |
| TC0107 | Lipoprotein, putative (intragenic) | 126475 | A | — (21/21 [100]) | — (16/16 [100]) | del→ |
| TC0124 | Transcription-repair-coupling factor (trcF) | 151212 | T | T | — (8/20 [40]) | del→FS |
| TC0138 | mraY phospho-N-acetylmuramoylpentapeptide transferase | 169449 | — | TTT (17/17 [100]) | — | ins→Phe |
| TC0155 | 3′(2′), 5′-bisphosphate nucleotidase, putative | 187432 | G | A (15/15 [100]) | G | SNP, Met→Ile |
| TC0168 | Ribosomal protein L34 (rpmH) | 200671 | G | C (30/30 [100]) | C (17/17 [100]) | SNP, Cys→Ser |
| TC0341 | ABC transporter, permease protein, putative | 403623 | T | — (27/27 [100]) | — (14/14 [100]) | del→FS |
| TC0341 | ABC transporter, permease protein, putative | 403626 | — | C (26/26 [100]) | C (13/13 [100]) | ins→FS |
| TC0341 | ABC transporter, permease protein, putative | 403652 | T | C (22/25 [88]) | C (15/16 [94]) | SNP, Ser→Pro |
| TC0342 | ABC transporter, permease protein, putative | 404884 | A | — (21/22 [95]) | — (13/13 [100]) | del→FS |
| TC0408 | Conserved hypothetical protein | 468932 | G | T (18/18 [100]) | T (32/32 [100]) | SNP, Trp→Leu |
| TC0412 | Conserved hypothetical protein | 473118 | A | A | — (13/13 [100]) | del→FS |
| TC0412 | Conserved hypothetical protein | 473705 | G | — (20/20 [100]) | G | del→FS |
| TC0708 | Conserved hypothetical protein | 846475 | G | — (30/30 [100]) | — (23/23 [100]) | del→FS |
| TC0727 | 60-kDa outer membrane protein | 866121 | G | T (21/21 [100]) | T (19/19 [100]) | SNP, Gly→Cys |
| TC0832 | DNA polymerase III alpha subunit (dnaE) | 967487 | A | A | — | del→FS |
| TC0867 | Hypothetical protein | 1004276 | T | C | T | SNP, Ser→Pro |
Published C. muridarum Nigg sequence (28).
Plaque-purified isolate from our stock C. muridarum Nigg strain (Nigg2).
Plaque-purified isolate from our stock C. muridarum Weiss strain.
The detection rate of the mutation is expressed as the number of mutations present out of the total number of sequences assessed for that isolate (percentage). —, no nucleotide at the corresponding position. There were 5 unique mutations and 11 shared mutations for Nigg2, and there were 4 unique mutations and 11 shared mutations for MoPn Weiss.
FS, frameshift mutation; ins, insertion of a single nucleotide; del, deletion of a single nucleotide; →, result.
A total of five nonsynonymous SNPs or indels that were unique to our MoPn Nigg isolate (we termed this isolate Nigg2) compared to the published MoPn Nigg sequence were identified. Five SNPs or indels unique to MoPn Weiss compared to the published MoPn Nigg sequence were identified. In addition, there were 11 total SNPs or indels that were shared between our MoPn Nigg2 and MoPn Weiss isolates compared to the published MoPn Nigg sequence. Eight of these mutations resulted in a putative frameshift and thus a concomitant putative change in protein expression and/or function. Nine mutations resulted in a single amino acid change, and three were in an intragenic region preceding TC0107. A total of two synonymous SNPs were identified in the MoPn Weiss and MoPn Nigg2 isolates compared to the published MoPn Nigg sequence (data not shown).
We conclude from this finding that the two plaque-purified isolates that were sequenced varied sufficiently in their genomes to account for the biological differences observed in the stock populations used in our studies. However, it should be noted that one must be careful not to ascribe any differences in virulence or immunity in these studies to the identified genetic variations, because it cannot be asserted with reasonable assurance that these isolates are representative of the entire population within our stock preparations. To verify this, one would need to repeat the in vivo studies using the plaque-purified clonal isolates that were sequenced.
DISCUSSION
Our first observations that the MoPn Nigg and Weiss strains may possess disparate characteristics came from the heterotypic challenge experiments shown in Fig. 1. At first, it was counterintuitive to us that if these strains were truly different, homotypic challenge but not a heterotypic one should yield a second infection because, in general, antigenic similarity would yield a more protective immune response. However, when we observed that regardless of the primary infection, any inoculation with MoPn Weiss was far more likely to yield a second infection, we began to suspect that the best explanation was that MoPn Weiss was simply more virulent. Thus, the more virulent MoPn Weiss strain was able to establish a second infection upon challenge, regardless of the strain used in primary infection. The hypothesis of greater virulence was confirmed by each of our subsequent ID50 estimations for the urogenital tract and our LD50 estimations and morbidity curves for the respiratory tract. Also, the observation that seroconversion rates were higher for low-dose infections with the MoPn Weiss strain in both urogenital and respiratory tract infections than for mice identically inoculated with MoPn Nigg indicates that the Weiss strain is better able to establish even subclinical infections than the Nigg strain. It would be interesting to know if isolates of these strains vary significantly in their affinities for cells or selected tissues and/or in their abilities to disseminate beyond the initial site of inoculation.
Regardless of the virulence differences between these two strains, our challenge results indicate that the two isolates are likely to be very closely related on an antigenic level. This is because in any challenge infection between the two strains, the infectious burden was far lower than what we had observed either for a primary infection with MoPn of either strain or, as we previously observed, for challenges with the less-related human serovariants (24). Indeed, not every mouse becomes reinfected even when challenged with MoPn Weiss, thereby indicating solid protective immunity in some animals. Thus, the minimal infectious burden after challenge would imply a high degree of antigenic similarity between the two strains. Note that the observation of heterologous protection and the concomitant lack of homologous protection is not unique to mouse chlamydial infections (30).
There is an additional caution that should be exercised when interpreting our present comparative genomic data: all of our in vitro and in vivo characterizations and comparisons of MoPn Nigg and MoPn Weiss were done prior to plaque purification and subsequent genomic sequencing. We believe it to be likely that our stocks of MoPn Nigg and Weiss are heterogeneous. Hence, the genomic characterization of the plaque-purified populations that we reported herein may or may not be completely representative of our stocks as a whole. It is certainly possible that further clonal isolates within our stocks exist, which vary significantly with regard to in vitro growth characteristics and in vivo virulence properties, and that perhaps canonical “pathotypes” may be found. The importance of the comparative genomic aspects of this study, therefore, is the confirmation that significant genomic differences exist within our stock isolates of MoPn Nigg and MoPn Weiss rather than the specific identification of individual genetic variances that could account for virulence. The latter will necessitate the isolation and characterization of multiple isolates with mutations similar to those which we have sequenced or, perhaps, the identification of common genotypic correlates of virulence among several isolates.
More importantly, we believe that the current findings provide a unique opportunity to study several facets of chlamydia-host interactions. To this end, the recent findings of Kari et al., who utilized human isolates of the trachoma serovariants of Chlamydia trachomatis, are quite interesting (14). In a manner similar to that reported here, by comparing closely related strains, those investigators were able to correlate virulence differences to the presence or absence of just a single nucleotide. This is highly important to the field in that for the first time, one can begin to directly correlate specific chlamydial factors to virulence rather than merely making inferences related to orthologs derived from the genome. There were distinct similarities in their observations of separate serovar A isolates and our current findings for MoPn isolates. For example, the main differences in virulence in both human C. trachomatis and C. muridarum in our studies correlated to replication rates or “burst size,” as termed by Kari et al. This difference was also noted and described in detail previously by Miyairi et al. (17). Considering both the current report and that described previously by Kari et al., it appears that the simplest explanation for virulence across chlamydia species relates to how rapidly the pathogen is able to reproduce itself in the host. Higher replication rates will yield a better ability to establish infection, the production of more viable EBs, and, thus, the induction of a more vigorous and potentially damaging inflammatory response in the host. To this end, when virulence is considered along the spectrum of human chlamydial pathogens, one can certainly correlate more virulent species, such as Chlamydia psittaci, as being faster-replicating strains, whereas less virulent strains such as C. pneumoniae are generally slower-replicating strains. A difference between our findings and those of Kari et al. is that the more virulent serovariant A isolate of C. trachomatis produced larger plaques, whereas our more virulent C. muridarum isolate, MoPn Weiss, produced smaller plaques and smaller inclusions, although there were certainly variances observed in plaque sizes within our stock populations.
Other important recent and related studies include those of O'Connell and Nicks, who derived C. muridarum Nigg isolates that have been cured of the native plasmid (23). Plasmid-deficient strains sustain lower urogenital tract infections similarly to those of the parental strain but do not ascend into the upper tract to the same degree and thus cause a significantly lower degree of pathological immune responses (22). Interestingly, the plasmid-deficient strains induce immune responses that are protective against challenge with the parental strain and thus bring into question the possibility of an attenuated vaccine. How our findings correlate with those of Kari et al. and the observations of O'Connell and coworkers regarding the role of the MoPn plasmid remains to be determined. No doubt, many hope that the recent report of Binet and Maurelli will translate into a tractable method of mutating chlamydiae for the purpose of studying virulence and immunity in chlamydial infections (2). In the meantime, studies such as those by Kari et al. and O'Connell et al. and our current results reported herein are likely to be of high importance to the field.
After their original isolation, the origins of these MoPn isolates are somewhat murky. Several MoPn isolates reported in the literature in the late 1930s and early 1940s were described in a similar time frame and were subsequently shared among several investigators. Careful scrutiny of their origins reveals these isolates are likely distinct isolates (as reviewed in detail in reference 27). Based on reports by Hilleman (12, 13) in which he compared both the Nigg (then the “Atherton II strain”) and Weiss (then the “Chicago strain”) strains, it seems clear that the investigators originally working with MoPn recognized that there were indeed two isolates, and these isolates were apparently maintained as such in several subsequent studies. Thus, in what we believe to be the more likely scenario, multiple variants are found in any chlamydial infection in nature. The two different isolates indicate that different variants were numerically dominant in the different animals from which they were collected at two different time points. The implications for human disease are obvious. We believe it to be likely that individuals are infected with multiple variant populations and that the pathogenesis of the disease may be defined by the sum total of the variants or by whichever variant becomes numerically dominant. One could envision shifts in the populations during sexual or vertical transmission or mechanical transfer in ocular infections, since the inoculating doses may be relatively small, allowing for shifts in the variant populations. While not discounting variances in the host response, this could, to some extent, help to explain why chlamydial disease manifestations are more severe in some individuals but not others.
However, one cannot rule out other possibilities altogether. Selective pressures represented in continuous culture in disparate mammalian cell lines or in embryonated yolk sacs in vitro in laboratories employing various culture methodologies may have amplified mutations in one strain or the other. To our knowledge, since the first isolations of the pathogen were reported and since the subsequent advent of mammalian cell culture in the 1960s, essentially no investigators have reported the sequential passage of these strains in the natural host, and all have opted for the more convenient and reliable cell culture methodology for stock propagation, usually using human cervical adenocarcinoma cell lines (HeLa or HeLa 229 cells) or mouse fibroblasts (McCoy cells). Cell culture is likely a highly permissive growth scenario for chlamydial pathogens. One could speculate that in vivo passage would select for or maintain the more virulent variants within a population, that is, those who are better able to survive within the natural host. Additionally, no one has reportedly clonally isolated and expanded MoPn Nigg or MoPn Weiss until the recent reports of O'Connell et al., and no one has determined virulence differences within clonal isolates (23). Lastly, one cannot discount the possibility that one or more isolates were comingled during passage over the ensuing years and that our stock populations represent a mixture thereof.
It is interesting that MoPn was not originally isolated from the urogenital tract but appeared to be either an endemic respiratory tract or, more likely, gastrointestinal tract pathogen (15, 27). We have also shown that MoPn can be passed from mouse to mouse housed in the same cage, likely by the oral route (6). The possibility that MoPn maybe a gastrointestinal pathogen is not surprising in that colonies of laboratory mice were maintained under far less stringent conditions than what is standard for today's laboratory. This means that MoPn is not likely to be a natural pathogen of the murine urogenital tract but rather that it is a pathogen of either of the respiratory or gastrointestinal tract. Certainly, genetic variation of chlamydiae according to tissue tropism is not unheard of (4, 9), and we feel that it is likely that MoPn variants that are identified subsequent to this report may exhibit such tropisms.
History aside, the importance of our findings as well as those of Kari et al. and O'Connell et al. is that in the current era of genomics and transcriptomics, the opportunity now exists to compare closely related chlamydial strains that vary in their immunobiological and pathogenic properties and to make inferences with regard to the function of specific genes and then study the functions of the variable gene products in the natural host in greater detail. We have an opportunity similar to that that reported previously by Kari et al., with the notable exception that MoPn has the obvious advantages of the mouse as a model and natural host, which will allow us to study chlamydial immunobiology, pathogenesis, and host-pathogen interactions in a more detailed manner. These types of investigations will ultimately lead to greater understandings of virulence, immunity, and host-parasite interactions in this important human pathogen.
Acknowledgments
This work was supported by Public Health Service grants AI49354 (K.H.R.) and AI59650 (R.G.R.). Funding for genomic sequencing was provided by a grant from the Chicago College of Osteopathic Medicine.
We thank James Molder and Julius Schachter for their personal communication regarding the history of the MoPn strains and reviewing the accuracy of this account prior to submission of the manuscript.
Editor: V. J. DiRita
Footnotes
Published ahead of print on 26 May 2009.
REFERENCES
- 1.Barron, A. L., H. J. White, R. G. Rank, B. L. Soloff, and E. B. Moses. 1981. A new animal model for the study of Chlamydia trachomatis genital infections: infection of mice with the agent of mouse pneumonitis. J. Infect. Dis. 14363-66. [DOI] [PubMed] [Google Scholar]
- 2.Binet, R., and A. T. Maurelli. 2009. Transformation and isolation of allelic exchange mutants of Chlamydia psittaci using recombinant DNA introduced by electroporation. Proc. Natl. Acad. Sci. USA 106292-297. doi: 10.1073/pnas.0806768106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caldwell, H. D., and J. Schachter. 1982. Antigenic analysis of the major outer membrane protein of Chlamydia spp. Infect. Immun. 351024-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caldwell, H. D., H. Wood, D. Crane, R. Bailey, R. B. Jones, D. Mabey, I. Maclean, Z. Mohammed, R. Peeling, C. Roshick, J. Schachter, A. W. Solomon, W. E. Stamm, R. J. Suchland, L. Taylor, S. K. West, T. C. Quinn, R. J. Belland, and G. McClarty. 2003. Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J. Clin. Investig. 1111757-1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cotter, T. W., G. S. Miranpuri, K. H. Ramsey, C. E. Poulsen, and G. I. Byrne. 1997. Reactivation of chlamydial genital tract infection in mice. Infect. Immun. 652067-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cotter, T. W., K. H. Ramsey, G. S. Miranpuri, C. E. Poulsen, and G. I. Byrne. 1997. Dissemination of Chlamydia trachomatis chronic genital tract infection in gamma interferon gene knockout mice. Infect. Immun. 652145-2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De La Maza, L. M., S. Pal, A. Khamesipour, and E. M. Peterson. 1994. Intravaginal inoculation of mice with the Chlamydia trachomatis mouse pneumonitis biovar results in infertility. Infect. Immun. 622094-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Everett, K. D., R. M. Bush, and A. A. Andersen. 1999. Emended description of the order Chlamydiales, proposal of Parachlamydiaceae fam. nov. and Simkaniaceae fam. nov., each containing one monotypic genus, revised taxonomy of the family Chlamydiaceae, including a new genus and five new species, and standards for the identification of organisms. Int. J. Syst. Bacteriol. 49415-440. [DOI] [PubMed] [Google Scholar]
- 9.Gieffers, J., L. Durling, S. P. Ouellette, J. Rupp, M. Maass, G. I. Byrne, H. D. Caldwell, and R. J. Belland. 2003. Genotypic differences in the Chlamydia pneumoniae tyrP locus related to vascular tropism and pathogenicity. J. Infect. Dis. 1881085-1093. [DOI] [PubMed] [Google Scholar]
- 10.Gonnert, R. 1941. Die Bronchopneumoniae, eine neue Viruskrankheit der Maus. Zentralbl. Backteriol. 147161-173. [Google Scholar]
- 11.Gordon, F. B., G. Freeman, and J. M. Clampit. 1938. A pneumonia-producing filtrable agent from stock mice. Proc. Soc. Exp. Biol. Med. 39450-453. [Google Scholar]
- 12.Hilleman, M. R. 1945. Immunological studies on the psittacosis-lymphgranuloma group of viral agents. J. Infect. Dis. 7696-114. [Google Scholar]
- 13.Hilleman, M. R., and F. B. Gordon. 1944. Immunologic relations of the psittacosis-lymphogranuloma venereum group of viral agents. Proc. Soc. Exp. Biol. Med. 56159-161. [Google Scholar]
- 14.Kari, L., W. M. Whitmire, J. H. Carlson, D. D. Crane, N. Reveneau, D. E. Nelson, D. C. Mabey, R. L. Bailey, M. J. Holland, G. McClarty, and H. D. Caldwell. 2008. Pathogenic diversity among Chlamydia trachomatis ocular strains in nonhuman primates is affected by subtle genomic variations. J. Infect. Dis. 197449-456. [DOI] [PubMed] [Google Scholar]
- 15.Karr, H. V. 1943. Study of a latent pneumotropic virus of mice. J. Infect. Dis. 43108-116. [Google Scholar]
- 16.Margulies, M., M. Egholm, W. E. Altman, S. Attiya, J. S. Bader, L. A. Bemben, J. Berka, M. S. Braverman, Y. J. Chen, Z. Chen, S. B. Dewell, L. Du, J. M. Fierro, X. V. Gomes, B. C. Godwin, W. He, S. Helgesen, C. H. Ho, G. P. Irzyk, S. C. Jando, M. L. Alenquer, T. P. Jarvie, K. B. Jirage, J. B. Kim, J. R. Knight, J. R. Lanza, J. H. Leamon, S. M. Lefkowitz, M. Lei, J. Li, K. L. Lohman, H. Lu, V. B. Makhijani, K. E. McDade, M. P. McKenna, E. W. Myers, E. Nickerson, J. R. Nobile, R. Plant, B. P. Puc, M. T. Ronan, G. T. Roth, G. J. Sarkis, J. F. Simons, J. W. Simpson, M. Srinivasan, K. R. Tartaro, A. Tomasz, K. A. Vogt, G. A. Volkmer, S. H. Wang, Y. Wang, M. P. Weiner, P. Yu, R. F. Begley, and J. M. Rothberg. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437376-380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyairi, I., O. S. Mahdi, S. P. Ouellette, R. J. Belland, and G. I. Byrne. 2006. Different growth rates of Chlamydia trachomatis biovars reflect pathotype. J. Infect. Dis. 194350-357. [DOI] [PubMed] [Google Scholar]
- 18.Morrison, R. P., and H. D. Caldwell. 2002. Immunity to murine chlamydial genital infection. Infect. Immun. 702741-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moulder, J. W., T. P. Hatch, C.-C. Kuo, J. Schachter, and J. Storz. 1984. Genus Chlamydia, p. 729-739. In N. R. Krieg (ed.), Bergey's manual of systematic bacteriology. Williams & Wilkins, Baltimore, MD.
- 20.Nigg, C. 1942. An unidentified virus which produces pneumonia and systemic infection in mice. Science 9549-50. [DOI] [PubMed] [Google Scholar]
- 21.Nigg, C., and M. D. Eaton. 1944. Isolation from normal mice of a pneumotropic virus which forms elementary bodies. J. Exp. Med. 79497-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Connell, C. M., R. R. Ingalls, C. W. Andrews, Jr., A. M. Scurlock, and T. Darville. 2007. Plasmid-deficient Chlamydia muridarum fail to induce immune pathology and protect against oviduct disease. J. Immunol. 1794027-4034. [DOI] [PubMed] [Google Scholar]
- 23.O'Connell, C. M., and K. M. Nicks. 2006. A plasmid-cured Chlamydia muridarum strain displays altered plaque morphology and reduced infectivity in cell culture. Microbiology 1521601-1607. [DOI] [PubMed] [Google Scholar]
- 24.Ramsey, K. H., T. W. Cotter, R. D. Salyer, G. S. Miranpuri, M. A. Yanez, C. E. Poulsen, J. L. DeWolfe, and G. I. Byrne. 1999. Prior genital tract infection with a murine or human biovar of Chlamydia trachomatis protects mice against heterotypic challenge infection. Infect. Immun. 673019-3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramsey, K. H., W. J. Newhall, and R. G. Rank. 1989. Humoral immune response to chlamydial genital infection of mice with the agent of mouse pneumonitis. Infect. Immun. 572441-2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rank, R. G. 1999. Models of immunity, p. 239-295. In R. S. Stephens (ed.), Chlamydia: intracellular biology, pathogenesis, and immunity. ASM Press, Washington, DC.
- 27.Rank, R. G. 2006. Chlamydial diseases, p. 325-348. In J. Fox, S. Barthold, C. Newcomer, A. Smith, F. Quimby, and M. Davisson (ed.), The mouse in biomedical research, 2nd ed. Academic Press, San Diego, CA.
- 28.Read, T. D., R. C. Brunham, C. Shen, S. R. Gill, J. F. Heidelberg, O. White, E. K. Hickey, J. Peterson, T. Utterback, K. Berry, S. Bass, K. Linher, J. Weidman, H. Khouri, B. Craven, C. Bowman, R. Dodson, M. Gwinn, W. Nelson, R. DeBoy, J. Kolonay, G. McClarty, S. L. Salzberg, J. Eisen, and C. M. Fraser. 2000. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 281397-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swenson, C. E., E. Donegan, and J. Schachter. 1983. Chlamydia trachomatis-induced salpingitis in mice. J. Infect. Dis. 1481101-1107. [DOI] [PubMed] [Google Scholar]
- 30.Thirumalapura, N. R., H. L. Stevenson, D. H. Walker, and N. Ismail. 2008. Protective heterologous immunity against fatal ehrlichiosis and lack of protection following homologous challenge. Infect. Immun. 761920-1930. [DOI] [PMC free article] [PubMed] [Google Scholar]