

Abstract
EAL domain-based cyclic di-GMP (c-di-GMP)-specific phosphodiesterases play important roles in bacteria by regulating the cellular concentration of the dinucleotide messenger c-di-GMP. EAL domains belong to a family of (β/α)8 barrel fold enzymes that contain a functional active site loop (loop 6) for substrate binding and catalysis. By examining the two EAL domain-containing proteins RocR and PA2567 from Pseudomonas aeruginosa, we found that the catalytic activity of the EAL domains was significantly altered by mutations in the loop 6 region. The impact of the mutations ranges from apparent substrate inhibition to alteration of oligomeric structure. Moreover, we found that the catalytic activity of RocR was affected by mutating the putative phosphorylation site (D56N) in the phosphoreceiver domain, with the mutant exhibiting a significantly smaller Michealis constant (Km) than that of the wild-type RocR. Hydrogen-deuterium exchange by mass spectrometry revealed that the decrease in Km correlates with a change of solvent accessibility in the loop 6 region. We further examined Acetobacter xylinus diguanylate cyclase 2, which is one of the proteins that contains a catalytically incompetent EAL domain with a highly degenerate loop 6. We demonstrated that the catalytic activity of the stand-alone EAL domain toward c-di-GMP could be recovered by restoring loop 6. On the basis of these observations and in conjunction with the structural data of two EAL domains, we proposed that loop 6 not only mediates the dimerization of EAL domain but also controls c-di-GMP and Mg2+ ion binding. Importantly, sequence analysis of the 5,862 EAL domains in the bacterial genomes revealed that about half of the EAL domains harbor a degenerate loop 6, indicating that the mutations in loop 6 may represent a divergence of function for EAL domains during evolution.
The cyclic dinucleotide cyclic di-GMP (c-di-GMP) has emerged as a major bacterial messenger for mediating a variety of cellular functions that range from virulence expression and biofilm formation (5, 14, 30). The cellular concentration of c-di-GMP is controlled by the GGDEF domain proteins with diguanylate cyclase (DGC) activity and the EAL domain proteins with c-di-GMP-specific phosphodiesterase (PDE) activity. GGDEF domains catalyze the synthesis of c-di-GMP from GTP, whereas EAL domains catalyze the hydrolysis of c-di-GMP to generate the linear 5′-pGpG. Although a family of HD-GYP domain proteins has also been found as c-di-GMP-specific PDEs, the overwhelmingly large number of genes encoding the EAL domains in bacterial genomes suggests that the EAL domains are the major PDEs for maintaining the cellular c-di-GMP concentration. Remarkably, multiple copies of EAL domain-encoding genes are usually found in bacterial cells, with as many as 21 in Pseudomonas aeruginosa and 32 in Vibrio cholerae. Although many of the EAL domains were found to function as PDE domains for c-di-GMP degradation, emerging evidence suggests that some EAL domains function as ligand- or protein-binding domains without catalytic activity (24, 28, 40).
The detailed structure and catalytic mechanism of the EAL domains have started to be elucidated recently. The crystal structures of two proteins with EAL domains, TdEAL and YkuI, have been determined (Protein Data Bank accession nos. 2BAS, 2R6O, and 2w27) (23). EAL domains adopt a (β/α)8 barrel fold that contains two extended strands, including an antiparallel strand. The (β/α)8 barrel fold, first found in triosephosphate isomerase, has been observed in a diversity of enzymes that include many hydrolyases and isomerases (34). Similar to other (β/α)8 barrel fold enzymes, the catalytic residues of the EAL domain are located at the C-terminal ends of the β-strands and the beginning of the β→α loops connecting the β-strands and α-helices. In the proposed mechanism, EAL domains catalyze the hydrolysis of c-di-GMP by using a Mg2+ ion and a general base catalyst (Glu) for generating the nucleophilic H2O (28). The catalytic mechanism is supported by the crystal structure of the YkuI-substrate binary complex (Protein Data Bank accession no. 2w27) and the model of the TdEAL-substrate complex (23, 28). Both structures showed that the EAL domains bind c-di-GMP in such a configuration that the scissile phosphorus-oxygen bond aligns linearly with the attaching water and the general base catalyst. The catalytic mechanism can account for the lack of catalytic activity for most known inactive EAL domains, with the loss of enzymatic activity arising from the absence of the general base catalyst and/or the residues that coordinate the Mg2+ ion (28).
It is well-known that many (β/α)8 barrel fold enzymes contain a flexible active site loop between the β6 strand and α6 helix (34). Despite the diverse reactions catalyzed by (β/α)8 barrel fold enzymes, this extended loop, often referred to as loop 6, plays an important role as a functional lid for substrate sequestering, solvent exclusion, and product release (15). The loop was found to facilitate substrate binding and conformational transition in tryptophan synthase (3, 4) and functions as a lid for substrate sequestering during catalysis in inosine 5′-monophosphate dehydrogenase (22). Notably, it was shown that the loop sways from the active site in the nonactive structure of ribulose-1,5-bisphosphate carboxylase but folds over to shield the active site from the solvent in the activated structure (21). Similar functions have also been proposed for loop 6 in other (β/α)8 barrel fold enzymes, such as triosephosphate isomerase and phosphoriboxyl anthranilate isomerase (15, 25, 26). Hence, it seems that the functional role of loop 6 has been well preserved in (β/α)8 barrel fold enzymes during evolution. The (β/α)8 barrel folded EAL domains also contain an eight-residue loop between the β6 strand and α6 helix that seems to be critical for catalysis. Schmidt and coworkers (32) first noticed that the catalytically active EAL domains seem to contain a conserved motif that was later confirmed to contain loop 6 [DFG(T/A)GYSS] and one of the residues (Asp) for Mg2+ binding (28, 32). We previously noticed that mutation of the essential catalytic residues is usually accompanied by the degeneration of loop 6 in catalytically inactive EAL domains (28). Moreover, we observed that the mutation of a residue interacting with loop 6 in the EAL domain-containing RocR abolished enzymatic activity, which led us to postulate a critical role for loop 6 in catalysis (28).
To elucidate the precise role played by loop 6 in c-di-GMP hydrolysis, we examined three EAL domain-containing proteins that include RocR, PA2567, and A. xylinus DGC2. The residues of loop 6 [DFG(A/T)SYSS] in RocR and PA2567 are well conserved, as observed in other catalytically active EAL domains. We show that mutations in the loop 6 region in RocR and PA2567 had significant effect on the structure and catalysis of the EAL domain. By using the method of hydrogen-deuterium (H/D) exchange-coupled mass spectrometry, we demonstrated that a single remote mutation in the phosphoreceiver domain of RocR caused correlated changes in loop 6 conformation and catalytic properties. We further show that the catalytic activity of the inactive EAL domain of A. xylinus DGC2 can be recovered by restoring loop 6. The functional roles of loop 6 in EAL domains in substrate binding and catalysis were discussed in conjunction with the structural data for two EAL domains.
MATERIALS AND METHODS
Protein cloning, expression, and purification.
The cloning, expression, and purification of RocR and PA2567 have been described previously (28). All mutants were generated using the site-directed mutagenesis II kit (Stratagene) according to the manufacturer's instruction manual. The mutant proteins were expressed and purified following the same procedure for the wild-type proteins. The proteins were flash frozen in liquid N2 and stored in a −80°C freezer after the measurement of concentration by the Bradford method.
For the full-length A. xylinus diguanylate cyclase 2 (DGC2), a gene construct encoding the full-length DGC2 gene from A. xylinus was commercially synthesized by GenScript Inc., amplified by PCR, and cloned into the expression vector pET26b(+) (Novagen) between the NdeI and XhoI restriction sites. The plasmid harboring the gene construct and the C-terminal His6 tag-encoding sequence were transformed into Escherichia coli strain BL21(DE3). For protein expression, 2 ml of inoculum from the cell stock was added to 1 liter of LB medium. The bacterial culture was grown at 37°C until it reached an optical density (600 nm) of 0.8 before being induced with 0.8 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 16°C for ca. 12 h. After centrifugation, the pellets were lysed in 20 ml lysis buffer, which consists of 50 mM Tris-Cl (pH 8.0), 200 mM NaCl, 5% glycerol, 1% β-mercaptoethanol, and 1 mM phenylmethylsulfonyl fluoride. After centrifugation at 25,000 rpm for 30 min, the supernatant was filtered and then incubated with 2 ml of Ni2+-nitrilotriacetic acid resin (Qiagen) for 30 min. The resin was washed with 50 ml of W1 buffer (lysis buffer with 20 mM imidazole) and 50 ml of W2 buffer (lysis buffer with 50 mM imidazole). The proteins were eluted using a step gradient method with the elution buffer containing 50 mM Tris (pH 8.0), 200 mM NaCl, 5% glycerol, and 200, 300, and 500 mM imidazole. After sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis, fractions with purity higher than 95% were pooled together and desalted using a PD-10 column (GE Healthcare). Proteins were first concentrated using an Amicon concentrator (Millipore) to ca. 5 mg/ml before being assayed for enzymatic activity.
For the stand-alone EALDGC2 domain, an expression vector encoding 306 to 574 amino acids of A. xylinus DGC2 was constructed by amplifying the corresponding gene fragment by high-fidelity PCR and cloning into pET-28(b+) (Novagen) between the NdeI and XhoI restriction sites. The plasmid harboring the gene construct was transformed into E. coli strain BL21(DE3). The protein was expressed and purified in the same procedure as full-length A. xylinus DGC2. The triple mutant (N473K476I478 → D473T476Y478) was generated using the site-directed mutagenesis II kit (Stratagene) according to the manufacturer's instruction manual. Mutant protein was expressed and purified following the same procedure described above.
Steady-state enzymatic activity assay.
The substrate c-di-GMP used for kinetic measurement was produced enzymatically by using an engineered thermophilic DGC protein (27). The measurement of the steady-state kinetic parameters was carried out by monitoring the formation of the product 5′-pGpG as described previously (28). The turnover number (kcat) and Michealis constant (Km) used for estimating enzyme efficiency and substrate binding were obtained by fitting the initial velocities at various substrate concentrations to the Michaelis-Menten equation with the exception of the two mutants that exhibit substrate inhibition. The kinetic data for the two mutants were fit using a model that assumes the substrate can bind to the enzyme at a productive and a nonproductive (or inhibitory) binding site (Fig. 1) .
FIG. 1.

Kinetic model for mutants that exhibit substrate inhibition. The model assumes that the substrate can bind to the enzyme at a productive binding site and a nonproductive (inhibitory) binding site. Abbreviations: E, enzyme; P, product; S, substrate; kc, rate constant for the chemical step.
The curves were obtained by fitting the kinetic data to the following equation derived from the model based on the steady-state and rapid equilibrium assumptions. In the following equation, Ki is the inhibition constant, α and β are the factors by which the Km, Ki, and Vmax change when the second substrate is bound at the nonproductive site, v is initial velocity, and [S] is the concentration of the substrate.
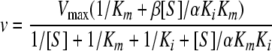 |
(1) |
Size exclusion chromatography.
Gel filtration was performed at 4°C using the AKTA FPLC (fast protein liquid chromatography) system (GE Healthcare) equipped with a Superdex 200 HR 16/60 column (Amersham Biosciences). The buffer used for gel filtration is comprised of 20 mM Tris-HCl (pH 8.0), 0.5 M NaCl, 5% glycerol, and 0.5 mM dithiothreitol.
Amide hydrogen-deuterium exchange by mass spectrometry.
The apparatus for H/D exchange was similar to the setup described elsewhere (13, 19, 29). The purified RocR or D56N mutant was exchanged into 100 mM sodium phosphate buffer (pH 7.0) that contains 150 mM sodium chloride and 1 mM dithiothreitol. The peptides generated by pepsin digestion were first identified by a tandem mass spectrometry experiment. The H/D exchange reactions were initiated by adding 10 μl of newly thawed protein solution to 90 μl of D2O. After incubation for various time lengths (1/3, 1, 5, 15, 30 and 60 min), the exchange reaction was rapidly quenched by lowering the pH to 2.4 and lowering the temperature to 0°C, and followed by pepsin digestion. The resulting peptides were passed through a homemade capillary reverse-phase C18 high-performance liquid chromatography (HPLC) column and eluted into the quadruple time-of-flight mass spectrometer (Applied Biosystems/MDX Sciex QSTAR Elite system) equipped with an IonSpray source. The peptides were eluted at a flow rate of 20 μl/min using a step gradient (7.5, 10, 12.5, 15, 17.5, 20, 22.5, 25, 30, 40, and 80% acetonitrile in 0.1% formic acid). The time from the initiation of digestion to the elution of the last peptide was approximately 22 min. Analyst QS software was used for spectrum analysis and data extraction, and the H/D exchange data were processed by using the program HX-Express (42). Zero time point control or the “artifactual in-exchange” control was performed by adding the quenching buffer to the protein solution before exposure to D2O (13). Measured peptide masses were corrected for artifactual in-exchange at t = 0, normalized to 100% D2O, and corrected for back exchange following the empirical method described by Hoofnagle et al. (13). The time-dependent H/D exchange data were fit by nonlinear least-squares fitting to the equation shown below.
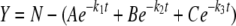 |
(2) |
where N is the total number of deuterons incorporated over the observed course for each peptide, and A, B, and C correspond to the number of amides exchanging with the rate constants k1, k2, and k3, respectively (13) (20). The number of nonexchanging amides is calculated from the total number of the backbone amides (NH) in the peptide, excluding proline residues.
Bioinformatic analysis and structural modeling.
The Stockholm-format alignment of 5,862 sequences in the EAL (PF00563) family was downloaded from the PFAM database (http://pfam.sanger.ac.uk//family/EAL). Two PERL scripts were used to search for sequences that contain the essential catalytic residues (equivalent to E175, N233, E265, D295, and E352 in RocR) and the motif DFG(A/S/T)(A/G)(Y/F)(S/T)(S/T/G/A/N) that constitutes loop 6. On the basis of the search results, the 5,862 sequences were divided into four groups as we discuss below in the Discussion. For homology modeling, the structural model for the EAL domain of RocR was constructed with the coordinates of the TdEAL structure (Protein Data Bank accession no. 2R6O) as the template in SWISS-MODEL (1). The structural model for the response receiver (RR) domain of RocR was built using the structure of the RR domain of CheB (Protein Data Bank accession no. 1A2O) as the template.
RESULTS
Catalytically active EAL domains require four residues for binding the essential Mg2+ ion and a general base catalyst for activating the nucleophilic water (28). In addition, a conserved loop, loop 6, which contains the motif DFG(T/A)GYSS, was found in most known active EAL domains (32). Considering that a large number of EAL domains in the genomes contain a degenerate loop 6, elucidation of the functional role of loop 6 may yield clues to the functions of these EAL domains.
Effects of mutations in the loop 6 region.
The P. aeruginosa protein RocR is a response regulator that contains a RR domain and a catalytically active EAL domain (18, 28). RocR catalyzes the hydrolysis of c-di-GMP with a turnover number (kcat) of 0.67 ± 0.03 s−1 and Michaelis constant (Km) of 3.2 ± 0.3 μM under the enzymatic assay conditions. We previously found that the mutation of Glu268, a highly conserved residue that was suggested to stabilize loop 6, significantly altered the catalytic properties of RocR (Fig. 2) (28). The E268A mutation reduced the enzyme activity to below the measurable level, whereas the E268Q mutation decreased Km to 0.31 ± 0.1 μM and caused a 446-fold reduction in kcat. We found that the mutation of Phe297, a residue on loop 6 (DF297GASYSS), reduced kcat to 0.02 ± 0.1 s−1 and Km to 0.73 ± 0.2 μM (Table 1) . Size exclusion chromatography showed that mutant enzymes with the E268A mutation, enzymes with the F297A mutation, and the majority of enzymes with the E268Q mutation formed high-molecular-weight oligomer (HMWO) and eluted in the void volume (data not shown). The change in oligomeric state for the three mutants indicated that loop 6 is crucial for maintaining the quaternary structure of RocR. This is in line with the observation that loop 6 is located at the dimer interface as we discuss later. In addition, the side chain of loop residue Ser302 (DFGAGYSS302) interacts with Glu268 through hydrogen bonding according to the EALRocR structural model and the TdEAL structure (Fig. 2A). Remarkably, the elimination of the single hydrogen bond by the S302A mutation caused the enzyme to exhibit strong substrate inhibition kinetics with an inhibition constant of 10.9 ± 3.0 μM without altering the oligomeric structure (Fig. 2B). The substrate inhibition strongly suggested that binding of c-di-GMP is sensitive to the change of the loop conformation. Finally, the first residue of the loop (Asp296) may be involved in the binding of c-di-GMP according to the structural model (28). The D296A mutation caused significant changes in catalytic parameters without altering the oligomeric structure, by causing a decrease in kcat by 33.5-fold and an increase of Km to 8.6 ± 2.8 μM.
FIG. 2.

Loop 6 of RocR and the effects of mutations on catalysis. (A) Structural model of EALRocR with the residues of loop 6 and Glu268 highlighted. The hydrogen bonds formed between Glu268 and the loop residues Gly300 and Ser302 are represented by the broken lines. The Mg2+ ion is shown as the ball. (B) Effects of the mutations in the loop 6 region on the steady-state kinetics of RocR. The curves were generated by fitting the data to the Michaelis-Menten equation with the exception of the S302A mutant, for which the curve was generated by fitting the kinetic data to equation 1.
TABLE 1.
Steady-state kinetic parameters for RocR and its mutantsa
| RocR enzyme | kcat (s−1) | Km (μM) | kcat/Km (s−1 μM−1) | Ki (μM) |
|---|---|---|---|---|
| Wild-type | 0.67 ± 0.03 | 3.2 ± 0.3 | 0.21 ± 0.02 | |
| E268A mutant | NDb | ND | ND | |
| E268Q mutant | (1.5 ± 0.1) × 10−3 | 0.3 ± 0.1 | (4.8 ± 0.5) × 10−3 | |
| S302A mutant | 0.14 ± 0.07 | 5.5 ± 2.6 | (2.5 ± 2.0) × 10−2 | 10.9 ± 3.0 |
| D296A mutant | (2.1 ± 0.3) × 10−2 | 8.6 ± 2.8 | (2.7 ± 0.9) × 10−3 | |
| D56N D296A double mutant | (1.2 ± 0.3) × 10−2 | 9.4 ± 2.3 | (2.1 ± 0.6) × 10−3 | |
| D56N mutant | 0.13 ± 0.01 | 0.2 ± 0.03 | 0.65 ± 0.1 | |
| F297A mutant | 0.02 ± 0.01 | 0.7 ± 0.2 | (2.8 ± 1.6) × 10−2 |
Conditions for RocR and its mutants were 100 mM Tris buffer (pH 8.0) (23°C), 20 mM KCl, and 25 mM MgCl2.
ND, not determined due to inactivity or extreme low activity (<105-fold less active than wild-type RocR).
A large number of EAL domain proteins contain a widely occurring GGDEF-EAL didomain unit. PA2567 from P. aeruginosa is such a protein; PA2567 contains a catalytically active EAL domain along with the GAF and GGDEF domains (28, 35) (Fig. 3). Site-directed mutagenesis and kinetic measurements were performed to determine whether loop 6 plays similar roles in PA2567. Surprisingly, the single mutations of Phe493 and Ser498, which are the equivalents of Phe297 and Ser302 in RocR, caused the protein to form inclusion bodies during protein expression. Meanwhile, the mutation of Glu464, the equivalent of Glu268 in RocR, caused strong substrate inhibition at high substrate concentration, with an inhibition constant of 30.4 ± 12.0 μM (Fig. 4 and Table 2). The mutation E464A did not perturb the oligomeric state of the dimeric PA2567 according to size exclusion chromatography. The results from the study of PA2567 and RocR seemed to suggest that loop 6 plays crucial roles in stabilizing the overall protein structure and participating in catalysis, probably through mediating substrate binding.
FIG. 3.
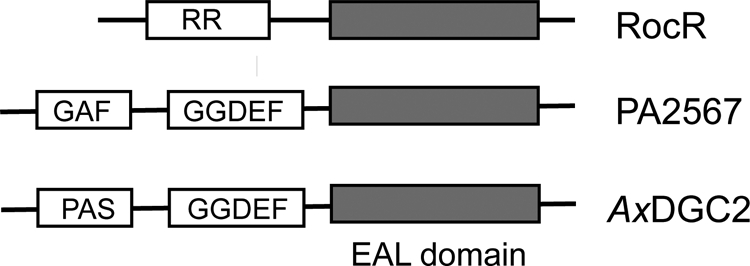
Domain organization of the RocR protein, the PA2567 protein from P. aeruginosa PAO-1, and the DGC2 protein from A. xylinus (AxDGC2).
FIG. 4.

Effect of the E464A mutation on the steady-state kinetics of PA2567. The curves were generated by fitting the kinetic data of the wild-type and mutant enzyme to the Michaelis-Menten equation and equation 1, respectively.
TABLE 2.
Steady-state kinetic parameters for PA2567 and its mutantsa
| PA2567 enzyme | kcat (s−1) | Km (μM) | kcat/Km (s−1 μM−1) | Ki(μM) |
|---|---|---|---|---|
| Wild-type | 0.39 ± 0.03 | 5.2 ± 1.3 | (7.5 ± 2.0) × 10−2 | |
| E464A mutant | 1.1 ± 0.5 | 13.0 ± 5.0 | (8.4 ± 5.0) × 10−2 | 30.4 ± 12.0 |
| S493A mutant | NDb | ND | ND | |
| F498A mutant | ND | ND | ND |
Conditions for PA2567 and its mutant were 100 mM Tris buffer (pH 8.0) (23°C), 50 mM KCl, and 10 mM MgCl2.
ND, not determined due to inactivity caused by protein instability.
Effect of the D56N mutation on catalytic activity and loop 6 conformation in RocR.
Because the small phosphor donors acetyl phosphate, carbamoyl phosphate, and phosphoramidite did not phosphorylate RocR in our in vitro phosphorylation assay, we were not able to examine the effect of phosphorylation on the catalytic activity of the EAL domain. In an effort to probe how the modification of the putative phosphorylation site (Asp56) may affect EAL domain activity, we found that the D56N mutation caused significant changes in the steady-state kinetic parameters (Table 1). The most significant change is in the Michaelis constant Km, which is 0.20 ± 0.03 μM for the D56N mutant, representing a 16-fold decrease from that of RocR (3.2 ± 0.3 μM). Meanwhile, the D56N mutant exhibits a kcat value only slightly smaller (0.13 s−1) than that of RocR (0.67 s−1). To probe the structural changes in the mutant that account for the small Km, the method of H/D exchange-coupled mass spectrometry was used, because this method has been used increasingly as a sensitive tool for detecting changes in protein conformation and mobility (13). H/D exchange of RocR and the mutant D56N enzyme were performed by following an established protocol (6, 13, 19, 20). In total, 21 pairs of peptides generated from pepsin digestion were identified by a tandem mass spectrometry experiment and chosen for H/D exchange analysis. The 21 pairs of peptides encompass 93% of the protein sequences for RocR and the D56N mutant, including peptides 1 to 6 from the RR domain, peptide 7 from the linker region, and peptides 8 to 21 from the EAL domain (Fig. 5A). The deuteration pattern of each pair of peptides from RocR and the D56N mutant was compared by examining the time-dependent deuteration plots. While about two-thirds of the peptides exhibited similar H/D exchange patterns, 8 out of 21 pairs of peptides exhibited notable differences in exchange rate and final deuteration level as we discuss below.
FIG. 5.

Comparison of amide H/D exchange between RocR and the D56N mutant. (A) Sequence coverage map of RocR with the 21 peptides generated from pepsin digestion represented by the bars below the sequence. The peptides from the mutant exhibiting decrease in deuteration are colored blue, whereas the peptides showing significant and moderate increase in deuteration are colored red and yellow, respectively. (B) Structural models of the EAL and REC domains of RocR. The peptides exhibiting decrease in deuteration are colored blue, and the peptides showing significant and moderate increase in deuteration are colored red and yellow, respectively. The linker between the RR and EAL domains is represented by the brown broken line. (C) Time-dependent H/D exchange plots for the peptides in the EAL domain that exhibit changes in deuteration (RocR [•] and D56N mutant [○]). The curves were generated by fitting the data to equation 2 to obtain the total number of incorporated deuterons (N) as well as the exchange rate constants k1, k2, and k3. (D) Time-dependent H/D exchange plots for the peptides in the phosphoreceiver (REC) domain and linker region that exhibit changes in deuteration (RocR ([•] and D56N mutant [○]). The phosphorylated site (Asp56) and two putative residues (Ser83 and Phe105) involved in signal transduction are shown as sticks in panel B.
In the EAL domain, 4 out of the 14 peptides in the D56N mutant exhibited different deuteration pattern from their counterparts in RocR (Fig. 5B and C). The most prominent one is peptide 16, which exhibited faster exchange and higher deuteration level in the mutant. Peptide 16 includes four residues (LAMD295) from the β6 strand that contains the conserved Asp295 for Mg2+ coordination and the D296FGAGYSS motif that constitutes loop 6. In addition to peptide 16, two peptides (peptides 9 and 19) showed moderate elevation in deuteration in the mutant. Peptide 9 contains the first two β strands of the (β/α)8 barrel and forms part of the substrate-binding pocket, whereas peptide 19 encompasses half of the α7 helix residing at the dimer interface. The fourth peptide (peptide 11) contains helix α2 and forms a lid on the top of the substrate-binding pocket. Peptide 11 in the mutant showed slightly faster exchange but a lower level of deuteration at the end of the exchange (Fig. 5C). The resulting crossover of the two exchange curves is likely to reflect a change in protein mobility (20). These results support a picture in which the D56N mutation induces rather localized structural changes in the EAL domain, with the most significant changes restricted to the region near loop 6 and the end of the β6 strand.
Since Asp296 is most likely to be the residue directly interacting with the substrate and the D296A mutation increases Km to 8.6 ± 2.8 μM, we further examine whether the effect of the D56N mutation on catalysis is exerted through Asp296. The D56N D296A double mutant was prepared and found to exhibit kinetic parameters similar to those of the D296A mutant, with kcat of (1.2 ± 0.3) × 10−2 s−1 and elevated Km of 9.4 ± 2.8 μM. Together with the H/D exchange results, the resemblance of the catalytic properties of the double mutant to the D296A mutant, rather than the D56N mutant, suggested that the “regulatory signal” originated from the RR domain probably exerts its effect on substrate binding and catalysis through Asp296 and the adjacent residues in loop 6.
In addition to the four peptides in the EAL domain, four peptides residing in the RR domain and linker region also exhibited altered H/D exchange patterns (Fig. 5D). Most notably, the peptide spanning the linker region and the end of the α5′ helix (peptide 7) showed a significant increase in deuteration in the mutant protein. The deuteration level for peptide 7 is already relatively high (75%) in comparison with other peptides in RocR, indicating a flexible and solvent-exposed linker region. The deuteration level of peptide 7 increased to 90% for the D56N mutant, suggesting that the solvent accessibility of the linker region has been further increased. Moreover, the two peptides (peptides 1 and 4) that contain parts of the α1′ and α4′ helices showed lower exchange rates and lower deuteration levels in the mutant, whereas the adjacent peptide, peptide 6, which contains the β5′ strand, part of the α5′ helix, and the α5′-β5′ loop shows moderate increases in deuteration and exchange rate. Together, the changes in solvent accessibility in the RR domain indicated that the most significant structural changes caused by the mutation D56N occur within the linker and the α4′-β5′-α5′ region.
Recovery of the c-di-GMP-specific phosphodiesterase activity of an inactive EAL domain.
DGC2 is a cytoplasmic protein involved in the regulation of cellulose biosynthesis in Acetobacter xylinus (37). A. xylinus DGC2 contains a PAS-like domain at the N terminus and the GGDEF-EAL didomain at the C terminus (Fig. 3). It was found that the GGDEF domain of A. xylinus DGC2 is an active DGC domain capable of synthesizing c-di-GMP and that the EAL domain is catalytically inactive for hydrolyzing c-di-GMP (37). Examination of the sequence of A. xylinus DGC2 suggested that the EAL domain contains the essential general base catalyst (Glu529) and the residues (Glu350, Asn409, Glu441, and Asp472) for Mg2+ binding, and thus, is a potentially active PDE domain (28). Indeed, we found that A. xylinus DGC2 was capable of hydrolyzing several nonphysiological PDE substrates, such as thymidine-5-monophosphate-p-nitrophenyl ester (thymidine-pNPP) and bis-(p-nitrophenyl) phosphate (bis-pNPP) (Y. Qi, F. Rao, and Z.-X. Liang, submitted for publication). The stand-alone EAL domain of A. xylinus DGC2 (DGC2306-574) was cloned and expressed to further determine whether the activity of the EAL domain is repressed by the PAS and GGDEF domains. We found that the stand-alone EAL domain was active toward the nonphysiological substrates described above, but still not c-di-GMP.
Although it contains all the catalytic residues for Mg2+ ion binding and general base catalysis, the EAL domain of A. xylinus DGC2 contains a highly degenerate loop 6, consisting of N473FGKGITVL481, in contrast to the DFG(T/A)GYSS loop in active EAL domains. The observed catalytic activity toward the synthetic substrates indicated that the lack of activity toward c-di-GMP is probably caused by the binding of c-di-GMP in an unproductive configuration. To examine whether the loss of activity is due to the degeneration of loop 6, we partially “restored” the loop by mutating three residues in the stand-alone EAL domain. We found that the triple mutant (Asn473Lys476Ile478 → Asp473Thr476Tyr478) of the EAL domain was capable of hydrolyzing c-di-GMP (Fig. 6A). Steady-state kinetic characterization revealed that the triple mutant catalyzes the hydrolysis of c-di-GMP with a kcat of (5.9 ± 0.2) × 10−4 s−1 and Km of 13.0 ± 1.2 μM under the enzymatic assay conditions (Fig. 6B). The observed catalytic activity toward nonphysiological substrates for the wild-type A. xylinus DGC2 and the recovered activity toward c-di-GMP for the EAL domain provided further support for the proposed roles of the catalytic residues (28). In addition, the observations also suggested that the degeneration of loop 6 is at least partially responsible for the lack of activity against c-di-GMP for A. xylinus DGC2, and maybe some other EAL domain proteins as well.
FIG. 6.

Enzymatic activity assay for the EAL domain of A. xylinus DGC2. (A) HPLC analysis of the enzymatic activity of the full-length A. xylinus DGC2, stand-alone EAL domain, and the triple mutant with three mutations (Gln473Lys476Ile478 → Asp473Thr476Tyr478) in the EAL domain. The enzymes were incubated with c-di-GMP and Mg2+ for 90 min. Abs (mAU), absorbance (milliabsorbance units). (B) Steady-state kinetics of the triple mutant of the stand-alone EAL domain. The curve was generated by fitting the data to the Michaelis-Menten equation.
DISCUSSION
It is common to find flexible loops in the vicinity of the active sites of enzymes. The loops play important roles in substrate binding and positioning, as well as shielding the substrate from the bulk solvent. The results of extensive structural and biochemical studies suggested that a functional loop (loop 6) has been preserved during evolution in (β/α)8 barrel fold enzymes (15, 34). Despite the diverse reactions catalyzed by (β/α)8 barrel fold enzymes, flexible loop 6 plays various functional roles assisting catalysis by undergoing significant structural changes during substrate binding and catalysis. The results presented here seem to support an equally important role for loop 6 in the hydrolysis of c-di-GMP for the (β/α)8 barrel folded EAL domains.
The single mutations in the loop 6 region exerted profound impact on the structure and enzymatic activity of RocR. The mutation of the loop residue Phe297 and the loop-stabilizing Glu268 caused the formation of HMWO and significantly changed both kcat and Km. The fact that the Glu268 and Phe297 mutations increased the propensity of RocR to form HMWO suggested that loop 6 plays a critical structural role in maintaining the quaternary structure of the protein. The change in oligomeric state is likely to be a result of disruption of the dimer interface, as inferred from the observation that loop 6 is located at the dimer interface in the crystal structure of TdEAL and EALYkuI. The observed antiparallel loop-loop interaction between the residues from loop 6 may stabilize the dimeric structure (23). It is conceivable that the mutation of Glu268 or the residues on the loop may disrupt the loop-loop interaction and result in the dissociation of the dimer and the formation of HMWO. On the other hand, the mutation of Asp296 and Ser302, two residues of loop 6 in RocR, led to significant changes in catalytic parameters without altering the oligomeric structure. The S302A mutant exhibited strong substrate inhibition with an inhibition constant (Ki) of 10.9 ± 3.0 μM, while for the D296A mutant, Km (8.6 ± 2.8 μM) was greater and kcat (0.02 s−1) was smaller. The observed substrate inhibition and greater Km indicated that the perturbation of the loop could lead to the alteration of the binding affinity for c-di-GMP. Similar results were obtained with the GGDEF-EAL didomain-containing PA2567. The mutation of Glu464, the equivalent of Glu268 in RocR, resulted in strong substrate inhibition, whereas the other two mutations seemed to affect protein folding, presumably due to the destabilization of the dimer structure. The effects of the mutations in RocR and PA2567 support the view that loop 6 plays important structural and catalytic roles in EAL domains.
The comparison of the H/D exchange pattern between the peptides from RocR and the D56N mutant revealed intriguing structural changes in both the RR and EAL domains as an effect of the single mutation. The most significant and intriguing changes in the EAL domain are in loop 6 and the end of β6 strand (Fig. 5B and C). The enhanced H/D exchange reflects increased solvent accessibility in loop 6 region resulting from conformational change. The slightly higher deuteration for the dimerization helix α7 (peptide 19), along with the great increase in solvent accessibility in the linker region (peptide 7), probably reflects a rearrangement of the dimer interface. Such an interface conformation change could be coupled to the local conformational change in loop 6, since part of loop 6 is located at the interface as well. The coupling of domain rearrangement and movement of Asp295 has been suggested to be critical for controlling EAL domain activity by comparing the structures of YkuI and TdEAL (23). Together with the change in Km caused by the D56N mutation, the observed change in solvent accessibility seemed to suggest that catalysis is sensitive to conformational changes in the substrate-binding pocket, especially in the vicinity of loop 6. Moreover, the comparison of H/D exchange patterns also revealed significant changes in solvent accessibility in the linker and the α4′-β5′-α5′ region of the RR domain. Interestingly, structural and biochemical studies have shown that the α4′-β5′-α5′ region undergoes conformational changes upon activation by phosphorylation or the binding of the phosphoryl mimic beryllium fluoride (BeF3−) in other RR domains (8, 12). The observed conformational changes in the α4′-β5′-α5′ face raised the tantalizing possibility that the D56N mutant might bear some resemblance to the activated form of RocR. However, this remains speculative before the comparison between the mutant and phosphorylated RocR proteins in the future. Another underlying implication of these observations is that the control of catalysis through loop 6 might be used by EAL domains as a regulatory mechanism.
In contrast to RocR and PA2567, the EAL domain of A. xylinus DGC2 represents one of the EAL domains that contains all the essential residues for Mg2+ binding and general base catalysis but has a highly degenerate loop 6. The observation that DGC2 is active against nonphysiological PDE substrates indicated that the loss of activity toward c-di-GMP is most likely due to alteration of substrate specificity. This idea is further supported by the recovery of activity for the triple mutant of EALDGC2 domain by partially “restoring” the degenerate loop. Although the catalytic rate for the triple mutant is low compared to those of RocR and PA2567, the small Km of the triple mutant (13.0 ± 1.2 μM) indicates that the triple mutant has reasonable binding affinity for c-di-GMP. These observations confirmed the importance of loop 6 in catalysis and further suggested that the restoration of loop 6 enables the EAL domain to bind c-di-GMP in a productive configuration for hydrolysis.
The functional roles of loop 6 in EAL domains can be rationalized in light of the structural data for TdEAL and the YkuI-substrate complex. Although the catalytic activity has not been experimentally validated, the TdEAL domain is most likely to be an active PDE domain with a conserved loop 6 and catalytic residues. In contrast, the EAL domain of YkuI, which contains a degenerate loop 6 (NIGKESS), is catalytically inactive despite its ability to bind c-di-GMP (23). The comparison of the structures of the active (TdEAL) and inactive (YkuI) EAL domain structures revealed some intriguing differences for loop 6 (Fig. 7A). One of the major differences is that the “anchor” residue Glu125 (Glu268 in RocR) is no longer tucked under loop 6 in YkuI. As a result, the loop adopts a very different conformation with the residues shifted away from the catalytic Mg2+ ion. The catalytically indispensable residue Asp152 (Asp295 of RocR) in YkuI is too far away to coordinate the catalytic Mg2+ ion as suggested by Minasov and coworkers (23). In addition, the residue Asn153 (Asp296 in RocR) that is involved in the binding of c-di-GMP is now pulled away from the substrate. On the basis of these observations, we propose that loop 6 is crucial not only for stabilizing the dimer interface but also for the binding of Mg2+ ions and c-di-GMP. The conformational change in loop 6 caused by mutation or regulatory signal will impact catalysis in one of the following ways. First, the conformational change in the loop can affect the dimerization of the protein, which would result in a change of oligomeric structure and even protein stability. Second, binding of c-di-GMP can be affected as a result of the alternation of the interaction between c-di-GMP and the loop. Both structural and biochemical data suggested that the loop may interact with c-di-GMP directly (or indirectly through a second Mg2+ ion [28]) through the conserved Asp (Asp296 of RocR). Hence, mutations that affect the interactions between loop 6 and substrate or that impede the ability of loop 6 to undergo conformational change for effective substrate binding would affect catalysis. Third, change in the loop conformation can severely hamper catalysis by dislocating the essential Mg2+ ion-binding residue (Asp295 in RocR). The reduced binding affinity for Mg2+ can have catastrophic effect on the catalytic activity as demonstrated by the detrimental effect of the D295N mutation in RocR (28). Given the sensitivity of the catalytic activity to the loop conformation and the position of the Mg2+-binding residue, we postulate that some EAL domains could be regulated through controlling the conformation of loop 6.
FIG. 7.

Summary of EAL domains. (A) Comparison of loop 6 in active and inactive EAL domains. Loop 6 of TdEAL (Protein Data Bank accession no. 2r6o) and YkuI (Protein Data Bank accession no. 2w27) were highlighted in orange and cyan, respectively. The Mg2+ ion in the TdEAL structure is shown as a green ball, and c-di-GMP is shown as a stick. The corresponding residues of Asp295, Asp296, and Glu268 of RocR are highlighted and labeled (Asp646, Asp 647, and Glu619 for TdEAL; Asp152, Asn153, and Glu125 for YkuI). (B) Comparison of the sequences of the loop among characterized EAL domains. The EAL domains shown are from the following proteins: PA2567, BifA, and FimX from Pseudomonas aeruginosa (indicated by the Pa suffix after the hyphen and protein) (16, 17, 31); TdEAL from Thiobacillus denitrificans (TdEAL-Td); VieA from Vibrio cholerae (VieA-Vc) (38); CC3396 from Crescentus caulobacter (CC3396-Cc) (9); YcgF, YahA, Dos, YciR, CsrD, and YegE from Escherichia coli (indicated by the Ec suffix after the hyphen and protein) (11, 32, 36, 40, 41); BphG from Rhodobacter sphaeroides (BphG-Rs) (39); PdeA1, DGC1, DGC2, and DGC3 from Gluconacetobacter xylinus (indicated by the Gx suffix after the hyphen and protein) (7, 37); HmsP from Yersinia pestis (Hmsp-Yp) (2); GcpC from Salmonella enterica (GcpC-Se) (10); STM1344 and STM3375 from Salmonella enterica serotype Typhimurium (indicated by the St suffix after the hyphen and protein)(33); and LapD from Pseudomonas fluorescens Pf0-1 (LapD-Pf) (24). The sequences were aligned using MultAlin (http://bioinfo.genopole-toulouse.prd.fr/multalin/multalin.html), and the figure was generated using ESPript 2.2 (http://espript.ibcp.fr/ESPript/ESPript/). (C) Pie chart summary of the classification of the 5,862 EAL domains from bacterial genomes according to the conservation of catalytic residues and loop 6.
Comparison of the sequences of EAL domains revealed that all the characterized inactive EAL domains harbor a degenerate loop 6 (Fig. 7B). Further sequence analysis of the 5,862 EAL domains found in bacterial genomes revealed that 4,809 of them contain the catalytic residues that include the general base catalyst and four essential residues for Mg2+ coordination. Among the 4,809 EAL domains, 2,895 of them contain a conserved loop 6 [DFG(A/S/T)(G/A)(Y/F)(S/A/T)(S/A/G/V/T)] and 1,914 of them contain a degenerate loop 6 (Fig. 7C) (see supplemental material). The 2,895 EAL domains are likely to be catalytically active PDE domains, whereas the functions of the 1,914 EAL domains with a degenerate loop 6 remain uncertain, as exemplified by the EAL domains of YkuI and A. xylinus DGC2. The EAL domains of YkuI and A. xylinus DGC2 were found to be catalytically inactive toward c-di-GMP by in vitro enzymatic assay (Qi et al., submitted). However, it remains to be seen whether the EAL domain can be “activated” by the putative regulatory domains in YkuI and A. xylinus DGC2. In addition to the 4,809 EAL domains, 1,053 EAL domains were found to lack one or more of the essential catalytic residues (Fig. 7C). Among the 1,053 EAL domains, the majority of them (990) do not contain a conserved loop 6. These 1,053 EAL domains are most likely to be catalytically inactive. This postulation is supported by previously characterized inactive EAL domains as well as two recently reported EAL domain proteins. The EAL domain of the protein LapD from Pseudomonas fluorescens Pf0-1 contains a highly degenerate loop 6 (QRFGGRFSM) and lacks an essential residue for Mg2+ binding. It was proposed that LapD functions as a c-di-GMP-binding receptor rather than a PDE domain (24). The E. coli protein YcgF contains an EAL domain that was initially assumed to be a c-di-GMP-hydrolyzing domain. However, the EAL domain contains a Met residue at the position of the general base catalyst and a highly degenerate loop 6 (QVGRDLQI). Recent studies showed that the EAL domain does not exhibit PDE activity but participates in protein-protein interaction instead (40). Given the strong correlation between the divergencs of loop 6 sequences and catalytic activity, we hope that the classification of EAL domains described here can provide some assistance in elucidating the evolution and biological functions of the versatile EAL domains.
Supplementary Material
Acknowledgments
This work is supported by Singapore Biomedical Research Council through a BMRC grant (06/1/22/19/464) and the Ministry of Education of Singapore through a URC grant (RG151/06).
We thank Thomas Lee (University of Wisconsin, Madison) for his kind assistance with the HPLC column packing for our H/D exchange experiment.
Footnotes
Published ahead of print on 17 April 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Arnold, K., L. Bordoli, J. Kopp, and T. Schwede. 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22195-201. [DOI] [PubMed] [Google Scholar]
- 2.Bobrov, A. G., V. Kirillina, and R. D. Perry. 2007. Regulation of biofilm formation in Yersinia pestis. Adv. Exp. Med. Biol. 603201-210. [DOI] [PubMed] [Google Scholar]
- 3.Brzovic, P. S., C. C. Hyde, E. W. Miles, and M. F. Dunn. 1993. Characterization of the functional role of a flexible loop in the alpha-subunit of tryptophan synthase from Salmonella typhimurium by rapid-scanning, stopped-flow spectroscopy and site-directed mutagenesis. Biochemistry 3210404-10413. [DOI] [PubMed] [Google Scholar]
- 4.Brzovic, P. S., Y. Sawa, C. C. Hyde, E. W. Miles, and M. F. Dunn. 1992. Evidence that mutations in a loop region of the alpha-subunit inhibit the transition from an open to a closed conformation in the tryptophan synthase bienzyme complex. J. Biol. Chem. 26713028-13038. [PubMed] [Google Scholar]
- 5.Camilli, A., and B. L. Bassler. 2006. Bacterial small-molecule signaling pathways. Science 3111113-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ceccarelli, C., Z. X. Liang, M. Strickler, G. Prehna, B. M. Goldstein, J. P. Klinman, and B. J. Bahnson. 2004. Crystal structure and amide H/D exchange of binary complexes of alcohol dehydrogenase from Bacillus stearothermophilus: insight into thermostability and cofactor binding. Biochemistry 435266-5277. [DOI] [PubMed] [Google Scholar]
- 7.Chang, A. L., J. R. Tuckerman, G. Gonzalez, R. Mayer, H. Weihouse, G. Volman, D. Amikam, M. Benziman, and M. A. Gilles-Gonzalez. 2001. Phosphodiesterase A1, a regulator of cellulose synthesis in Acetobacter xylinum, is a heme-based sensor. Biochemistry 403420-3426. [DOI] [PubMed] [Google Scholar]
- 8.Cho, H. S., S. Y. Lee, D. L. Yan, X. Y. Pan, J. S. Parkinson, S. Kustu, D. E. Wemmer, and J. G. Pelton. 2000. NMR structure of activated CheY. J. Mol. Biol. 297543-551. [DOI] [PubMed] [Google Scholar]
- 9.Christen, M., B. Christen, M. Folcher, A. Schauerte, and U. Jenal. 2005. Identification and characterization of a cyclic di-GMP-specific phosphodiesterase and its allosteric control by GTP. J. Biol. Chem. 28030829-30837. [DOI] [PubMed] [Google Scholar]
- 10.Garcia, B., C. Latasa, C. Solano, F. G. Portillo, C. Gamazo, and I. Lasa. 2004. Role of the GGDEF protein family in Salmonella cellulose biosynthesis and biofilm formation. Mol. Microbiol. 54264-277. [DOI] [PubMed] [Google Scholar]
- 11.Girgis, H. S., Y. Liu, W. S. Ryu, and S. Tavazoie. 2007. A comprehensive genetic characterization of bacterial motility. PLOS Genet. 31644-1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guhaniyogi, J., T. Wu, S. S. Patel, and A. M. Stock. 2008. Interaction of CheY with the C-terminal peptide of CheZ. J. Bacteriol. 1901419-1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoofnagle, A. N., K. A. Resing, and N. G. Ahn. 2003. Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 321-25. [DOI] [PubMed] [Google Scholar]
- 14.Jenal, U., and J. Malone. 2006. Mechanisms of cyclic-di-GMP signaling in bacteria. Annu. Rev. Genet. 40385-407. [DOI] [PubMed] [Google Scholar]
- 15.Joseph, D., G. A. Petsko, and M. Karplus. 1990. Anatomy of a conformational change: hinged “lid” motion of the triosephosphate isomerase loop. Science 2491425-1428. [DOI] [PubMed] [Google Scholar]
- 16.Kazmierczak, B. I., M. B. Lebron, and T. S. Murray. 2006. Analysis of fimX, a phosphodiesterase that governs twitching motility in Pseudomonas aeruginosa. Mol. Microbiol. 601026-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuchma, S. L., K. M. Brothers, J. H. Merritt, N. T. Liberati, F. M. Ausubel, and G. A. O'Toole. 2007. BifA, a cyclic-di-GMP phosphodiesterase, inversely regulates biofilm formation and swarming motility by Pseudomonas aeruginosa PA14. J. Bacteriol. 1898165-8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kulasekara, H. D., I. Ventre, B. R. Kulasekara, A. Lazdunski, A. Filloux, and S. Lory. 2005. A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol. Microbiol. 55368-380. [DOI] [PubMed] [Google Scholar]
- 19.Liang, Z. X., T. Lee, K. A. Resing, N. G. Ahn, and J. P. Klinman. 2004. Thermal-activated protein mobility and its correlation with catalysis in thermophilic alcohol dehydrogenase. Proc. Natl. Acad. Sci. USA 1019556-9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang, Z. X., I. Tsigos, T. Lee, V. Bouriotis, K. A. Resing, N. G. Ahn, and J. P. Klinman. 2004. Evidence for increased local flexibility in psychrophilic alcohol dehydrogenase relative to its thermophilic homologue. Biochemistry 4314676-14683. [DOI] [PubMed] [Google Scholar]
- 21.Lundqvist, T., and G. Schneider. 1991. Crystal structure of activated ribulose-1,5-bisphosphate carboxylase complexed with its substrate, ribulose-1,5-bisphosphate. J. Biol. Chem. 26612604-12611. [PubMed] [Google Scholar]
- 22.McMillan, F. M., M. Cahoon, A. White, L. Hedstrom, G. A. Petsko, and D. Ringe. 2000. Crystal structure at 2.4 A resolution of Borrelia burgdorferi inosine 5′-monophosphate dehydrogenase: evidence of a substrate-induced hinged-lid motion by loop 6. Biochemistry 394533-4542. [DOI] [PubMed] [Google Scholar]
- 23.Minasov, G., S. Padavattan, L. Shuvalova, J. S. Brunzelle, D. J. Miller, A. Basle, C. Massa, F. R. Collart, T. Schirmer, and W. F. Anderson. 24 February 2009. Crystal structures of YkuI and its complex with second messenger c-di-GMP suggests catalytic mechanism of phosphodiester bond cleavage by EAL domains. J. Biol. Chem. doi: 10.1074/jbc.M808221200. [DOI] [PMC free article] [PubMed]
- 24.Newell, P. D., R. D. Monds, and G. A. O'Toole. 13 February 2009. LapD is a bis-(3′,5′)-cyclic dimeric GMP-binding protein that regulates surface attachment by Pseudomonas fluorescens Pf0-1. Proc. Natl. Acad. Sci. USA doi: 10.1073/pnas.0808933106. [DOI] [PMC free article] [PubMed]
- 25.Pompliano, D. L., A. Peyman, and J. R. Knowles. 1990. Stabilization of a reaction intermediate as a catalytic device: definition of the functional role of the flexible loop in triosephosphate isomerase. Biochemistry 293186-3194. [DOI] [PubMed] [Google Scholar]
- 26.Priestle, J. P., M. G. Grütter, J. L. White, M. G. Vincent, M. Kania, E. Wilson, T. S. Jardetzky, K. Kirschner, and J. N. Jansonius. 1987. Three-dimensional structure of the bifunctional enzyme N-(5′-phosphoribosyl) anthranilate isomerase-indole-3-glycerol-phosphate synthase from Escherichia coli. Proc. Natl. Acad. Sci. USA 845690-5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rao, F., S. Pasunooti, Y. Ng, W. Zhuo, L. Lim, W. Liu, and Z.-X. Liang. 2009. Enzymatic synthesis of c-di-GMP using a thermophilic diguanylate cyclase. Anal. Biochem. 389138-142. [DOI] [PubMed] [Google Scholar]
- 28.Rao, F., Y. Yang, Y. Qi, and Z.-X. Liang. 2008. Catalytic mechanism of cyclic di-GMP-specific phosphodiesterase: a study of the EAL domain-containing RocR from Pseudomonas aeruginosa. J. Bacteriol. 1903622-3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Resing, K. A., A. N. Hoofnagle, and N. G. Ahn. 1999. Modeling deuterium exchange behavior of ERK2 using pepsin mapping to probe secondary structure. Am. Soc. Mass. Spectrom. 10685-702. [DOI] [PubMed] [Google Scholar]
- 30.Romling, U., M. Gomelsky, and M. Y. Galperin. 2005. c-di-GMP: the dawning of a novel bacterial signalling system. Mol. Microbiol. 57629-639. [DOI] [PubMed] [Google Scholar]
- 31.Ryan, R. P., Y. Fouhy, J. F. Lucey, L. C. Crossman, S. Spiro, Y.-W. He, L.-H. Zhang, S. Heeb, M. Camara, P. Williams, and J. M. Dow. 2006. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. USA 1036712-6717. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Schmidt, A. J., D. A. Ryjenkov, and M. Gomelsky. 2005. The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J. Bacteriol. 1874774-4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simm, R., A. Lusch, A. Kader, M. Andersson, and U. Romling. 2007. Role of EAL-containing proteins in multicellular behavior of Salmonella enterica serovar Typhimurium. J. Bacteriol. 1893613-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sterner, R., and B. Hocker. 2005. Catalytic versatility, stability and evolution of the (β/α)8-barrel enzyme fold. Chem. Rev. 1054038-4055. [DOI] [PubMed] [Google Scholar]
- 35.Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J. Hickey, F. S. L. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L. Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L. Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D. Spencer, G. K. S. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. E. W. Hancock, S. Lory, and M. V. Olson. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406959-964. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki, K., P. Babitzke, S. R. Kushner, and T. Romeo. 2006. Identification of a novel regulatory protein (CsrD) that targets the global regulatory RNAs CsrB and CsrC for degradation by RNase E. Genes Dev. 202605-2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tal, R., H. C. Wong, R. Calhoon, D. Gelfand, A. L. Fear, G. Volman, R. Mayer, P. Ross, D. Amikam, H. Weinhouse, A. Cohen, S. Sapir, P. Ohana, and M. Benziman. 1998. Three cdg operons control cellular turnover of cyclic di-GMP in Acetobacter xylinum: genetic organization and occurrence of conserved domains in isoenzymes. J. Bacteriol. 1804416-4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamayo, R., A. D. Tischler, and A. Camilli. 2005. The EAL domain protein VieA is a cyclic diguanylate phosphodiesterase. J. Biol. Chem. 28033324-33330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tarutina, M., D. A. Ryjenkov, and M. Gomelsky. 2006. An unorthodox bacteriophytochrome from Rhodobacter sphaeroides involved in turnover of the second messenger c-di-GMP. J. Biol. Chem. 28134751-34758. [DOI] [PubMed] [Google Scholar]
- 40.Tschowri, N., S. Busse, and R. Hengge. 2009. The BLUF-EAL protein YcgF acts as a direct anti-repressor in a blue-light response of Escherichia coli. Genes Dev. 23522-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weber, H., C. Persavento, G. Possling, G. Tischendorf, and R. Hengge. 2006. Cyclic-di-GMP-mediated signalling within the sigma(S) network of Escherichia coli. Mol. Microbiol. 621014-1034. [DOI] [PubMed] [Google Scholar]
- 42.Weis, D. D., I. J. Kass, and J. R. Engen. 2006. Semi-automated analysis of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass. Spectrom. 171700-1703. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.