

Abstract
Type III polyketide synthases (PKSs) synthesize a variety of aromatic polyketides in plants, fungi, and bacteria. The bacterial genome projects predicted that probable type III PKS genes are distributed in a wide variety of gram-positive and -negative bacteria. The gram-positive model microorganism Bacillus subtilis contained the bcsA-ypbQ operon, which appeared to encode a type III PKS and a methyltransferase, respectively. Here, we report the characterization of bcsA (renamed bpsA, for Bacillus pyrone synthase, on the basis of its function) and ypbQ, which are involved in the biosynthesis of aliphatic polyketides. In vivo analysis demonstrated that BpsA was a type III PKS catalyzing the synthesis of triketide pyrones from long-chain fatty acyl-coenzyme A (CoA) thioesters as starter substrates and malonyl-CoA as an extender substrate, and YpbQ was a methyltransferase acting on the triketide pyrones to yield alkylpyrone methyl ethers. YpbQ thus was named BpsB because of its functional relatedness to BpsA. In vitro analysis with histidine-tagged BpsA revealed that it used broad starter substrates and produced not only triketide pyrones but also tetraketide pyrones and alkylresorcinols. Although the aliphatic polyketides were expected to localize in the membrane and play some role in modulating the rigidity and properties of the membrane, no detectable phenotypic changes were observed for a B. subtilis mutant containing a whole deletion of the bpsA-bpsB operon.
Type III polyketide synthases (PKSs), represented by a plant chalcone synthase (CHS), are the condensing enzymes that catalyze the synthesis of aromatic polyketides in plants, fungi, and bacteria (2). CHS catalyzes the decarboxylative condensation of p-coumaroyl-coenzyme A (p-coumaroyl-CoA), called a starter substrate, with three malonyl-CoAs, called extender substrates, and synthesizing a tetraketide intermediate. The synthesized tetraketide intermediate was cyclized and aromatized by CHS and resulted in naringenin chalcone. Like CHS, most of the type III PKSs catalyze the condensation of a starter substrate with several molecules of an extender substrate and cyclization. There are many type III PKSs that differ in these specificities.
Until recently, type III PKSs were discovered only from plants. In 1999, the first bacterial type III PKS, RppA, was discovered. RppA catalyzes the condensation of five malonyl-CoAs to synthesize 1,3,6,8-tetrahydroxynaphthalene, which is a precursor of hexahydroxyperylenequinone melanin in the actinomycete Streptomyces griseus (4). Since then, the genome projects of various bacteria have revealed that type III PKSs are widely distributed in a variety of bacteria. For example, ArsB and ArsC, both of which are type III PKSs in Azotobacter vinelandii, catalyze the synthesis of alkylresorcinols and alkylpyrones, respectively, which are essential for encystment as the major lipids in the cyst membrane (5). In S. griseus, the srs operon consisting of srsA, srsB, and srsC is responsible for the synthesis of methylated phenolic lipids derived from alkylresorcinols and alkylpyrones (6). The function of each of the operon members is that SrsA is a type III PKS responsible for the synthesis of phenolic lipids alkylresorcinol and alkylpyrones, SrsB is a methyltransferase acting on the phenolic lipids to yield alkylresorcinol methyl ethers, and SrsC is a hydroxylase acting on the alkylresorcinol methyl ethers. The phenolic lipids synthesized by the Srs enzymes confer resistance to β-lactam antibiotics (6). Therefore, it is suggested that phenolic lipids play an important role as minor components in the biological membrane in various bacteria. In fact, srsAB- and srsABC-like operons are distributed widely in both gram-positive and -negative bacteria (see Fig. S1 in the supplemental material). However, most of these type III PKSs have not been characterized.
Bacillus subtilis is one of the best-characterized gram-positive bacteria. BcsA, which stands for bacterial chalcone synthase, was annotated as a homologue of type III PKS in B. subtilis (3). As described in this paper, however, this annotation needs correction. We renamed the gene bpsA (for Bacillus pyrone synthase). Moreover, the functional unknown gene ypbQ is located next to bpsA. YpbQ, consisting of 168 amino acid residues, contained an isoprenylcysteine carboxyl methyltransferase (ICMT) domain of the ICMT family members, which are unique membrane proteins that are involved in the posttranslational modification of oncogenic proteins (23). Therefore, the bpsA and ypbQ genes were predicted to form an operon, just like srsA and srsB in the srs operon in S. griseus. We therefore named ypbQ, a thus-far functionally unknown gene, bpsB.
In this study, we characterized the functions of BpsA and BpsB by in vivo and in vitro experiments. The in vivo experiments revealed that the overexpression of bpsA in B. subtilis led to the production of triketide pyrones, and the co-overexpression of bpsA and bpsB led to the production of triketide pyrone methyl ethers. The in vitro analysis showed that BpsA produced triketide pyrones and a small amount of tetraketide pyrones and tetraketide resorcinols from long-chain fatty acyl CoA thioesters as starter substrates and malonyl-CoA as an extender substrate. Therefore, BpsA is a type III PKS that is responsible for the synthesis of alkylpyrones, and BpsB is a methyltransferase that acts on the alkylpyrones to yield alkylpyrone methyl ethers. BpsB is the first enzyme found to methylate alkylpyrones. Furthermore, we attempted to analyze the biological function of the aliphatic polyketides by disrupting the bpsA and bpsB genes, but no distinct phenotypic changes were detected under laboratory conditions.
MATERIALS AND METHODS
Chemicals.
[2-14C]malonyl-CoA was purchased from American Radiolabeled Chemicals (St. Louis, MO). The nonlabeled CoA esters 13-methyltetradecanoic acid, 12-methyltetradecanoic acid, 14-methylpentadecanoic acid, 15-methylhexadecanoic acid, and 14-methylhexadecanoic acid were purchased from Sigma. N-acetylcysteamine (NAC) was supplied by Aldrich. 13-Methyltetradecanoyl-NAC, 12-methyltetradecanoyl-NAC, 14-methylpentadecanoyl-NAC, 15-methylhexadecanoyl-NAC, and 14-methylhexadecanoyl-NAC were chemically synthesized as described by Oguro et al. (17).
Bacterial strains, plasmids, and media.
Escherichia coli JM109 and BL21(DE3), plasmids pUC19 and pColdI, restriction enzymes, and other DNA-modifying enzymes used for DNA manipulation were purchased from Takara Biochemicals (Shiga, Japan). B. subtilis strain 168 was obtained from H. Kada (Mizkan, Aichi, Japan). For the expression of bpsA and bpsB in B. subtilis, pWH1530 (MoBiTec, Florida) was used. E. coli and B. subtilis were cultured in Luria-Bertani (LB) medium. All PCRs were conducted by using the chromosomal DNA of B. subtilis 168 as the template. The absence of undesired alterations during PCR was checked by nucleotide sequencing. Plasmids were introduced by transformation into B. subtilis 168 by using Spizizen's minimal medium (1, 11).
Construction of pHO1.
For the construction of pWH1530-bpsA (pHO1) containing bpsA under the control of the xylA promoter in pWH1530, a 1.1-kb DNA fragment containing the bpsA coding region was amplified by PCR with primer I (5′-GTTGGATCCACTAGTATGCAAAGAGGTGATCGCATGGC-3′, with the BamHI site shown by underlining, the SpeI site shown by the italic letters, and the start codon of bpsA shown by the boldface letters) and primer II (5′-ACAGCATGCATGGCGATCAACAACCAAAAC-3′, with the SphI site shown by underlining). The fragment was cloned between the BamHI and SphI sites of pUC19, resulting in pUC19-bpsA. The SpeI-SphI fragment excised from pUC19-bpsA was cloned between the SpeI and SphI sites of pWH1530, resulting in pHO1.
Construction of pHO2.
For the construction of pWH1530-bpsA/bpsB (pHO2), containing bpsA and bpsB under the control of the xylA promoter in pWH1530, a 1.6-kb DNA fragment containing bpsA and bpsB coding regions was amplified by PCR with primer I and primer III (5′-TAAGCATGCTGAGAATGATTTTCAATGGC-3′, with the SphI site underlined). The fragment was cloned between the BamHI and SphI sites of pUC19, resulting in pUC19-bpsA/bpsB. The SpeI-SphI fragment excised from pUC19-bpsA/bpsB was cloned between the SpeI and SphI sites of pWH1530, resulting in pHO2.
Construction of pColdI-bpsA.
A 1.1-kb DNA fragment containing the bpsA coding region was amplified by PCR with primer IV (5′-CGCGCATGCCATATGGCGTTTATTTTATCCATTGGA-3′, with the SphI site underlined; the nucleotide sequence [CGCATG] covering the ATG start codon of bpsA was changed to CATATG to create an NdeI site and is in italic letters) and primer V (5′-CGCGGATCCGAATGATTTTCAATCGCTAT-3′, with the BamHI site underlined). The amplified fragment was cloned between the SphI and BamHI sites of pUC19, resulting in pUC19-bpsA2. The NdeI-BamHI fragment excised from pUC19-bpsA2 was cloned between the NdeI and BamHI sites of pColdI, resulting in pColdI-bpsA.
Analysis of lipids produced by B. subtilis.
B. subtilis 168 harboring pHO1, pHO2, or pWH1530 was inoculated into 100 ml of LB medium containing 10 μg/ml tetracycline and grown at 37°C. After 2 h, 5 mg/ml of xylose was added to induce the xylA promoter, and the culture was continued for a further 8 h. Cells were harvested by centrifugation and resuspended in 0.1 M Tris-HCl (pH 8.0). After sonication, the cell broth was adjusted to pH 1.0 with 6 M HCl and extracted with ethyl acetate. After evaporation to dryness, the residue was dissolved in 20 μl of methanol for reverse-phase high-performance liquid chromatography (HPLC), liquid chromatography-atmospheric pressure chemical ionization mass spectrometry (LC-APCIMS), and LC-APCI tandem mass spectrometry (LC-APCIMS/MS) analyses. The conditions for analytical HPLC were the following: a Pegacil B C4 column (4.6 by 250 mm; Senshu Scientific, Tokyo, Japan) was eluted with a linear gradient of 50 to 100% CH3CN in water (each containing 0.1% acetic acid) at a flow rate of 1.0 ml/min. UV absorbance was detected at 280 nm. LC-APCIMS analysis was carried out by using an Esquire High-Capacity Trap Plus system (Bruker Daltonics, Bremen, Germany) equipped with a Pegasil B C4 column (4.6 by 250 mm). The elution conditions were equivalent to those for the HPLC analysis. Triketide pyrones 2d to 2i were identified by comparing their retention times in HPLC and their LC-MS and MS/MS spectra to those obtained from the triketide pyrones that had been prepared by ArsC, a type III PKS from Azotobacter vinelandii (5). The triketide pyrones produced by ArsC were confirmed to yield the fragment patterns of [M−H]− and [M−CO2−H]− ions, as determined by LC-APCIMS and LC-APCIMS/MS analyses. The spectrometric data are summarized in the supplemental material.
Large-scale preparation and isolation of lipids.
B. subtilis 168 harboring pHO2 was inoculated to 1.5 liters of 20× LB medium containing 10 μg/ml tetracycline and 0.01% antifoam silicone (vol/vol) (Wako, Osaka, Japan) and grown at 37°C. After 2 h, 5 mg/ml of xylose was added, and the culture was continued for a further 24 h. After cultivation, the cells obtained by centrifugation were extracted by ethyl acetate by the same method as that used for the analytical scale. The extraction was washed with brine, dried with Na2SO4, and evaporated to dryness. The crude materials were dissolved in a small amount of chloroform and flash-chromatographed on silica gel using chloroform-methanol (80:20, vol/vol) as an eluent. The eluate was evaporated and dissolved in methanol for reverse-phase preparative HPLC. The methoxy triketide pyrones (3d to 3i) were purified by reverse-phase preparative HPLC using a Pegasil C4 column (10 by 250 mm; Senshu). They were eluted with a linear gradient of 70 to 100% CH3CN in water (each containing 0.1% acetic acid) at a flow rate of 3.0 ml/min. The collected fractions were lyophilized to give 3h plus 3i (1.8 mg), 3f plus 3g (0.3 mg), and 3d plus 3e (3.5 mg) as white solids.
Structure determination.
The structures of 3h, 3i, 3d, and 3e were determined by proton and carbon nuclear magnetic resonance (NMR) spectroscopy, with the aid of heteronuclear multiple quantum correlation, heteronuclear multiple bond correlation (HMBC), and LC-APCIMS, LC-APCIMS/MS, and high-resolution mass spectroscopy (HR-MS) analyses. The structures of 3f and 3g were determined by proton NMR, LC-APCIMS, LC-APCIMS/MS, and HR-MS analyses. HR-MS was measured on a JEOL (Tokyo, Japan) Accu TOF T-100 equipped with electrospray ionization (in positive ion mode). The NMR and MS data are summarized in the supplemental material.
Production and purification of BpsA.
For the production of histidine-tagged BpsA, E. coli BL21(DE3) harboring pColdI-bpsA was inoculated into LB medium containing 100 μg/ml ampicillin and incubated at 37°C. After the optical density at 600 nm had reached 0.4, the cells were kept at 15°C for 30 min. The culture was continued for a further 24 h in the presence of 0.25 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cells were harvested by centrifugation and resuspended in 10 mM Tris-HCl (pH 8.0) and 145 mM NaCl and were disrupted by sonication. A crude cell lysate was prepared by the removal of cell debris by centrifugation at 10,000 × g for 20 min. BpsA was purified by using a nickel-nitrilotriacetic acid spin column (Qiagen, Valencia, CA) according to the manual from the manufacturer, except for adding 10% glycerol to each buffer. The purified histidine-tagged protein was dialyzed against 10 mM Tris-HCl (pH 8.0), 500 mM NaCl, and 10% glycerol.
In vitro BpsA reaction.
The standard reaction mixture contained 100 μM [2-14C]malonyl-CoA, 100 μM starter CoA or starter NAC, 100 mM Tris-HCl (pH 7.5), and 41.4 μg of BpsA in a total volume of 100 μl. Reactions were incubated at 30°C for 30 min before being quenched with 20 μl of 6 M HCl. The products were extracted with ethyl acetate, and the organic layer was evaporated to dryness. The residual material was dissolved in 15 μl of methanol for thin-layer chromatography (TLC) and LC-APCIMS analyses. Silica gel 60 WF254 TLC plates (Merck, Darmstadt, Germany) were developed in benzene-acetone-acetic acid (85:15:1, vol/vol/vol), and the 14C-labeled compounds were detected by using a BAS-MS imaging plate (Fuji Film, Tokyo, Japan). The products from the in vitro BpsA reaction were identified by comparing their LC-MS and MS/MS spectra to those obtained from the authentic standards that had been prepared by ArsB and ArsC, type III PKSs from A. vinelandii (5).
Kinetic parameters were determined as follows. The reaction mixture contained 100 mM Tris-HCl (pH 7.5), 100 μM malonyl-CoA, and 10.4 μg of BpsA in a total volume of 200 μl. The concentration of 15-methylhexadecanoyl-NAC was varied between 0.1 to 5 μM. We confirmed that the formation of a triketide pyrone as the product was linear during the assay, which implies that the analysis was carried out under steady-state conditions. After the reaction mixture had been preincubated at 30°C for 5 min, the reactions were initiated by adding the substrates. The reactions were continued for 60 s. The reactions were stopped with 20 μl of 6 M HCl, and the material in the mixture was extracted with ethyl acetate. The conditions for analytical HPLC are described above. Steady-state parameters were determined by fitting the curve to v = Vmax[S]/(Km + [S]), where Vmax is the maximum rate of metabolism, v is the initial velocity of formation of 4-hydroxy-6-(14′-methylpentadecyl)-2-pyrone (2e) by BpsA, and S is the concentration of 15-methylhexadecanoyl-NAC.
Construction of mutant strain bpsA-bpsB::cat.
Oligonucleotide primers were used to amplify the bpsAU fragment (bpsAUF, 5′-TCGTGACAGGCTCTGTTGTT-3′; bpsAUR, 5′-CTATTGCCGGGCGATCACCTCTTTGCATAC-3′) and the bpsBD fragment (bpsBDF, 5′-GCTCCAGATCTAGCGATTGAAAATCATTCTCAACC-3′; bpsBDR, 5′-TCATCATGGCGGTCAATTTG-3′). The chloramphenicol resistance gene of pCBB31 (9) was amplified by PCR using primers catF (5′-CCGGCAATAGTTACCCTTAT-3′) and catR (5′-GATCTGGAGCTGTAATATAAAAACC-3′). The fragments obtained were used simultaneously as the template for PCR amplification using primers bpsAUF and bpsBDR. The resulting fragment was used to transform Bacillus subtilis 168, and chloramphenicol-resistant transformants were selected. Correct integration was confirmed by PCR amplification.
Tests of phenotypic changes of mutant bpsA-bpsB::cat.
Sensitivity to cell wall-targeting antibiotics of the wild-type strain B. subtilis 168 and mutant bpsA-bpsB::cat was tested by growing the strains on LB agar medium at 30°C for 18 h. The antibiotics added were penicillin G at 1 ng/ml, 0.05, or 0.1 μg/ml, vancomycin at 0.2, 0.5, or 1 μg/ml, and cephalosporin C at 5, 7, or 10 μg/ml. The heat stress survival of the wild-type and bpsA-bpsB::cat strains was examined in LB liquid medium; the cells were grown at 37°C until the optical density at 600 nm reached 0.2, preadapted to heat by transferring them to a shaking water bath set at 48°C for 60 min with gentle shaking, and challenged at 54°C for 1, 2, 3, and 5 h or at 56°C for 10 min. After the heat shock treatment, portions of the cells were diluted and plated on LB agar.
RESULTS AND DISCUSSION
Organization and distribution of bpsA-bpsB operons in a wide variety of bacteria.
The gene organization of the 1.6-kb bpsA and bpsB genes is shown in Fig. 1. The bpsA and bpsB genes were thought to form an operon, because the stop codon of bpsA is located at three nucleotides upstream from the start codon of bpsB and because not only in B. subtilis but also in many bacteria, bpsA and bpsB genes almost always are neighbors on the chromosomes (see Fig. S1 in the supplemental material). pbuX, encoding a protein homologous to a xanthine permease, which is located 73 nucleotides upstream of bpsA, appeared not to be functionally linked to the bpsA-bpsB operon.
FIG. 1.
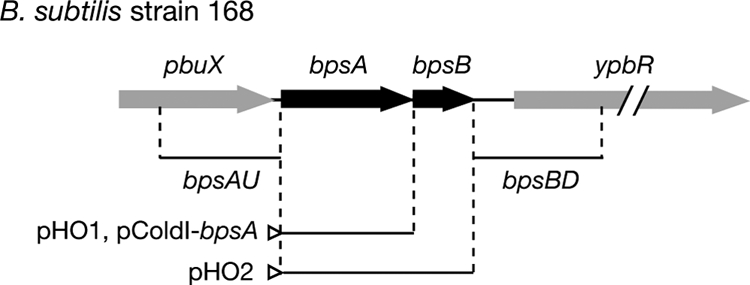
Genetic organization of the bpsA-bpsB operon in B. subtilis. The open triangles indicate the promoters on the vectors. The bpsAU and bpsBD fragments were used for gene disruption to construct mutant bpsA-bpsB::cat.
BpsA, consisting of 365 amino acid residues, shared 35% amino acid sequence identity with SrsA in S. griseus, a type III PKS that catalyzes C-methylated alkylresorcinol synthesis from long-chain fatty acyl-CoAs (6). In addition, a Cys-His-Asn catalytic triad, which is crucial for the decarboxylative condensation activity of all type III PKSs, was conserved (2). The amino acid alignment of BpsA with other type III PKSs, including the catalytic triads, is shown in Fig. S2 in the supplemental material.
BpsB, consisting of 168 amino acid residues, shared 38% amino acid sequence identity to SrsB in S. griseus, a methyltransferase that catalyzes the methylation of alkylresorcinol. bpsB homologues are present in various bacterial species, such as Mycobacterium, and form an operon with a type III PKS gene in most cases.
In vivo analysis of bpsA and bpsB.
To clarify the functions of BpsA and BpsB, we constructed two plasmids for overexpression. pHO1 carrying bpsA and pHO2 carrying bpsA and bpsB were constructed by using pWH1530 and were introduced by transformation into B. subtilis 168. These genes were under the control of the xylose-inducible xylA promoter. After the B. subtilis cells had been grown in the presence of xylose, cell extracts were prepared and analyzed by HPLC. Under the conditions employed, B. subtilis harboring only the empty pWH1530 vector produced no detectable aliphatic polyketides (Fig. 2). On the other hand, B. subtilis strains overexpressing either bpsA or bpsA and bpsB produced several lipids, giving multiple peaks on HPLC (Fig. 2).
FIG. 2.

HPLC chromatograms of the lipid fractions prepared from cell extracts of B. subtilis harboring the empty vector pWH1520, pHO1 carrying bpsA, and pHO2 carrying both bpsA and bpsB. Below the chromatograms are the chemical structures of triketide pyrones (2d to 2i) produced by B. subtilis carrying bpsA and triketide pyrone methyl ethers (3d to 3i) produced by B. subtilis carrying both bpsA and bpsB.
We analyzed the lipids 2d to 2i (Fig. 2) by LC-APCIMS and LC-APCIMS/MS analyses. The mass spectra of 2 h, 2i, 2f, 2g, 2d, and 2e showed molecular ions at m/z 307, 307, 321, 321, 335, and 335, respectively, in a negative ion mode. On the basis of the [M−H]− ions of these compounds, 2d to 2i were predicted to be alkylpyrones. Moreover, the MS/MS spectra of 2 h, 2i, 2f, 2g, 2d, and 2e showed fragments of m/z 263, 263, 277, 277, 291, and 291, corresponding to [M−CO2−H]− ions, respectively, which indicated the presence of an α-pyrone ring (20). Thus, 2d to 2i were predicted to be triketide pyrones. Because major fatty acids contained in B. subtilis cells are 12-methyltridecanoic acid (iso-C14; 4%), 13-methyltetradecanoic acid (iso-C15; 14%), 12-methyltetradecanoic acid (anteiso-C15; 33%), n-hexadecanoic acid (n-C16; 6%), 14-methylpentadecanoic acid (iso-C16; 11%), 15-methylhexadecanoic acid (iso-C17; 15%), and 14-methylhexadecanoic acid (anteiso-C17; 10%) (10), the alkyl chains of the triketide pyrones 2d to 2i presumably were derived from these long-chain fatty acids. We then attempted to identify these compounds by comparing their retention times on HPLC, MS, and MS/MS spectra to those obtained from the triketide pyrones that had been prepared by ArsC, a type III PKS from A. vinelandii. ArsC produces alkylpyrones by using long-chain fatty acyl CoA thioesters as starter substrates and malonyl-CoA as the extender substrate (5). The triketide pyrones that were prepared by using branched-chain fatty acyl NAC thioesters (NAC is a mimic of CoA) or straight-chain fatty acyl CoA thioesters as starter substrates yielded the fragment patterns of [M−H]− and [M−CO2−H]− ions, as determined by LC-APCIMS and LC-APCIMS/MS analyses (see structural data in the supplemental material). By comparing the triketide pyrones produced by ArsC, we showed that B. subtilis harboring pHO1 produced 4-hydroxy-6-(11′-methyltridecyl)-2-pyrone (2h), 4-hydroxy-6-(12′-methyltridecyl)-2-pyrone (2i), 4-hydroxy-6-(13′-methyltetradecyl)-2-pyrone (2f), 4-hydroxy-6-pentadecyl-2-pyrone (2g), 4-hydroxy-6-(13′-methylpentadecyl)-2-pyrone (2d), and 4-hydroxy-6-(14′-methylpentadecyl)-2-pyrone (2e) (see structural data in the supplemental material).
We next analyzed the products of B. subtilis harboring pHO2 by HPLC and LC-APCIMS analyses (Fig. 2). The mass spectra of the products had molecular ions at 323 (3h and 3i), 337 (3f), 337 (3g), and 351 (3d and 3e). On the basis of [M+H]+ ions of these compounds, 3d to 3i were predicted to be methyl ether compounds of alkylpyrones 2d to 2i. To determine the structures of lipids 3d to 3i by NMR and HR-MS analyses, the lipids were separated into 3h and 3i, 3f and 3g, and 3d and 3e by HPLC equipped with a preparative C4 column. Their structures were determined as 4-methoxy-6-(11′-methyltridecyl)-2-pyrone (3h), 4-methoxy-6-(12′-methyltridecyl)-2-pyrone (3i), 4-methoxy-6-(13′-methylpentadecyl)-2-pyrone (3d), and 4-methoxy-6-(14′-methylpentadecyl)-2-pyrone (3e) by NMR and HR-MS analyses (see structural data in the supplemental material). The structures of 3f and 3g were determined as 4-methoxy-6-(13′-methyltetradecyl)-2-pyrone and 4-methoxy-6-pentadecyl-2-pyrone by proton NMR and HR-MS analyses, respectively (see structural data in the supplemental material). HMBC analysis showed the linkage of the methoxy group to the carbon at position 4 (Fig. 3), which clearly indicated that these alkylpyrone methyl ethers were derived from α-pyrones. The pyrone methyl ether moieties of compounds 3f and 3g were identified by comparing the proton NMR spectra to those of 3h, 3d, 3i, and 3e. The proton NMR spectra of the pyrone methyl ether moieties of 3f and 3g were the same as those for 3h, 3d, 3i, and 3e, showing that 3f and 3g possessed the same pyrone methyl ether moiety. These results were consistent with those for the in vivo products of BpsA. Therefore, BpsA was predicted to be a type III PKS responsible for triketide pyrone synthesis from branched and straight long-chain fatty acyl CoA thioesters as starter substrates and malonyl-CoA as an extender substrate, and BpsB was a methyltransferase acting on alkylpyrones (Fig. 4).
FIG. 3.
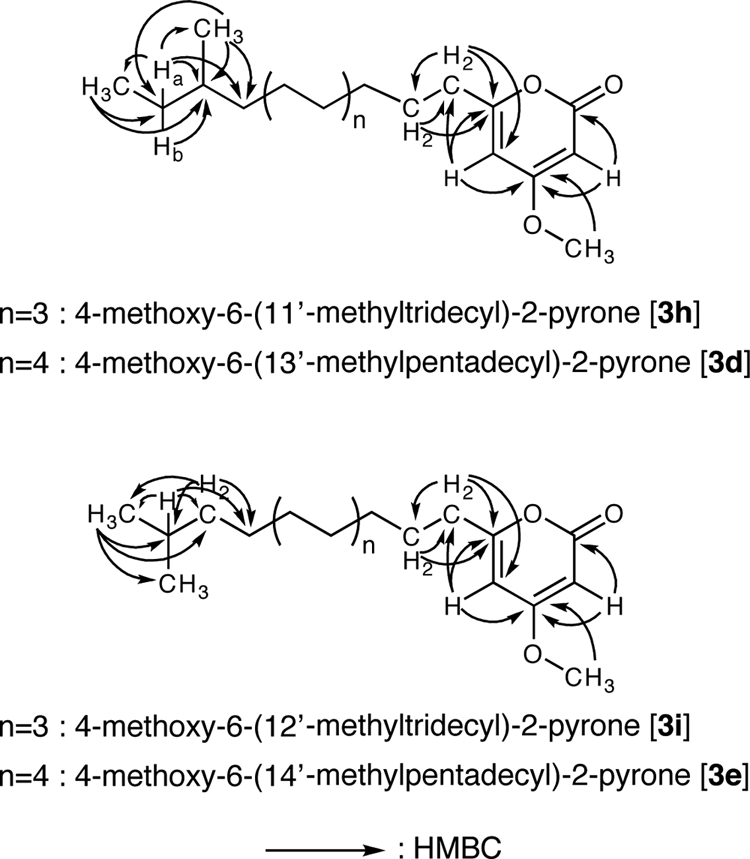
Key HMBC correlations of compounds 3h, 3d, 3i, and 3e. HMBC analysis showed that 3h and 3d were α-pyrone methyl ethers possessing an anteiso-alkyl chain, and 3i and 3e were α-pyrone methyl ethers possessing an iso-alkyl chain.
FIG. 4.

Summary of the reactions of BpsA and BpsB. BpsA catalyzes the synthesis of alkylpyrones and alkylresorcinols. The main in vivo products produced by B. subtilis carrying bpsA are triketide pyrones, and those produced by B. subtilis carrying both bpsA and bpsB are their methyl ethers. This major pathway is highlighted with thick arrows and squares. Small amounts of tetraketide pyrones and resorcinols also are produced as by-products.
In vitro analysis of recombinant BpsA protein.
From the in vivo experiments described above, BpsA was predicted to be a type III PKS responsible for triketide pyrone synthesis. To reconstruct this reaction in vitro, we placed bpsA under the control of the cspA promoter on the pColdI vector to produce BpsA as a fusion with a His tag at its N terminus. The histidine-tagged BpsA protein, purified with a nickel-nitrilotriacetic acid spin column, gave a single protein band at a position of ∼43 kDa on SDS-polyacrylamide gel electrophoresis (Fig. 5A). From in vivo analysis, the major product of BpsA was 4-hydroxy-6-(14′-methylpentadecyl)-2-pyrone (2e). Compound 2e was expected to be synthesized from 15-methylhexadecanoyl-CoA or 15-methylhexadecanoyl-ACP as a starter substrate and malonyl-CoA as an extender substrate. Neither 15-methylhexadecanoyl-CoA nor 15-methylhexadecanoyl-ACP was commercially available, therefore we synthesized NAC, a mimic of CoA and ACP, to check the presumed in vitro synthesis. As expected, the incubation of BpsA with 15-methylhexadecanoyl-NAC and malonyl-CoA resulted in the production of 4-hydroxy-6-(14′-methylpentadecyl)-2-pyrone (2e) (data not shown). In addition to this major product, BpsA produced a small amount of an alkylresorcinol (data not shown).
FIG. 5.
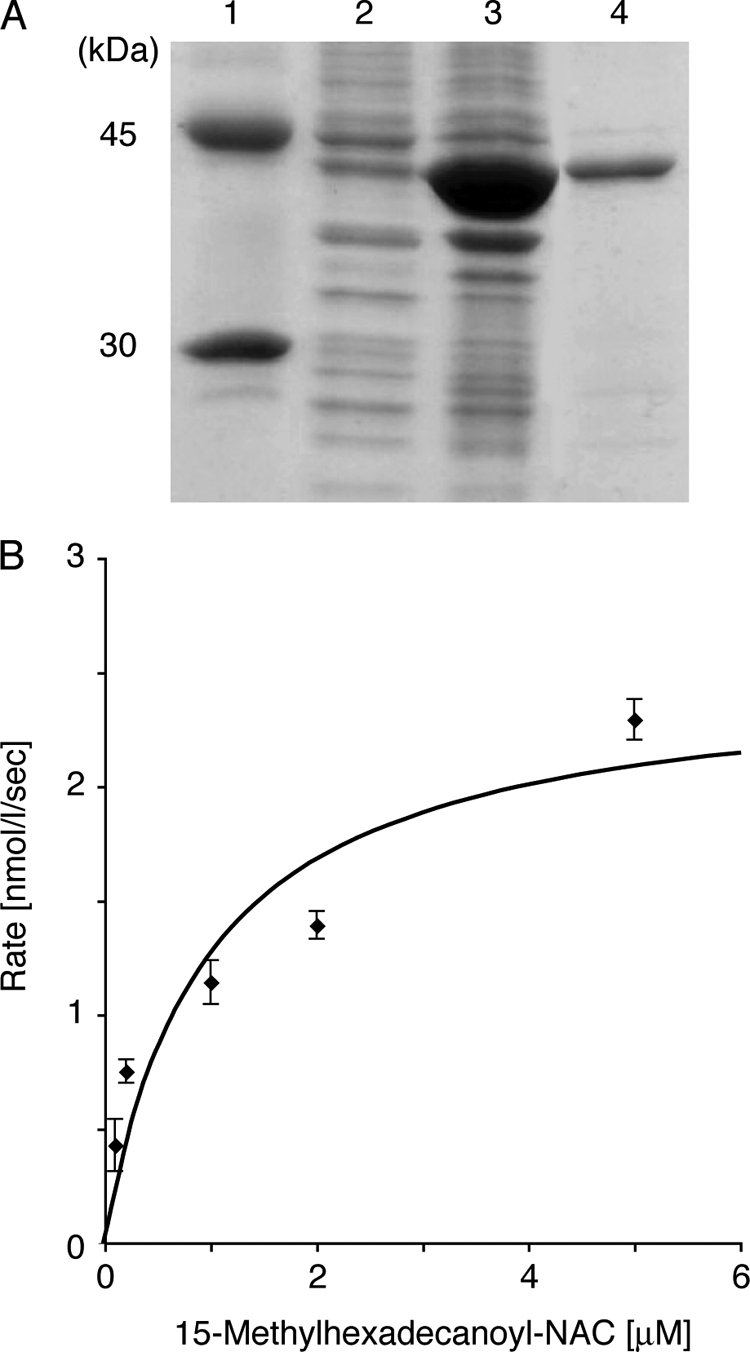
SDS-polyacrylamide gel electrophoresis of histidine-tagged BpsA used for the in vitro assays (A) and kinetic analysis of the enzyme (B). (A) SDS-polyacrylamide gel electrophoresis of histidine-tagged BpsA purified from E. coli BL21(DE3) harboring pColdI-bpsA. Lane 1, size markers including ovalbumin (45 kDa) and carbonic anhydrase (30 kDa); lane 2, the soluble fraction of the crude extract; lane 3, the insoluble fraction; and lane 4, histidine-tagged BpsA purified with a Ni-nitrilotriacetic acid spin column. (B) The 15-methylhexadecanoyl-NAC concentration profile for the synthesis of 4-hydroxy-6-(14′-methylpentadecyl)-2-pyrone (2e) from 15-methylhexadecanoyl-NAC and malonyl-CoA are shown. The data were obtained from three independent experiments.
We determined the temperature and pH dependence of BpsA for triketide pyrone synthesis using 15-methylhexadecanoyl-NAC as a starter substrate. BpsA showed a temperature optimum at 30°C and a pH optimum at 7.5. The kinetic properties of BpsA were determined under the optimum reaction conditions. The Km and kcat values for 15-methylhexadecanoyl-NAC were calculated to be 0.98 ± 0.02 μM and 4.90 × 10−3 ± 0.12 × 10−3 s−1, respectively (Fig. 5B).
Generally, type III PKSs show broad starter substrate specificity (2). We determined whether BpsA accepts acyl-CoA esters possessing C2 to C22 straight-chain alkyl moieties. BpsA accepted the C4 to C22 CoA esters and produced the corresponding triketide pyrones (Fig. 6). Moreover, this radio-TLC analysis revealed that BpsA produced not only triketide pyrones but also tetraketide pyrones and alkylresorcinols from the C8 to C20 esters (Fig. 6). BpsA produced 6a, 6b, 6c, and 6g from the C16 to C22 esters (Fig. 6), but we could not determine their structures because of the very small amounts of the compounds produced. By carefully examining the HPLC analysis of the ethyl acetate extract of B. subtilis harboring pHO1, several phenolic lipids were observed by LC-APCIMS in addition to compounds 2d to 2i, but the structures of these compounds could not be identified because the amounts were too small.
FIG. 6.

Substrate specificity of BpsA, as determined by radio-TLC analysis of the products synthesized by BpsA from various acyl-CoA starter substrates and [2-14C]malonyl-CoA. The starter substrates used were acetyl-CoA (1p) (lane 2), butyryl-CoA (1o) (lane 4), hexanoyl-CoA (1n) (lane 6), octanoyl-CoA (1m) (lane 8), decanoyl-CoA (1l) (lane 10), lauroyl-CoA (1k) (lane 12), myristoyl-CoA (1j) (lane 14), palmitoyl-CoA (1g) (lane 16), stearoyl-CoA (1c) (lane 18), arachidoyl-CoA (1b) (lane 20), and behenyl-CoA (1a) (lane 22). The products were identified as triketide pyrones (2a to 2o), tetraketide pyrones (4j to 4l), and resorcinols (5b to 5m). The structures of the four compounds (6a, 6b, 6c, and 6g) could not be determined.
Analysis of the possible biological function of aliphatic polyketides.
Recently, we revealed that the bacterial phenolic lipids that are synthesized by type III PKSs have some important roles in the membrane/cell wall (5, 6). The amphiphilic phenolic lipids alkylresorcinols and alkylpyrones are readily incorporated into phospholipid bilayers and biological membrane, thereby causing considerable changes in their structure and properties (12). In fact, the alkylresorcinols are fractionated mainly in the membrane/cell wall fraction (6). In A. vinelandii, alkylresorcinols and alkylpyrones, major components in the cyst membrane that are produced by the type III PKSs, ArsB and ArsC, are essential for cyst formation (5). The phenolic lipids synthesized by the Srs enzymes in S. griseus confer resistance to β-lactam antibiotics (6). Therefore, we speculate that aliphatic polyketides play some roles in the membrane in B. subtilis.
We constructed a mutant in which both bpsA and bpsB were deleted (mutant bpsA-bpsB::cat) to observe the possible phenotypic changes. Mutant bpsA-bpsB::cat grew normally in LB liquid and on agar medium, showing that the aliphatic polyketides had no detectable effects on growth. First, we compared the metabolic profiles of the lipid fraction of the wild-type and mutant bpsA-bpsB::cat strains by LC-APCIMS analysis. Although we carefully examined the data, both strains produced no detectable amounts of triketide pyrone derivatives.
The biological role of phenolic lipids observed for A. vinelandii and S. griseus prompted us to examine the biological roles of BpsA and BpsB. We examined their sensitivity to β-lactam antibiotics. However, no difference in sensitivity to the antibiotics of the mutant bpsA-bpsB::cat was observed between the wild-type and mutant strains (data not shown). We next examined the survival activity against a heat stress, because the transcription of bpsA was increased by a heat shock, as determined by microarray analysis (BSORF Bacillus subtilis genome database [http://bacillus.genome.jp/]). In B. subtilis, the sigB-dependent general stress genes and heat shock proteins, such as the GroESL and DnaK chaperone machines, and the proteins of the Clp families of chaperones and proteases contribute to coping with heat stresses (8, 13, 14, 16, 21, 22). The preadaptation of B. subtilis by exposure to 48°C induces the heat-specific protection systems mentioned above (8, 22). After the wild-type and mutant bpsA-bpsB::cat strains had been preadapted by exposure to 48°C for 1 h, they were challenged by heat treatment at 54 or 56°C, and their survival rates were quantified. However, the survival rates of mutant bpsA-bpsB::cat and the wild-type strain were the same (data not shown). In this study, we could not observe any other phenotypic differences between the wild-type strain and the mutant bpsA-bpsB::cat. In addition, we could not observe the triketide pyrone derivatives by HPLC analysis of the ethyl acetate extract prepared from the wild-type cell exposed to 48°C, although the transcription of bpsA increased upon a heat shock by microarray analysis (BSORF Bacillus subtilis genome database). Further analysis of the biological function of alkylpyrones and alkylpyrone methyl ethers in B. subtilis is now in progress.
The catalytic properties of BpsA and BpsB.
The present in vivo and in vitro experiments have demonstrated clearly that BpsA is the alkylpyrone synthase that catalyzes the synthesis of triketide pyrones from long-chain fatty acyl CoA thioesters and malonyl-CoA. BpsA was found to possess broad substrate specificity and accept branched and nonbranched fatty acyl CoA thioesters with alkyl chain lengths of C4 to C22. The in vivo analysis revealed that the amounts of the alkylpyrones produced by the action of BpsA reflect the ratio of the fatty acids present in B. subtilis. The de novo products by bacterial type II fatty acid synthases are released as ACP thioesters (18), and the CoA thioesters of long-chain fatty acids probably are absent in vivo. In addition, the direct transfer of the acyl moiety attached to the ACP domain of type I fatty acid synthase to type III PKS, ArsB and ArsC, was demonstrated in vitro (15). Furthermore, SCO7671, a type III PKS in Streptomyces coelicolor A3(2), accepts an acyl moiety from both hexanoyl-CoA and hexanoyl-ACP (7). Therefore, BpsA may directly accept the acyl moiety of acyl-ACP that is synthesized by the type II fatty acid synthesis system. The in vitro reaction of BpsA with fatty acyl-AcpA, which is an ACP responsible for type II fatty acid synthesis in B. subtilis, is our future work.
PKS18, a type III PKS in Mycobacterium tuberculosis, exhibits a broad starter substrate specificity and incorporates C6 to C20 CoA esters to produce tri- and tetraketide pyrones. Sankaranarayanan et al. (19) discovered a novel substrate binding tunnel in PKS18 that accommodates an alkyl moiety, as observed by the X-ray crystallography of an enzyme-myristic acid complex, and found that the side chains of the amino acids Thr144, Cys205, and Ala209 were crucial in determining the cavity volume. These residues correspond to Thr113, Cys174, and Gln179 of BpsA. We suppose that a similar binding tunnel accommodating various fatty acyl CoA esters as starter substrates exists in BpsA.
BpsB is the first reported enzyme that is responsible for catalyzing the methylation of alkylpyrones. BpsB also may act on alkylresorcinols, because it shows significant similarity to SrsB catalyzing the methylation of alkylresorcinols (6).
Regarding a possible role of the aliphatic polyketides produced by the BpsA-BpsB enzyme system, contrary to our expectation, mutant bpsA-bpsB::cat showed no detectable phenotypic changes. A possible biological function of the alkylpyrones in B. subtilis therefore remains unknown. The wide distribution of a bpsA-bpsB operon in gram-positive and -negative bacteria and the distinct roles of the phenolic lipids in A. vinelandii (5) and S. griseus (6) suggest important, but thus far overlooked, role of the phenolic lipids probably incorporated in the membrane. There also was no significant difference in the lipids produced between mutant bpsA-bpsB::cat and the wild-type strains, and the production of alkylpyrones was not detected in the wild-type strain. The amounts of the pyrones may be under the detection limits, or the triketide pyrone methyl ethers may be modified further by some enzymes existing in B. subtilis. Therefore, the compounds synthesized by BpsA and its tailoring enzymes are mysterious. The genome sequence revealed that in the neighborhood of the bpsA-bpsB operon, there are no genes that appear to encode enzymes able to modify the alkylpyrones or its methyl ethers. Further study will reveal their biological functions.
Supplementary Material
Acknowledgments
G. Akanuma was supported by the Japan Society for the Promotion of Science. This work was supported, in part, by a research grant from the New Energy and Industrial Technology Development Organization of Japan and a Grant-in-Aid for Scientific Research on Priority Area Applied Genomics from Monkasho.
Footnotes
Published ahead of print on 22 May 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Anagnostopoulos, C., and J. Spizizen. 1961. Requirements for transformation in Bacillus subtilis. J. Bacteriol. 81741-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Austin, M. B., and J. P. Noel. 2003. The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 2079-110. [DOI] [PubMed] [Google Scholar]
- 3.Capuano, V., N. Galleron, P. Pujic, A. Sorokin, and S. D. Ehrlich. 1996. Organization of the Bacillus subtilis 168 chromosome between kdg and the attachment site of the SPβ prophage: use of Long Accurate PCR and yeast artificial chromosomes for sequencing. Microbiology 1423005-3015. [DOI] [PubMed] [Google Scholar]
- 4.Funa, N., Y. Ohnishi, I. Fujii, M. Shibuya, Y. Ebizuka, and S. Horinouchi. 1999. A new pathway for polyketide synthesis in microorganisms. Nature 400897-899. [DOI] [PubMed] [Google Scholar]
- 5.Funa, N., H. Ozawa, A. Hirata, and S. Horinouchi. 2006. Phenolic lipid synthesis by type III polyketide synthases is essential for cyst formation in Azotobacter vinelandii. Proc. Natl. Acad. Sci. USA 1036356-6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Funabashi, M., N. Funa, and S. Horinouchi. 2008. Phenolic lipids synthesized by type III polyketide synthase confer penicillin resistance on Streptomyces griseus. J. Biol. Chem. 28313983-13991. [DOI] [PubMed] [Google Scholar]
- 7.Grüschow, S., T. J. Buchholz, W. Seufert, J. S. Dordick, and D. H. Sherman. 2007. Substrate profile analysis and ACP-mediated acyl transfer in Streptomyces coelicolor type III polyketide synthases. Chembiochem 8863-868. [DOI] [PubMed] [Google Scholar]
- 8.Höper, D., U. Völker, and M. Hecker. 2005. Comprehensive characterization of the contribution of individual SigB-dependent general stress genes to stress resistance of Bacillus subtilis. J. Bacteriol. 1872810-2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Imamura, D., K. Kobayashi, J. Sekiguchi, N. Ogasawara, M. Takeuchi, and T. Sato. 2004. spoIVH (ykvV), a requisite cortex formation gene, is expressed in both sporulating compartments of Bacillus subtilis. J. Bacteriol. 1865450-5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaneda, T. 1977. Fatty acids of the genus Bacillus: an example of branched-chain preference. Bacteriol. Rev. 41391-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawamura, F., H. Saito, and Y. Ikeda. 1980. Bacteriophage φ1 as a gene-cloning vector in Bacillus subtilis. Mol. Gen. Genet. 180259-266. [DOI] [PubMed] [Google Scholar]
- 12.Kozubek, A., and J. H. P. Tyman. 1999. Resorcinolic lipids, the natural non-isoprenoid phenolic amphiphiles and their biological activity. Chem. Rev. 991-25. [DOI] [PubMed] [Google Scholar]
- 13.Krüger, E., U. Völker, and M. Hecker. 1994. Stress induction of clpC in Bacillus subtilis and its involvement in stress tolerance. J. Bacteriol. 1763360-3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li, M., and S. Wong. 1992. Cloning and characterization of the groESL operon from Bacillus subtilis. J. Bacteriol. 1743981-3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyanaga, A., N. Funa, T. Awakawa, and S. Horinouchi. 2008. Direct transfer of starter substrates from type I fatty acid synthase to type III polyketide synthases in phenolic lipid synthesis. Proc. Natl. Acad. Sci. USA 105871-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Msadek, T., F. Kunst, and G. Rapoport. 1994. MecB of Bacillus subtilis, a member of the ClpC ATPase family, is a pleiotropic regulator controlling competence gene expression and growth at high temperature. Proc. Natl. Acad. Sci. USA 915788-5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oguro, S., T. Akashi, S. Ayabe, H. Noguchi, and I. Abe. 2004. Probing biosynthesis of plant polyketides with synthetic N-acetylcysteamine thioesters. Biochem. Biophys. Res. Commun. 325561-567. [DOI] [PubMed] [Google Scholar]
- 18.Rock, C. O., and J. E. Cronan. 1996. Escherichia coli as a model for the regulation of dissociable (type II) fatty acid biosynthesis. Biochim. Biophys. Acta 13021-16. [DOI] [PubMed] [Google Scholar]
- 19.Sankaranarayanan, R., P. Saxena, U. B. Marathe, R. S. Gokhale, V. M. Shanmugam, and R. Rukmini. 2004. A novel tunnel in mycobacterial type III polyketide synthase reveals the structural basis for generating diverse metabolites. Nat. Struct. Mol. Biol. 11894-900. [DOI] [PubMed] [Google Scholar]
- 20.Saxena, P., G. Yadav, D. Mohanty, and R. S. Gokhale. 2003. A new family of type III polyketide synthases in Mycobacterium tuberculosis. J. Biol. Chem. 27844780-44790. [DOI] [PubMed] [Google Scholar]
- 21.Schulz, A., B. Tzschaschel, and W. Schumann. 1995. Isolation and analysis of mutants of the dnaK operon of Bacillus subtilis. Mol. Microbiol. 15421-429. [DOI] [PubMed] [Google Scholar]
- 22.Völker, U., B. Maul, and M. Hecker. 1999. Expression of the σB-dependent general stress regulon confers multiple stress resistance in Bacillus subtilis. J. Bacteriol. 1813942-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winter-Vann, A. M., and P. J. Casey. 2005. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer. 5405-412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.