

Abstract
Human vesicle-associated membrane protein-associated protein (VAP) subtype A (VAP-A) and subtype B (VAP-B) are involved in the regulation of membrane trafficking, lipid transport and metabolism, and the unfolded protein response. VAP-A and VAP-B consist of the major sperm protein (MSP) domain, the coiled-coil motif, and the C-terminal transmembrane anchor and form homo- and heterodimers through the transmembrane domain. VAP-A and VAP-B interact with NS5B and NS5A of hepatitis C virus (HCV) through the MSP domain and the coiled-coil motif, respectively, and participate in the replication of HCV. VAP-C is a splicing variant of VAP-B consisting of the N-terminal half of the MSP domain of VAP-B followed by the subtype-specific frameshift sequences, and its biological function has not been well characterized. In this study, we have examined the biological functions of VAP-C in the propagation of HCV. VAP-C interacted with NS5B but not with VAP-A, VAP-B, or NS5A in immunoprecipitation analyses, and the expression of VAP-C inhibited the interaction of NS5B with VAP-A or VAP-B. Overexpression of VAP-C impaired the RNA replication of the HCV replicon and the propagation of the HCV JFH1 strain, whereas overexpression of VAP-A and VAP-B enhanced the replication. Furthermore, the expression of VAP-C was observed in various tissues, whereas it was barely detected in the liver. These results suggest that VAP-C acts as a negative regulator of HCV propagation and that the expression of VAP-C may participate in the determination of tissue tropism of HCV propagation.
Hepatitis C virus (HCV) is a major causative agent of chronic liver disease and thus a major public health problem, infecting at least 3% of the world population (47). HCV infection proceeds to the persistent stage in approximately 80% of patients, leading to the development of cirrhosis in 20% to 50% of patients, of whom approximately 5% eventually develop hepatocellular carcinoma (12). HCV encompasses a single-stranded positive-sense RNA genome of approximately 9.6 kb, which encodes a large precursor polyprotein comprising approximately 3,000 amino acids (26). The structural proteins are cleaved from the N-terminal one-fourth of the polyprotein by the host signal peptidase and signal peptide peptidase (23, 32, 33), resulting in the maturation of the capsid protein, two envelope proteins and viroporin p7. The NS2 protease cleaves after the carboxyl terminus, and then NS3 cleaves the appropriate downstream positions to produce NS4A, NS4B, NS5A, and NS5B (8, 42), all of which form the replication complex along with several host proteins (5, 21). NS5B is the RNA-dependent RNA polymerase, which is a main enzymatic component of the replication complex of HCV (3), while NS5A is a membrane-anchored zinc-binding phosphoprotein that appears to possess diverse functions, including the suppression of host defense and the regulation of the virus's replication (1, 4, 6, 41), although its biological function remains unclear.
The NS5A protein has been shown to interact with several host proteins, including vesicle-associated membrane protein (VAMP)-associated protein (VAP) subtype A (VAP-A) (44) and subtype B (VAP-B) (9), FKBP8 (34), MyD88 (1), FBL2 (46), human butyrate-induced transcript 1 (hB-ind1) (40), and so on (25). VAP-A and VAP-B also bind to NS5B, although it remains unclear whether these interactions modulate HCV replication positively or negatively (9, 44). VAP-A and VAP-B have been shown to associate with the cytoplasmic face of the endoplasmic reticulum (ER) and the Golgi apparatus (38) and to consist of the major sperm protein (MSP) domain, the coiled-coil domain, and the transmembrane (TM) region, in that order (30, 39), as shown in Fig. 1A. VAP was originally reported as a protein binding to VAMP, which is a synaptic vesicle SNARE protein required for synaptic-vesicle fusion in the nematode Aplysia californica, and was designated the 33-kDa VAMP-associated protein, VAP-33 (39). Two mammalian homologues, VAP-A and VAP-B, were subsequently identified (30, 38). The transcription of VAP-A and VAP-B is ubiquitously detected in mammalian organs, including the heart, placenta, lung, liver, skeletal muscle, and pancreas (30), suggesting that VAP family proteins are involved in diverse cellular functions other than neurotransmitter release (30, 38, 49). Several VAP-interacting proteins share the FFAT motif (two phenylalanines in an acidic tract), which has the consensus amino acid sequence EFFDAXE, as determined by a comparison among oxysterol binding proteins (OSBPs), OSBP-related proteins (ORPs) (20), and the ceramide transport protein CERT (10, 19), contributing to the regulation of fatty acid metabolism. The interaction of VAP family proteins with other host proteins, including VAMP and tubulin, is independent of the FFAT motif (16, 36, 38, 50). The third subtype of VAP is VAP-C, which is an alternative spliced isoform of VAP-B, consisting of the N-terminal half of the MSP domain and the subtype-specific 29 amino acids (Fig. 1A). However, its tissue distribution and physiological function remain largely unknown.
FIG. 1.

VAP-C interacts with neither VAP-A nor VAP-B. (A) Structures of VAP family proteins. The MSP domain, the coiled-coil domain, and the TM region are indicated as MSP, CC, and TM, respectively. (B) Interaction among VAP family proteins. The expression plasmids encoding VAP proteins or empty vector (1 μg each) were transfected into 293T cells, FLAG-tagged VAP proteins coexpressed with EE-tagged VAP-A or VAP-B were immunoprecipitated (IP) with anti-FLAG antibody, and the resulting precipitates were examined by immunoblotting using anti-FLAG or anti-EE antibody. One percent of the volume of the lysate was used as an input control. The data in each panel are representative of the results of three independent experiments. +, present.
Glutathione S-transferase pulldown and immunoprecipitation analyses revealed that both VAP-A and VAP-B interact with NS5B and NS5A through the MSP domain and the coiled-coil domain, respectively (9, 44), and the MSP domains of VAP-A and VAP-B exhibit 82.3% homology. Although VAP-C possesses the N-terminal-half region of the MSP domain of VAP-B, the biological significance of VAP-C in the propagation of HCV has not yet been clarified. In this study, we examined the expression of VAP-C in human tissues and the effects of VAP-C expression on the RNA replication, translation, and particle formation of HCV.
MATERIALS AND METHODS
Cell lines.
Cells of the human hepatoma cell line Huh-7, cell line Huh7OK1, and embryonic kidney cell line 293T were maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma, St. Louis, MO) containing 10% fetal calf serum (FCS) and nonessential amino acids (NEAA), while Huh 9-13 cells, which possess a subgenomic HCV RNA replicon of genotype 1b (21), were cultured in DMEM supplemented with 10% FCS, NEAA, and 1 mg/ml G418. The Huh7OK1 cell line exhibits the highest efficiency of propagation of strain JFH1 virus, as described previously (35). All cell lines were cultured at 37°C in a humidified atmosphere with 5% CO2.
Antibodies.
Chicken anti-human VAP-B antibody was described previously (9). Rabbit anti-human VAP-C antibody was prepared by immunization using synthetic peptides of the amino acid residues from 86 to 98, QPHFSISPNWEGR, which region does not share the homology to VAP-A and VAP-B. The mouse monoclonal antibody to human VAP-A was purchased from BD Pharmingen (San Diego, CA). Mouse monoclonal antibodies to influenza virus hemagglutinin (HA) and the GluGlu (EE) tag were from Covance (Richmond, CA). Mouse and rabbit anti-FLAG antibodies and mouse anti-β-actin monoclonal antibody were from Sigma. Rabbit polyclonal antibody to NS5A was prepared as described previously (34). Mouse anti-NS5A monoclonal antibody was from Austral Biologicals (San Ramon, CA).
Plasmids.
A cDNA clone encoding NS5A was amplified from HCV genotype 1b strain J1 (9) (GenBank database accession number D89815) by PCR, using Pfu turbo DNA polymerase (Stratagene, La Jolla, CA). The fragments were then cloned into the appropriate sites in pEF-FLAG pGBK puro (13). The DNA fragment encoding NS5B of the J1 strain was generated by PCR and cloned into pCAGGs-PUR (31). The DNA fragment encoding human VAP-A was amplified by PCR from a human fetal-brain library (Clontech, Palo Alto, CA) and was introduced into pEF-FLAG pGBK puro and pEF-EE hygro (13), as described previously (9). A DNA fragment encoding VAP-C was amplified from cDNA of hepatoma cell line Huh-7 and was introduced into pEF-FLAG pGBK puro. Pro56-to-Ser (P56S) mutants of VAPs were generated by site-directed mutagenesis (11). All PCR products were confirmed by sequencing with an ABI Prism 3130 genetic analyzer (Applied Biosystems, Tokyo, Japan).
Transfection, immunoblotting, and immunoprecipitation.
Cells were seeded onto a six-well tissue culture plate 24 h before transfection. The plasmids were transfected into cells by liposome-mediated transfection using TransIT LT1 (Mirus Bio, Madison, WI). These transfected cells were harvested at 36 h posttransfection, washed three times with 1 ml of ice-cold phosphate-buffered saline (PBS), and suspended in 0.2 ml lysis buffer (20 mM Tris-HCl, pH 7.4, containing 135 mM NaCl and 1% Triton X-100) supplemented with protease inhibitor cocktail (Roche, Indianapolis, IN). The cell lysates were sonicated at 4°C for 5 min, incubated for 30 min at 4°C, and centrifuged at 15,000 rpm for 30 min at 4°C. The supernatant was subjected to immunoprecipitation analyses as described previously (27). The immunoprecipitated proteins were boiled in 30 μl of loading buffer and then subjected to sodium dodecyl sulfate-12.5% polyacrylamide gel electrophoresis. The proteins were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) and then reacted with primary antibody and secondary horseradish peroxidase-conjugated antibody. The immunocomplexes were visualized with Super Signal West Femto substrate (Pierce, Rockford, IL) and detected by using an LAS-3000 image analyzer (Fujifilm, Tokyo, Japan). The distribution of VAPs in human organs was determined by using premade human tissue lysates (Protein medleys; Clonetech), which are aliquots of various organ lysates prepared from samples from several people, and liver tissues obtained during surgery after approval of the ethical committee of Kyushu University Graduate School of Medicine.
Real-time PCR.
The HCV genomic RNA was determined by the method described previously (40). Total RNA was prepared from cells by using an RNeasy mini kit (Qiagen, Tokyo, Japan). First-strand cDNA was synthesized using an RNA LA PCR kit (Takara Bio, Inc., Shiga, Japan) and random primers. Expression of the appropriate gene was estimated by using platinum SYBR green quantitative PCR SuperMix UDG (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Fluorescent signals were estimated by using an ABI Prism 7000 system (Applied Biosystems). The 5′ untranslated region of HCV and the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA were amplified using primer pairs described previously (40). The amount of HCV genomic RNA was normalized with that of GAPDH mRNA.
Focus-forming assay.
The viral RNA of the JFH1 strain was introduced into the Huh7OK1 cell line according to the method of Zhong et al. (51). The culture supernatant was collected at 7 days posttransfection and used as the infectious HCV particles. Huh7OK1 cells in DMEM containing 10% FCS were seeded at 5 × 104 cells per well into a 24-well plate 12 h before infection. The cells were infected with the JFH1 strain at a multiplicity of infection (MOI) of 0.05 and incubated at 37°C for 2 h. The medium was replaced with fresh DMEM containing 10% FCS and NEAA at 2 h postinfection. The cells were fixed with 4% paraformaldehyde at 96 h postinfection and permeabilized with PBS containing 0.2% Triton X-100. These fixed and permeabilized cells were stained with the anti-NS5A mouse monoclonal antibody and Alexa Fluor (AF) 488-conjugated antibody to mouse immunoglobulin G (Molecular Probes, Eugene, OR). Clusters of infected cells stained with the NS5A antibody were derived from a single infectious focus, and virus titers were represented as focus-forming units/ml.
Quantification of the HCV core protein by ELISA.
The HCV core protein was quantified by using an Ortho HCV antigen enzyme-linked immunosorbent assay (ELISA) test (Ortho Clinical Diagnostics, Tokyo, Japan) according to the manufacturer's instructions. To determine the intracellular expression of core protein, Huh7OK1 cells were infected with the infectious HCV particles described above, lysed with the lysis buffer on ice, and applied to the ELISA after 100- to 10,000-fold dilution with PBS. Total protein was quantified by using a Micro BCA protein assay reagent kit (Pierce). The intracellular and extracellular levels of expression of the core protein were normalized by the total amount of protein.
Effect of the VAP expression on the cap-independent translational activity of the viral IRES.
The cDNA fragment encoding a firefly luciferase was excised from a pGL3 basic plasmid (Promega, Madison, WI) and introduced into the downstream region of the Renilla luciferase gene of pRL-CMV (cytomegalovirus) (Promega). Then, the cDNA fragments encoding the internal ribosome entry site (IRES) of the HCV strains Con1 and JFH1 were introduced between the Renilla and firefly luciferase genes, and the resulting plasmids were designated pRL-CMV-HCVCon1 and pRL-CMV-HCVJFH1, respectively (see Fig. 4A). The IRES region of HCV was replaced with that of poliovirus (PV) or encephalomyocarditis virus (EMCV), and the plasmids designated pRL-CMV-PV and pRL-CMV-EMCV, respectively (see Fig. 4B). Each reporter plasmid was introduced into Huh7OK1 cells that had been transfected with the expression plasmid encoding FLAG-green fluorescent protein (GFP), FLAG-VAP-A, FLAG-VAP-B, or FLAG-VAP-C 24 h previously, and cells were harvested at 48 h posttransfection. Luciferase activities in cells were measured by using a dual-luciferase reporter assay system (Promega). The activity of firefly luciferase was normalized with that of Renilla luciferase and represented as relative luciferase activity (RLU).
FIG. 4.
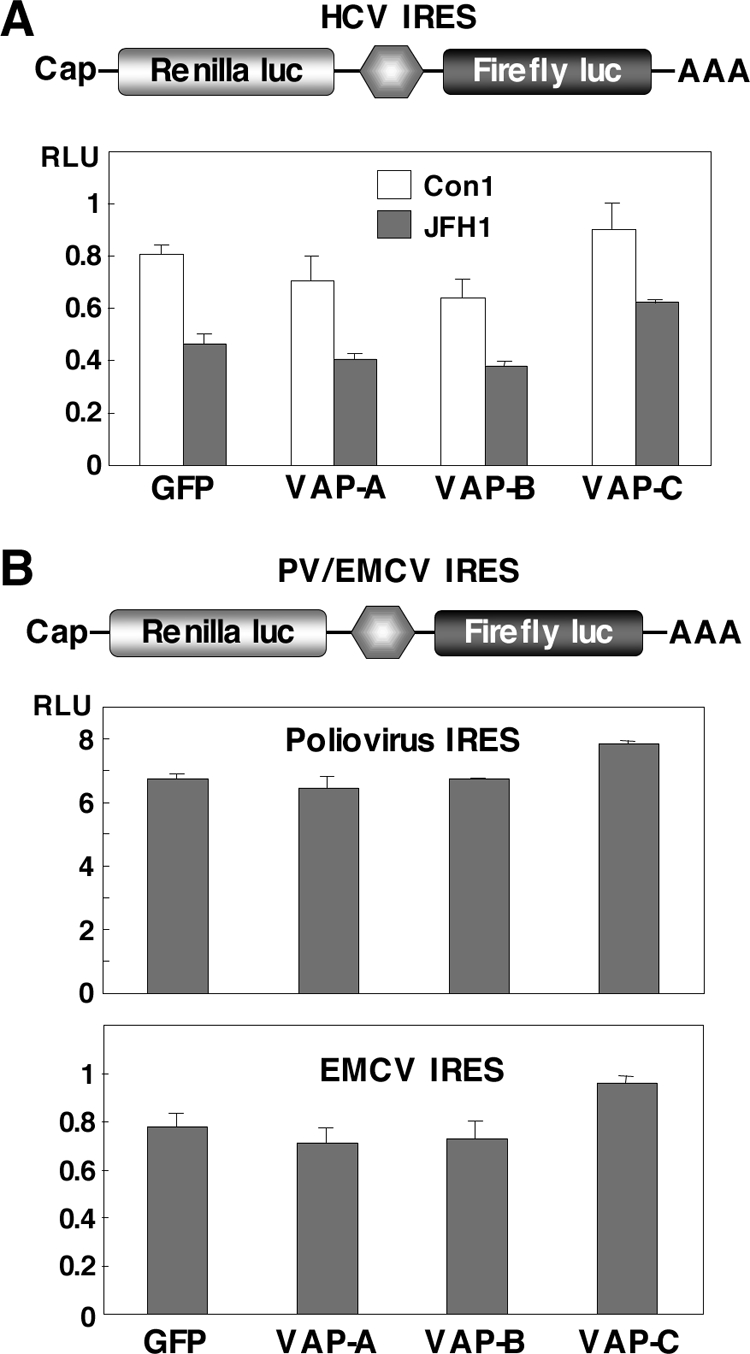
VAP-C exhibits no effect on the viral IRES-dependent translation. (A) Top: structure of a reporter plasmid encoding the Renilla luciferase gene under the control of the CMV promoter and the firefly luciferase gene under the HCV IRES, in order. Bottom: the reporter plasmid was introduced into Huh7OK1 cells 24 h after transfection of the expression plasmids encoding VAP-A, VAP-B, or VAP-C, the cells harvested at 48 h posttransfection, and the RLUs determined after standardization with the expression of Renilla luciferase. (B) Top: structure of a reporter plasmid encoding the Renilla luciferase gene under the control of the CMV promoter and the firefly luciferase gene under the PV or EMCV IRES, in order. Bottom: each of the reporter plasmids was introduced into Huh7OK1 cells, and the RLU values were determined as described for panel A. Data in this figure are shown as the means ± standard deviations.
Indirect immunofluorescence assay.
The Huh 9-13 cells were cultured on glass slides and transfected with the expression plasmids encoding FLAG-tagged VAPs, P56S VAP mutants, or empty vector. The resulting cells were fixed at 72 h posttransfection with 4% paraformaldehyde in PBS at room temperature for 30 min. After being washed twice with PBS, cells were permeabilized for 20 min at room temperature with PBS containing 0.25% saponin and blocked with PBS containing 1% bovine serum albumin (BSA-PBS) for 60 min at room temperature. The cells were then incubated with BSA-PBS containing rabbit anti-FLAG and mouse anti-NS5A antibodies at 37°C for 60 min, washed three times with PBS containing 1% Tween 20 (PBS-T), and incubated with BSA-PBS containing AF 488-conjugated goat anti-rabbit immunoglobulin G and AF 594-conjugated goat anti-mouse antibodies at 37°C for 60 min. Finally, the cells were washed three times with PBS-T and observed with a FluoView FV1000 laser-scanning confocal microscope (Olympus, Tokyo, Japan).
RESULTS
VAP-C interacts with neither VAP-A nor VAP-B.
The length of VAP-A was originally reported to be 242 amino acids but was recently corrected to 249 amino acids in the GenBank database due to the detection of 7 extra amino acids in the N terminus (Fig. 1A). VAP-C is a splicing variant of VAP-B that shares the N-terminal half of the MSP domain with VAP-B but lacks the coiled-coil motif and TM region (Fig. 1A). The region spanning residues 71 to 99 of VAP-C exhibits no homology to VAP-A and VAP-B, due to the frameshift. VAP-A and VAP-B form homo- or heterodimers via their TM domains, which is required for HCV replication (9, 44). To examine whether VAP-C is capable of interacting with VAP-A and VAP-B, FLAG-tagged VAP-A, -B, or -C was coexpressed with EE-tagged VAP-A or -B in 293T cells and was immunoprecipitated with the anti-FLAG antibody. Although EE-tagged VAP-A and VAP-B were coprecipitated with FLAG-tagged VAP-B and VAP-A, as reported previously, FLAG-VAP-C was precipitated with neither EE-VAP-A nor EE-VAP-B (Fig. 1B). These results indicate that VAP-C does not interact with VAP-A and VAP-B.
VAP-C binds to NS5B and interrupts the interaction of VAP-A and VAP-B with NS5B.
VAP-A and VAP-B were identified as NS5A-binding proteins by yeast two-hybrid screening (9, 44). The coiled-coil domains of VAP-A and VAP-B were involved in the binding to NS5A, contributing to the efficiency of HCV replication (9, 44). However, VAP-C does not have the coiled-coil domain (Fig. 1A) and, therefore, VAP-C was expected not to interact with NS5A. To examine whether or not interaction between VAP-C and NS5A actually occurred, HA-tagged NS5A was coexpressed with FLAG-tagged VAP-A, -B, or -C in 293T cells and was immunoprecipitated with anti-HA antibody (Fig. 2). The results showed that the expression level of FLAG-VAP-C in the transfected cells was comparable to that of FLAG-VAP-A or FLAG-VAP-B (Fig. 2A, left). Although FLAG-tagged VAP-A and VAP-B were coprecipitated with HA-NS5A, no precipitation of FLAG-VAP-C with NS5A was detected (Fig. 2A, right), indicating that VAP-C does not interact with NS5A.
FIG. 2.

VAP-C binds to NS5B but not NS5A and interrupts the interaction of VAP-A and VAP-B with NS5B. (A) The expression plasmids encoding NS5A or VAP proteins (1 μg each) were transfected into 293T cells after adjusting the total amounts of DNA to 2.0 μg with empty plasmid. HA-tagged NS5A was coexpressed with either FLAG-tagged VAP-A, VAP-B, or VAP-C in 293T cells and immunoprecipitated (IP) with anti-HA antibody, and the resulting precipitates were immunoblotted using anti-FLAG or anti-HA antibody. (B) The expression plasmids encoding NS5B or VAP proteins (1 μg each) were transfected into 293T cells after adjusting the total amounts of DNA to 3.0 μg with empty plasmid. HA-tagged NS5B was coexpressed with either FLAG-tagged VAP-A or VAP-B in the presence or absence of FLAG-tagged VAP-C in 293T cells and immunoprecipitated (IP) with anti-HA antibody, and the resulting precipitates were immunoblotted using anti-FLAG or anti-HA antibody. One percent of the lysate was used as an input control. The data in each panel are representative of the results of three independent experiments. +, present.
The RNA-dependent RNA polymerase NS5B was shown to interact with VAP-A through the MSP domain (44). The region spanning residues 1 to 70 of VAP-C is the same as the N-terminal-half region of the MSP domain of VAP-B and exhibits 77% homology to that of VAP-A (Fig. 1A). To examine whether VAP-C is capable of interacting with NS5B, as are VAP-A and VAP-B, HA-NS5B was coexpressed with FLAG-VAP-A, FLAG-VAP-B, or FLAG-VAP-C in 293T cells and was immunoprecipitated with anti-HA antibody (Fig. 2B). Although substantial amounts of FLAG-tagged VAP-A, VAP-B, and VAP-C were coexpressed, and although all three were coprecipitated with HA-NS5B at comparable levels, the interaction of HA-NS5B with FLAG-tagged VAP-A or VAP-B was impaired by the coexpression of VAP-C, while FLAG-VAP-C was coprecipitated with HA-NS5B instead of FLAG-tagged VAP-A or VAP-B. These results suggest that VAP-C is capable of binding to NS5B and that the expression of VAP-C interrupts the interactions of NS5B with VAP-A and VAP-B.
Expression of VAP-C impairs the replication of HCV.
VAP-A and VAP-B are known to support the replication of HCV RNA (2, 7). To examine the effect of VAP-C on the replication of HCV, FLAG-VAP-C was expressed in HCV replicon cells, Huh 9-13, in which a subgenomic HCV RNA of the genotype 1b strain Con1 was autonomously replicating. Huh 9-13 cells transfected with a plasmid encoding FLAG-VAP-C were harvested periodically up to 72 h posttransfection. The levels of replication of viral RNA and expression of NS5A were determined by real-time PCR and immunoblotting, respectively (Fig. 3). The expression of VAP-C reduced the intracellular RNA of the subgenomic HCV replicon in accordance with the incubation period after transfection with the expression plasmid of FLAG-VAP-C; the empty plasmid did not reduce the intracellular RNA (Fig. 3A). The expression of NS5A was gradually decreased and was undetectable at 72 h posttransfection, in contrast to the increase of VAP-C expression (Fig. 3B).
FIG. 3.

Expression of VAP-C impairs the replication of HCV. (A) HCV replicon cells (Huh 9-13) were transfected with 4 μg of the expression plasmids encoding FLAG-tagged VAP-C or empty vector, and the level of intracellular HCV RNA was determined at 0, 12, 24, 48, or 72 h posttransfection by real-time PCR after normalization with GAPDH mRNA. The value of HCV RNA at 0 h posttransfection in the cell line transfected with the empty plasmid is represented as 100%. Data in this panel are shown as means ± standard deviations. (B) Huh 9-13 cells were transfected with 4 μg of the plasmid encoding FLAG-tagged VAP-C or empty plasmid, and the levels of expression of NS5A, β-actin, and VAP-C were determined at 0, 12, 24, 48, or 72 h posttransfection by immunoblotting using anti-NS5A, anti-β-actin, or anti-FLAG tag antibody. (C) Huh 9-13 cells were transfected with 0 to 4 μg of the plasmids encoding FLAG-tagged VAP-A, VAP-B, or VAP-C or empty vector, and the level of intracellular HCV RNA was determined at 72 h posttransfection as described for panel A. Data in this panel are shown as means ± standard deviations. (D) Huh 9-13 cells treated as described for panel C were harvested at 72 h posttransfection, and the levels of expression of NS5A, β-actin, VAP-A, VAP-B, and VAP-C were determined by immunoblotting. The data in each panel are representative of the results of three independent experiments.
Next, to determine the effects of VAP-C expression on the replication of HCV, Huh 9-13 cells were transfected with 0 to 4 μg of the expression plasmid encoding VAP-A, VAP-B, or VAP-C and the replication of the subgenomic HCV RNA was determined at 48 h posttransfection. Although the HCV replicon cells transfected with 4 μg of a plasmid encoding FLAG-VAP-B exhibited enhancement of the RNA replication, those transfected with an equivalent amount of plasmid encoding FLAG-VAP-A or empty vector showed a slight reduction of HCV RNA replication. In contrast, the replicon cells transfected with a plasmid encoding FLAG-VAP-C exhibited a clear reduction of the HCV RNA replication in a dose-dependent manner (Fig. 3C). The expression of FLAG-tagged VAP-A, VAP-B, or VAP-C in the replicon cells was increased in correspondence with the amount of the transfected plasmid (Fig. 3D), and the expression of NS5A was suppressed in accordance with the expression of FLAG-VAP-C, whereas the expression of FLAG-VAP-A and FLAG-VAP-B exhibited no effect on the expression of NS5A. These results suggest that the expression of VAP-C impairs the replication of HCV RNA.
VAP-C exhibits no effect on the IRES-dependent translation.
The expression of VAP-C was shown to suppress the replication of the HCV RNA replication of the replicon cells. Next, to determine the effect of VAPs on the translation of HCV RNA, the reporter plasmid encoding the Renilla luciferase gene under the control of the CMV promoter and the firefly luciferase gene under the IRES of HCV, PV, or EMCV, in that order, was prepared as shown in Fig. 4. These reporter plasmids were introduced into Huh7OK1 cells 24 h after transfection of the expression plasmids encoding VAP-A, VAP-B, or VAP-C and harvested at 48 h posttransfection, and then the RLUs were determined. Although VAP-C exhibited a slight increase in the IRES-dependent translations of the HCV strains Con1 and JFH1, no significant effect of the expression of the VAPs on the HCV IRES-dependent translation was observed (Fig. 4A). Similarly, the expression of each of the VAPs in Huh7OK1 cells exhibited no significant effect on the IRES-dependent translation of PV or EMCV (Fig. 4B). These results indicate that the suppression of HCV RNA replication by the expression of VAP-C was not due to the suppression of the IRES-dependent translation of the viral RNA genome.
VAP-C impairs HCV propagation.
To examine the effect of VAP expression on HCV propagation, Huh7OK1 cells transfected with the expression plasmids encoding VAP-A, VAP-B, or VAP-C were infected with JFH1 virus, and the levels of production of the viral RNA, core protein, and infectious particles were determined at 96 h postinfection. The production of intracellular and extracellular viral RNA was increased up to 10 to 30 times and 2 to 3 times, respectively, by the expression of VAP-A or VAP-B whereas it was clearly decreased in a dose-dependent manner by the expression of VAP-C (Fig. 5A). Although the extracellular core protein was increased from 0.6 to 2.6 nmol/liter by the expression of VAP-A or VAP-B, as seen in the production of viral RNA, the intracellular core protein showed only a marginal increase (40 to 65 nmol/liter) (Fig. 5A). Although the reason for the discrepancy between the intracellular production of viral RNA and core protein is not known at the moment, some mechanisms other than RNA translation might be involved, because VAP expression exhibited no effect on the HCV IRES-dependent translation, as shown in Fig. 4A. In contrast to the enhancement of core protein production by the expression of VAP-A or VAP-B, the expression of VAP-C significantly reduced both the intracellular and extracellular expression of the core protein (Fig. 5A). Furthermore, the production of infectious particles in the culture supernatants of Huh7OK1 cells infected with JFH1 virus was slightly enhanced by the expression of VAP-A or VAP-B, whereas it was suppressed by the expression of VAP-C (Fig. 5A). To further confirm the effects of VAPs on the expression of HCV proteins, Huh7OK1 cells transfected with various amounts of the expression plasmids of VAP-A, VAP-B, or VAP-C and infected with the JFH1 virus were examined by immunoblotting (Fig. 5B). Although the expression of VAP-A or VAP-B exhibited no effect on NS5A expression, VAP-C expression clearly decreased the expression of NS5A in a dose-dependent manner. These results clearly indicate that the expression of VAP-C negatively regulates HCV propagation. Overexpression of VAP-C did not affect the endogenous expression of VAP-A or VAP-B (Fig. 5C), suggesting that suppression of HCV propagation by VAP-C is not due to the reduction of VAP-A or VAP-B expression.
FIG. 5.

VAP-C impairs HCV propagation but does not affect endogenous expression of VAP-A or VAP-B. Huh7OK1 cells transfected with 0 to 4 μg of plasmid encoding the FLAG-tagged VAP-A, VAP-B, or VAP-C or empty vector were infected with strain JFH1 at an MOI of 0.05 at 14 h posttransfection and then harvested at 96 h postinfection. (A) The intracellular and extracellular expression levels of viral RNA (top) and core protein (middle) were determined by real-time PCR and ELISA, respectively. Infectious viral titers in the culture supernatants were determined by focus-forming assay (bottom). Data in this panel are shown as the means ± standard deviations. (B) The expression levels of NS5A, β-actin, VAP-A, VAP-B, and VAP-C were determined by immunoblotting using anti-NS5A, anti-β-actin, or anti-FLAG tag antibody. (C) The embryonic kidney cell line (293T), the cured hepatoma cell line (Huh7OK1), and the replicon cell line (Huh 9-13) were transfected with 2 μg of the plasmid encoding FLAG-tagged VAP-C (+) or empty plasmid. In the case of the infected cells, Huh7OK1 cells were infected with strain JFH1 at an MOI of 0.05, reseeded onto the tissue culture plate at 96 h postinfection, and then transfected with 2 μg of the plasmids. These cells were harvested at 36 h posttransfection and examined by immunoblotting using antibodies to VAP-A, VAP-B, FLAG, NS5A, and β-actin. The data in each panel are representative of the results of three independent experiments.
Lack of VAP-C expression in human livers.
VAP-C consists of the first 70 amino acid residues of VAP-B and the subtype-specific 29 amino acid residues derived from frameshift (Fig. 1A). The VAP-C-specific antibody generated by immunization with the peptide corresponding to the residues from 86 to 98 clearly detected VAP-C but neither VAP-A nor VAP-B in cells transfected with expression plasmids encoding FLAG-tagged VAP-A, VAP-B, or VAP-C (Fig. 6A). To determine the distribution of VAPs in human organs, the pool lysates of various organs prepared from several people were examined by immunoblotting (Fig. 6B). Expression of VAP-A was detected clearly in the kidney, lung, prostate, and liver; slightly in the duodenum, uterus, vagina, and bladder; and barely in the small intestine and stomach. VAP-B was detected clearly in the bladder, kidney, and prostate and slightly in the duodenum, small intestine, uterus, vagina, and liver. Expression of VAP-C was detected clearly in the stomach, uterus, kidney, and bladder; slightly in the duodenum, small intestine, and prostate; and barely detected in the vagina, lung, and liver. Several bands smaller than the expected size of VAP-C were observed in the stomach, duodenum, small intestine, uterus, vagina, prostate, and bladder. Because the main target of HCV replication is thought to be the liver, we next examined the expression of VAPs in individual human liver samples. VAP-A and VAP-B were clearly detected in the liver tissues obtained from chronic hepatitis C patients and a healthy donor, but no expression of VAP-C was detected (Fig. 6C). These results suggest that the expression of VAP-C may participate in the determination of tissue tropism of HCV propagation.
FIG. 6.

Distribution of VAPs in human tissues. (A) Anti-VAP-C antibody specifically recognizes VAP-C. Human embryonic kidney 293T cells transfected with expression plasmid encoding FLAG-tagged VAP-A, VAP-B, or VAP-C or empty vector were harvested at 48 h posttransfection and examined by immunoblotting using anti-FLAG tag, anti-VAP-C, and anti-β-actin antibodies. (B) The premade human tissue lysates “Protein medleys” (20 μg each; Clonetech) were examined by immunoblotting using antibodies against VAP-A, VAP-B, VAP-C, or β-actin. (C) Expression of VAP family proteins in human liver tissues. Liver samples obtained from four hepatitis C patients (1 to 4) and one healthy donor (HD) were examined by immunoblotting as described above. The data in each panel are representative of the results of three independent experiments. PC indicates 293T cells transfected with expression plasmid encoding VAP-A, VAP-B, and VAP-C.
Substitution of Ser for Pro56 in VAPs leads to suppression of HCV replication.
A single mutation of Pro56 to Ser (P56S) of VAP-B has been reported to be highly associated with amyotrophic lateral sclerosis (ALS), and the P56S mutation of VAP-B but not of VAP-A has been shown to induce large aggregations of ER in culture cells and to sequester the wild-type protein into ubiquitinated inclusions (29, 37). To examine the effects on the replication of HCV of the P56S mutation in VAPs, FLAG-tagged VAP mutants were expressed in the HCV replicon cells. RNA replication of the subgenomic replicon in Huh 9-13 cells was impaired by the expression of each of the mutant VAPs (Fig. 7A, left). The expression of NS5A in the replicon cells was decreased by the expression of the mutant VAPs in a dose-dependent manner (Fig. 7A, right). Next, to examine the effect of the expression of the P56S VAP mutants on HCV propagation, Huh7OK1 cells expressing the FLAG-tagged VAP mutants were infected with JFH1 virus. The production of intracellular and extracellular viral RNA at 96 h postinfection was decreased by the expression of the P56S mutation in VAPs (Fig. 7B). Although the results of a previous study indicated that the expression of the P56S mutant of VAP-B but not that of VAP-A induced a large aggregation of ER in hamster ovary cell line CHO (37), the P56S mutants of VAP-A and VAP-B but not that of VAP-C exhibited accumulation of membranous aggregates in Huh 9-13 cells (Fig. 7C). These results indicate that the P56S mutation in both VAP-B and VAP-A induces aggregation of ER in human hepatoma cells, which in turn leads to the suppression of HCV propagation.
FIG. 7.

Substitution of Ser for Pro56 in VAPs leads to suppression of HCV replication. (A) Left: Huh 9-13 cells were transfected with 4 μg of the expression plasmids encoding FLAG-tagged P56S VAP mutants or empty vector, and the level of intracellular HCV RNA was determined at 72 h posttransfection by real-time PCR after normalization with GAPDH mRNA. The value for HCV RNA at 0 h posttransfection in the cell line transfected with the empty plasmid is represented as 100%. Data in this panel are shown as the means ± standard deviations. Right: Huh 9-13 cells were transfected with 0 to 4 μg of the FLAG-tagged P56S VAP mutant plasmids or empty vector, and the levels of expression of NS5A, β-actin, and the mutant VAPs were determined by immunoblotting at 72 h posttransfection. The data in each panel are representative of the results of three independent experiments. (B) Huh7OK1 cells transfected with 4 μg of the expression plasmids encoding FLAG-tagged P56S VAP mutants or empty vector were infected with strain JFH1 at an MOI of 0.05 at 14 h posttransfection, and the intracellular (left) and extracellular (right) expression levels of viral RNA were determined by real-time PCR after normalization with GAPDH mRNA at 96 h postinfection. Data in this panel are shown as the means ± standard deviations. (C) Levels of expression of wild-type VAPs, P56S mutant VAPs, and NS5A in Huh 9-13 cells at 72 h after transfection with the expression plasmids encoding FLAG-tagged VAPs or P56S VAP mutants were determined by immunofluorescent assay. The data in each panel are representative of the results of three independent experiments.
DISCUSSION
The replication of HCV has been shown to require several host proteins, including VAP-A/VAP-B (6, 9, 44), FBL2 (46), FKBP8 (34), hB-ind1 (40), Hsp90 (28, 34, 45), and cyclophilins (15, 48). VAP-A has been detected in a detergent-resistant membrane fraction that was shown to be capable of replicating HCV RNA in vitro, and the interaction of VAP-A with NS5A is required for the efficient replication of HCV genomic RNA (2, 7) and is modulated by the phosphorylation of NS5A (4, 6). VAP-B also participates in HCV replication through the formation of homo- and/or heterodimers with VAP-A (9). VAP-A and VAP-B form hetero- and homodimers through their TM regions and interact with NS5A and NS5B through the coiled-coil domain and MSP domain, respectively (9, 44). VAP-C is a splicing variant of VAP-B, consisting of the N-terminal half of VAP-B and the subtype-specific amino acid residues generated by the frameshift. However, the biological significance of VAP-C in the life cycle of HCV has not been determined. In this study, we have demonstrated that VAP-C is capable of binding to HCV NS5B but not to NS5A, VAP-A, and VAP-B due to the lack of the coiled-coil and TM regions. The expression of VAP-C inhibited the interaction of VAP-A and VAP-B with NS5B, impaired the RNA replication and particle formation of HCV, and was barely detected in human liver cells. These results suggest that VAP-C acts as a negative regulator for HCV propagation and is partly involved in the determination of the tissue specificity of HCV replication.
Overexpression of VAP-A but not of VAP-B inhibited the incorporation of the vesicular stomatitis virus (VSV) envelope glycoprotein G (VSV-G) into ER vesicles in CHO cells, resulting in impairment of membrane protein transport from the ER to the Golgi apparatus (37). VAP-B was shown to be involved in the unfolded protein response, which is an ER reaction to suppress the accumulation of misfolded proteins, and the expression of the P56S VAP-B mutant was suggested to nullify the unfolded protein response induced by VAP-B, to produce a large aggregation of ER, and to be involved in the development of ALS (17, 37). These data suggest that VAP-A and VAP-B possess different physiological functions; however, the contributions of the proteins to the life cycle of HCV have not been characterized. The expression of VAP-B but not of VAP-A resulted in an enhancement of the replication of the subgenomic HCV RNA of the genotype 1b strain Con1, whereas the expression of either VAP-A or VAP-B clearly enhanced viral RNA replication in cells infected with the genotype 2a strain JFH1 virus, suggesting that the contributions of VAP-A and VAP-B to viral RNA replication might differ among the genotypes of HCV. The expression of VAP-B or VAP-A enhanced RNA replication in the HCV replicon cells and the secretion of viral RNA, core protein, and infectious particles into the culture supernatants of Huh7OK1 cells infected with JFH1 virus, whereas the expression of these proteins had no effect on the expression of NS5A or on IRES-dependent translation. Thus, further studies will be needed to clarify the molecular mechanisms underlying the posttranslational enhancement of HCV production by the expression of VAP-A and VAP-B. In contrast to the expression of VAP-A and VAP-B, the expression of VAP-C clearly suppressed the RNA replication of both the genotype 1b RNA replicon cells and the genotype 2a strain JFH1 virus, by which both the expression of the viral proteins and the viral particle production were drastically impaired. Furthermore, the expression of the P56S mutants of VAP-A and VAP-B reduced RNA replication in HCV replicon cells and propagation of the JFH1 virus, probably due to the induction of aggregation of the ER. The reason why ER aggregation was induced by the expression of the P56S VAP-A mutant in Huh7 cells but not in CHO cells (17, 37) is not known at the moment.
The phosphorylation state of NS5A was suggested to control the interaction between VAP-A and NS5A and the replication efficiency of HCV RNA (6). Introduction of the adaptive mutations originally identified in the genotype 1b strain Con1 into NS5A of genotype 1a suppressed the hyperphosphorylation of NS5A, potentiated interaction with VAP-A, and enhanced the RNA replication (6). However, we have previously shown that NS5A of genotype 1a could bind to VAP-A and VAP-B at a level similar to that of genotype 1b despite the adaptive mutations (9). In this study, overexpression of each of the VAP proteins exhibited no effect on the mobility of NS5A in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Fig. 3 and 5), suggesting that there is no correlation between the VAP-dependent regulation of HCV propagation and the phosphorylation state of NS5A.
FKBP8 exhibits peptidyl prolyl cis-trans isomerase activity and interacts with NS5A and Hsp90 through the tetratricopeptide repeat (TPR) domain, and these interactions are suggested to be involved in the correct folding of the HCV replication complex (34). Treatment of cells with inhibitors of the ATPase activity of Hsp90, such as geldanamycin and its derivatives, impairs the RNA replication and particle production of HCV (28, 34, 45). The MSP domain of VAP-A was shown to interact with the TPR1 protein, which has a TPR domain and forms the chaperone complex with Hsp90 (22). Knockdown of the TPR1 protein or treatment with Hsp90 inhibitors in mammalian cells has been shown to inhibit the transport of VSV-G, leading to accumulation of the glycoprotein in the Golgi apparatus (22). The VAP-A- or VAP-B-induced enhancement of virus production might be attributable to the recruitment of Hsp90 into the replication complex through the interaction with the MSP domain.
VAP-A is well known to interact through the MSP domain with a number of mammalian and yeast proteins sharing the FFAT motif, including OSBPs, ORPs (20), and CERT (10, 19), and to be involved in the regulation of biosynthesis or trafficking of sterols and lipids. HCV replication and infection have been shown to be regulated by lipid components and to be capable of being inhibited by treatment with several inhibitors targeting lipid biosynthesis (14, 18). The intracellular membranous web structure observed in HCV replicon cells was shown to be resistant to detergent treatment, suggesting that the lipid raft-like structure abundant in cholesterol and sphingolipid is generated by the replication of HCV RNA (2, 24). Therefore, it might be feasible to speculate that VAP-A and VAP-B are involved in the construction of the HCV replication complex consisting of viral proteins and host cellular lipid components and that VAP-C interrupts the VAP-A and VAP-B functions and negatively regulates HCV propagation. Although the molecular mechanisms and the biological significance remain to be clarified, the MSP domain of VAP proteins was processed in human leukocytes and secreted into human serum (43). Further studies are needed to clarify the biogenesis and biological functions of the truncated VAP proteins in the replication of HCV.
In summary, we have shown that VAP-C is capable of suppressing the RNA replication and particle production of HCV by inhibiting the binding of VAP-A and VAP-B to NS5B through the N-terminal half of its MSP domain. The clear suppression of HCV propagation by the expression of VAP-C further suggests the possibility of developing a novel therapeutic measure to eliminate HCV by the exogenous expression of VAP-C in the hepatocytes of chronic hepatitis C patients.
Acknowledgments
We thank H. Murase for her secretarial work. We also thank R. Bartenschlager and T. Wakita for providing cell lines and plasmids.
This work was supported in part by grants-in-aid from the Ministry of Health, Labor, and Welfare; the Ministry of Education, Culture, Sports, Science, and Technology; the Global Center of Excellence Program; and the Foundation for Biomedical Research and Innovation.
Footnotes
Published ahead of print on 10 June 2009.
REFERENCES
- 1.Abe, T., Y. Kaname, I. Hamamoto, Y. Tsuda, X. Wen, S. Taguwa, K. Moriishi, O. Takeuchi, T. Kawai, T. Kanto, N. Hayashi, S. Akira, and Y. Matsuura. 2007. Hepatitis C Virus nonstructural protein 5A modulates Toll-like receptor-MyD88-dependent signaling pathway in the macrophage cell lines. J. Virol. 818953-8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aizaki, H., K. J. Lee, V. M. Sung, H. Ishiko, and M. M. Lai. 2004. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 324450-461. [DOI] [PubMed] [Google Scholar]
- 3.Behrens, S. E., L. Tomei, and R. De Francesco. 1996. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1512-22. [PMC free article] [PubMed] [Google Scholar]
- 4.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 2901972-1974. [DOI] [PubMed] [Google Scholar]
- 5.Egger, D., B. Wolk, R. Gosert, L. Bianchi, H. E. Blum, D. Moradpour, and K. Bienz. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 765974-5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans, M. J., C. M. Rice, and S. P. Goff. 2004. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl. Acad. Sci. USA 10113038-13043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao, L., H. Aizaki, J.-W. He, and M. M. C. Lai. 2004. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 783480-3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grakoui, A., D. W. McCourt, C. Wychowski, S. M. Feinstone, and C. M. Rice. 1993. Characterization of the hepatitis C virus-encoded serine proteinase: determination of proteinase-dependent polyprotein cleavage sites. J. Virol. 672832-2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamamoto, I., Y. Nishimura, T. Okamoto, H. Aizaki, M. Liu, Y. Mori, T. Abe, T. Suzuki, M. M. Lai, T. Miyamura, K. Moriishi, and Y. Matsuura. 2005. Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J. Virol. 7913473-13482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanada, K., K. Kumagai, S. Yasuda, Y. Miura, M. Kawano, M. Fukasawa, and M. Nishijima. 2003. Molecular machinery for non-vesicular trafficking of ceramide. Nature 426803-809. [DOI] [PubMed] [Google Scholar]
- 11.Ho, S. N., H. D. Hunt, R. M. Horton, J. K. Pullen, and L. R. Pease. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 7751-59. [DOI] [PubMed] [Google Scholar]
- 12.Hoofnagle, J. H. 2002. Course and outcome of hepatitis C. Hepatology 36S21-S29. [DOI] [PubMed] [Google Scholar]
- 13.Huang, D. C., S. Cory, and A. Strasser. 1997. Bcl-2, Bcl-XL and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene 14405-414. [DOI] [PubMed] [Google Scholar]
- 14.Ikeda, M., K. Abe, M. Yamada, H. Dansako, K. Naka, and N. Kato. 2006. Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology 44117-125. [DOI] [PubMed] [Google Scholar]
- 15.Inoue, K., T. Umehara, U. T. Ruegg, F. Yasui, T. Watanabe, H. Yasuda, J. M. Dumont, P. Scalfaro, M. Yoshiba, and M. Kohara. 2007. Evaluation of a cyclophilin inhibitor in hepatitis C virus-infected chimeric mice in vivo. Hepatology 45921-928. [DOI] [PubMed] [Google Scholar]
- 16.Kaiser, S. E., J. H. Brickner, A. R. Reilein, T. D. Fenn, P. Walter, and A. T. Brunger. 2005. Structural basis of FFAT motif-mediated ER targeting. Structure 131035-1045. [DOI] [PubMed] [Google Scholar]
- 17.Kanekura, K., I. Nishimoto, S. Aiso, and M. Matsuoka. 2006. Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8). J. Biol. Chem. 28130223-30233. [DOI] [PubMed] [Google Scholar]
- 18.Kapadia, S. B., and F. V. Chisari. 2005. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. USA 1022561-2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawano, M., K. Kumagai, M. Nishijima, and K. Hanada. 2006. Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J. Biol. Chem. 28130279-30288. [DOI] [PubMed] [Google Scholar]
- 20.Loewen, C. J., A. Roy, and T. P. Levine. 2003. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 222025-2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lohmann, V., F. Korner, J. Koch, U. Herian, L. Theilmann, and R. Bartenschlager. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285110-113. [DOI] [PubMed] [Google Scholar]
- 22.Lotz, G. P., A. Brychzy, S. Heinz, and W. M. Obermann. 2008. A novel HSP90 chaperone complex regulates intracellular vesicle transport. J. Cell Sci. 121717-723. [DOI] [PubMed] [Google Scholar]
- 23.McLauchlan, J., M. K. Lemberg, G. Hope, and B. Martoglio. 2002. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 213980-3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyanari, Y., M. Hijikata, M. Yamaji, M. Hosaka, H. Takahashi, and K. Shimotohno. 2003. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 27850301-50308. [DOI] [PubMed] [Google Scholar]
- 25.Moriishi, K., and Y. Matsuura. 2007. Host factors involved in the replication of hepatitis C virus. Rev. Med. Virol. 17343-354. [DOI] [PubMed] [Google Scholar]
- 26.Moriishi, K., and Y. Matsuura. 2003. Mechanisms of hepatitis C virus infection. Antivir. Chem. Chemother. 14285-297. [DOI] [PubMed] [Google Scholar]
- 27.Moriishi, K., T. Okabayashi, K. Nakai, K. Moriya, K. Koike, S. Murata, T. Chiba, K. Tanaka, R. Suzuki, T. Suzuki, T. Miyamura, and Y. Matsuura. 2003. Proteasome activator PA28gamma-dependent nuclear retention and degradation of hepatitis C virus core protein. J. Virol. 7710237-10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakagawa, S., T. Umehara, C. Matsuda, S. Kuge, M. Sudoh, and M. Kohara. 2007. Hsp90 inhibitors suppress HCV replication in replicon cells and humanized liver mice. Biochem. Biophys. Res. Commun. 353882-888. [DOI] [PubMed] [Google Scholar]
- 29.Nishimura, A. L., M. Mitne-Neto, H. C. Silva, A. Richieri-Costa, S. Middleton, D. Cascio, F. Kok, J. R. Oliveira, T. Gillingwater, J. Webb, P. Skehel, and M. Zatz. 2004. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75822-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishimura, Y., M. Hayashi, H. Inada, and T. Tanaka. 1999. Molecular cloning and characterization of mammalian homologues of vesicle-associated membrane protein-associated (VAMP-associated) proteins. Biochem. Biophys. Res. Commun. 25421-26. [DOI] [PubMed] [Google Scholar]
- 31.Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108193-199. [DOI] [PubMed] [Google Scholar]
- 32.Okamoto, K., Y. Mori, Y. Komoda, T. Okamoto, M. Okochi, M. Takeda, T. Suzuki, K. Moriishi, and Y. Matsuura. 2008. Intramembrane processing by signal peptide peptidase regulates the membrane localization of hepatitis C virus core protein and viral propagation. J. Virol. 828349-8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okamoto, K., K. Moriishi, T. Miyamura, and Y. Matsuura. 2004. Intramembrane proteolysis and endoplasmic reticulum retention of hepatitis C virus core protein. J. Virol. 786370-6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okamoto, T., Y. Nishimura, T. Ichimura, K. Suzuki, T. Miyamura, T. Suzuki, K. Moriishi, and Y. Matsuura. 2006. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 255015-5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okamoto, T., H. Omori, Y. Kaname, T. Abe, Y. Nishimura, T. Suzuki, T. Miyamura, T. Yoshimori, K. Moriishi, and Y. Matsuura. 2008. A single-amino-acid mutation in hepatitis C virus NS5A disrupting FKBP8 interaction impairs viral replication. J. Virol. 823480-3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pennetta, G., P. R. Hiesinger, R. Fabian-Fine, I. A. Meinertzhagen, and H. J. Bellen. 2002. Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron 35291-306. [DOI] [PubMed] [Google Scholar]
- 37.Prosser, D. C., D. Tran, P. Y. Gougeon, C. Verly, and J. K. Ngsee. 2008. FFAT rescues VAPA-mediated inhibition of ER-to-Golgi transport and VAPB-mediated ER aggregation. J. Cell Sci. 1213052-3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skehel, P. A., R. Fabian-Fine, and E. R. Kandel. 2000. Mouse VAP33 is associated with the endoplasmic reticulum and microtubules. Proc. Natl. Acad. Sci. USA 971101-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skehel, P. A., K. C. Martin, E. R. Kandel, and D. Bartsch. 1995. A VAMP-binding protein from Aplysia required for neurotransmitter release. Science 2691580-1583. [DOI] [PubMed] [Google Scholar]
- 40.Taguwa, S., T. Okamoto, T. Abe, Y. Mori, T. Suzuki, K. Moriishi, and Y. Matsuura. 2008. Human butyrate-induced transcript 1 interacts with hepatitis C virus NS5A and regulates viral replication. J. Virol. 822631-2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tellinghuisen, T. L., J. Marcotrigiano, and C. M. Rice. 2005. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435374-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomei, L., C. Failla, E. Santolini, R. De Francesco, and N. La Monica. 1993. NS3 is a serine protease required for processing of hepatitis C virus polyprotein. J. Virol. 674017-4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsuda, H., S. M. Han, Y. Yang, C. Tong, Y. Q. Lin, K. Mohan, C. Haueter, A. Zoghbi, Y. Harati, J. Kwan, M. A. Miller, and H. J. Bellen. 2008. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell 133963-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tu, H., L. Gao, S. T. Shi, D. R. Taylor, T. Yang, A. K. Mircheff, Y. Wen, A. E. Gorbalenya, S. B. Hwang, and M. M. Lai. 1999. Hepatitis C virus RNA polymerase and NS5A complex with a SNARE-like protein. Virology 26330-41. [DOI] [PubMed] [Google Scholar]
- 45.Ujino, S., S. Yamaguchi, K. Shimotohno, and H. Takaku. 2009. Heat-shock protein 90 is essential for stabilization of the hepatitis C virus non-structural protein NS3. J. Biol. Chem. 2846841-6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang, C., M. Gale, Jr., B. C. Keller, H. Huang, M. S. Brown, J. L. Goldstein, and J. Ye. 2005. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol. Cell 18425-434. [DOI] [PubMed] [Google Scholar]
- 47.Wasley, A., and M. J. Alter. 2000. Epidemiology of hepatitis C: geographic differences and temporal trends. Semin. Liver Dis. 201-16. [DOI] [PubMed] [Google Scholar]
- 48.Watashi, K., N. Ishii, M. Hijikata, D. Inoue, T. Murata, Y. Miyanari, and K. Shimotohno. 2005. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell 19111-122. [DOI] [PubMed] [Google Scholar]
- 49.Weir, M. L., A. Klip, and W. S. Trimble. 1998. Identification of a human homologue of the vesicle-associated membrane protein (VAMP)-associated protein of 33 kDa (VAP-33): a broadly expressed protein that binds to VAMP. Biochem. J. 333247-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weir, M. L., H. Xie, A. Klip, and W. S. Trimble. 2001. VAP-A binds promiscuously to both v- and tSNAREs. Biochem. Biophys. Res. Commun. 286616-621. [DOI] [PubMed] [Google Scholar]
- 51.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 1029294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]