

Abstract
Transneuronal spread of pseudorabies virus (PRV) is a multistep process that requires several virally encoded proteins. Previous studies have shown that PRV glycoprotein B (gB), a component of the viral fusion machinery, is required for the transmission of infection to postsynaptic, second-order neurons. We sought to identify the gB-mediated step in viral transmission. We determined that gB is not required for the sorting of virions into axons of infected neurons, anterograde transport, or the release of virions from the axon. trans or cis expression of gB on the cell surface was not sufficient for transneuronal spread of the virus; instead, efficient incorporation of gB into virions was required. Additionally, neuron-to-cell spread of PRV most likely does not proceed through syncytial connections. We conclude that, upon gB-independent release of virions at the site of neuron-cell contacts, the virion-incorporated gB/gH/gL fusion complex mediates entry into the axonally contacted cell by fusion of the closely apposed membranes.
Alphaherpesviruses, which constitute a subfamily of the family Herpesviridae, include the human pathogens herpes simplex virus (HSV) and varicella-zoster virus and the swine pathogen pseudorabies virus (PRV). These closely related pantropic, neuroinvasive viruses establish latency in the peripheral nervous systems of their natural hosts. During the normal course of infection, periodic viral reactivation leads to recurrent epithelial lesions (38). Although rare in the natural host, transneuronal spread of the virus from the peripheral to the central nervous system (CNS) results in death or debilitating disease, such as encephalitis or keratitis (50). Nonnatural hosts infected with PRV almost invariably experience viral spread to the CNS and succumb to infection (36).
Transneuronal spread of alphaherpesviruses is an incompletely understood multistep process that requires the concerted action of viral and cellular proteins. Following replication in the soma of an infected neuron, viral progeny may spread in the retrograde direction to the presynaptic cell or anterogradely to the postsynaptic cell. During anterograde spread of PRV, virus particles are sorted from the neuronal soma into the cognate axon. Upon entering the axonal compartment, virions are transported in a microtubule-dependent manner toward the synaptically connected cell (41). Recent in vitro evidence suggests that boutons en passant and axon termini serve as sites for PRV spread from the axon (13). Additionally, in vivo experiments demonstrate that the transneuronal spread of alphaherpesviruses is remarkably specific, occurring only between synaptically connected cells (15). This property has made alphaherpesviruses invaluable as neural circuit tracers in studies that aim to map the synaptic architecture of the CNS (14). However, the mechanisms that confer such specificity on the spread of infection are not well understood.
The study of mechanisms underlying PRV trafficking revealed that the virally encoded membrane proteins Us9, glycoprotein E (gE), and gI are required for the efficient sorting of virions from an infected neuronal cell body into its cognate axon (6, 26, 29, 44, 49). Therefore, in the absence of any of these proteins, infection cannot be transmitted efficiently from a presynaptic to a postsynaptic cell (3, 23). Another viral membrane protein required for the transneuronal spread of PRV is gB (2, 21). Along with gH and gL, this 913-amino-acid type I viral membrane protein is part of the viral fusion machinery, and it is essential for infection by free virions and for cell-to-cell spread in epithelial cultures (25, 35, 37). gB is the most highly conserved glycoprotein in the family Herpesviridae. X-ray crystallography of the HSV type 1 (HSV-1) gB ectodomain revealed a trimeric structure with a high degree of homology to fusion protein G of vesicular stomatitis virus (22). By homology to vesicular stomatitis virus fusion protein G, the ectodomain of gB is predicted to contain fusion loops; indeed, mutation of these regions in HSV-1 gB inhibits its fusion function (20). Mutagenesis of the gB cytoplasmic tail in HSV-1 and PRV revealed its role in the regulation of the fusion function, virion incorporation of gB, and interactions with cellular adaptor proteins (16, 32, 34, 48; summarized in reference 39). Tyrosine motif-mediated interaction of PRV gB with adaptor protein 2 leads to its clathrin-dependent internalization (48). In polarized epithelial cells, gB is targeted to the basolateral surface, presumably via interactions of its cytoplasmic tail with adaptor protein 1B. The basolateral sorting of gB is hypothesized to enhance the efficiency of direct cell-to-cell spread of the virus (16).
While its requirement for transneuronal spread is known, the function that gB performs in this process has not been identified. The block in the transmission of gB-null PRV infection from a neuron to an axonally contacted cell may occur during viral trafficking in the neuron, egress from the axon, or entry into the postsynaptic cell. We investigated whether gB participates in the axonal targeting of newly synthesized virions. Our imaging data revealed that gB is not required for axonal sorting of PRV, placing gB function downstream of Us9, gE, and gI. Further experiments showed that gB is not required for virion egress from the infected neuron and that neuron-to-cell spread of PRV does not proceed through syncytia. Importantly, incorporation of gB into virions was required for efficient spread of infection. We conclude that PRV virions are released from axons in a gB-independent manner and enter the postsynaptic cell at synaptic contacts by gB-mediated fusion of the closely apposed viral and cellular membranes.
MATERIALS AND METHODS
Cell lines and virus strains.
The porcine kidney epithelial cell line PK15 was purchased from the American Type Culture Collection and maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin. PK15 cells were infected in DMEM supplemented with 2% FBS, penicillin, and streptomycin. The gB-null viral strains were propagated, and their titers were determined in PK15 cells stably transfected with LP64e3, a plasmid expressing PRV gB under the control of the cytomegalovirus (CMV) promoter (Lisa Pomeranz). PRV Becker is a wild-type laboratory strain. PRV GS443 expresses VP26-green fluorescent protein (GFP) in a PRV Becker background (42). PRV HF22 is a gB-null strain that carries a kanamycin resistance cassette in the gB locus in a PRV Becker background (16). PRV 232 is a gB-null strain expressing VP26-GFP in a PRV Becker background; it was generated for this study by crossing PRV HF22 and PRV GS443 in gB-complementing cells. The resulting viral progeny were plated on gB-complementing cells, and green plaques were picked. The viral progeny were screened by plating each plaque on gB-complementing and noncomplementing cells. Recombinants that grew only on gB-complementing cells were picked for three subsequent rounds of purification. PRV 233 is a gB-null strain expressing diffusible GFP from the gG locus under the control of a CMV promoter. It was constructed by crossing PRV HF22 and PRV 151 in gB-complementing cells and screening for the desired recombinants as described above. Strain PRV gB-007 has been described previously (32).
Neuronal cultures.
Superior cervical ganglia (SCG) were isolated from embryonic-day-16 Sprague-Dawley rats (Hilltop Labs, Inc., Scottsdale, PA) and were cultured in compartmentalized chambers as described previously (5, 7). Neuronal medium consisted of neurobasal medium (Gibco) supplemented with 100 ng/ml nerve growth factor 2.5S (Invitrogen), 2% B27 (Gibco), 2 mM glutamine (Invitrogen), penicillin, and streptomycin; this medium was changed every 3 days. All animal work was done in accordance with the Institutional Animal Care and Use Committee of the Princeton University Research Board under protocol 1691.
Viral infection of compartmented neuronal cultures.
To determine the efficiency of neuron-to-cell spread of infection, gB-expressing PK15 cells were plated on top of neurite (N) compartment axons of 2-week-old compartmented neuronal cultures. Twenty-four hours postplating, the soma (S) compartment medium was removed, and a viral inoculum containing 105 PFU was adsorbed to the cell bodies for 1 h. Following adsorption, the conditioned medium was returned to the S compartment. The contents of the N compartment were collected by scraping the dish with a pipette tip. To determine whether virions undergo release from axons in the absence of epithelial cells, a viral inoculum was added to the S compartment, and the extracellular medium was collected from the N compartment 24 h postinfection. The N compartment was then treated for 1 min with a citrate solution (40 mM citrate, 10 mM KCl, 0.135 M NaCl [pH 3]), to inactivate extracellular virions, or with nonsupplemented DMEM. The solutions were washed away with three DMEM rinses, and the axons were scraped into fresh medium. All titers were determined on gB-expressing PK15 cells.
Antibodies.
The anti-PRV antibody Rb 134 was used at a 1:5,000 dilution (4). Monoclonal antibody M2 against PRV gB was used at a 1:800 dilution (19). The pooled anti-gE monoclonal antibodies M133, M138, and M156 were used at a 1:200 dilution (43). The secondary Alexa fluorophores (Molecular Probes) were used at a 1:400 dilution.
Immunofluorescence assays.
Dissociated SCG neurons were cultured and infected in compartmentalized chambers on Aclar (EM Sciences) strips, and immunofluorescence assays were performed as described previously (10). Briefly, each chamber compartment was rinsed once with phosphate-buffered saline (PBS) and fixed with 3.2% paraformaldehyde for 10 min. Chambers were removed following three rinses with PBS containing 3% bovine serum albumin (BSA), and the Aclar strips were trimmed to the size of a coverslip. The samples were permeabilized with PBS containing 3% BSA and 0.5% saponin (PBS-BSA-SAP) and were incubated for 1 h with primary antibodies diluted in PBS-BSA-SAP. Following three PBS-BSA-SAP rinses, secondary antibodies diluted in PBS-BSA-SAP were added for 1 h. Samples were then rinsed three times with PBS-BSA-SAP and once with distilled water. Then they were mounted, cell side up, on glass slides with Aqua-Poly/Mount; another drop of Aqua-Poly/Mount was placed over the samples, and a glass coverslip was placed over them. For cell surface staining, saponin was omitted from all solutions.
Live-cell imaging.
Dissociated SCG neurons were plated onto glass-bottom MatTek dishes and infected at a high multiplicity of infection for 16 h. Live-cell imaging was performed as described previously (10). Imaging was performed on a Perkin-Elmer RS3 spinning-disk confocal microscope with a 60× oil objective (numerical aperture, 1.3).
PEG fusion assay.
Polyethylene glycol (PEG)-induced fusion was performed essentially as described previously (37). Briefly, PEG solutions of 1.3 g/ml, 0.4 g/ml, and 0.2 g/ml were made by mixing melted PEG 8000 with warmed nonsupplemented DMEM. PEG was melted by autoclaving and was cooled to 60°C. DMEM was added to the PEG, and the solutions were immediately shaken. All solutions were kept at 37°C until use. Following a 1-h adsorption of viral samples to gB-expressing PK15 cells, fusion of virions with the cells was induced by adding 1.3 g/ml PEG solution over the cells for 15 s. The solution was then decanted and washed away with 0.4 g/ml PEG, followed by 0.2 g/ml PEG. The cells were rinsed three times with DMEM supplemented with 10% FBS, penicillin, and streptomycin and were incubated in the medium for 2 h. A semisolid overlay of medium containing 1% methylcellulose was added to the cells, which were then incubated at 37°C under 5% CO2. PEG was omitted from solutions used to treat control cells. PRV 233 plaques were captured using an inverted epifluorescence microscope (Nikon Eclipse TE300).
RESULTS
gB is not required for axonal sorting of PRV.
gB is required for the transneuronal spread of PRV. This requirement has been demonstrated previously in vivo and in vitro, but the gB-mediated step in the process has not been identified (2, 17, 21, 27). The viral membrane proteins Us9, gE, and gI are also required for the transneuronal spread of PRV and have been shown to mediate the axonal sorting of newly synthesized virions. Lyman and colleagues recently demonstrated that the viral protein Us9 strongly partitions with lipid rafts in infected neuron-like PC12 cells. Importantly, the association between Us9 and lipid rafts is required for the axonal sorting of viral particles (28). gB also strongly partitions with lipid rafts in PC12 cells and has a large cytoplasmic tail that interacts with cellular adaptor proteins (16, 28, 48). We therefore hypothesized that gB may participate, in concert with Us9, gE, and gI, in the sorting of virions into axons, possibly by recruiting appropriate cellular effectors via its cytoplasmic tail domain.
We used immunofluorescence assays to determine whether PRV proteins localize to axons of infected cells in the absence of gB. To avoid detection of the input inoculum, neurons were infected in compartmentalized cultures where the neuronal soma and axons were maintained in fluidically isolated compartments (Fig. 1). Dissociated SCG neurons were plated in the S compartment of a modified Campenot trichamber system. By 2 weeks postplating, axons emanating from the soma penetrated underneath the water-tight silicone grease barriers of the middle (M) compartment and reached the N compartment. An inoculum containing 105 PFU of either PRV Becker (wild-type strain) or the gB-null PRV strain HF22 was applied to the S compartment. Because gB is required for the entry of PRV into cells, the PRV HF22 stock was grown on gB-expressing PK15 cells. The progeny virions are genotypically gB null but phenotypically complemented and able to enter neurons; however, any virions newly replicated in the neuron do not contain gB. The compartmentalized cultures were processed for immunofluorescence against PRV at 24 h postinfection. Anti-PRV staining showed the presence of PRV structural proteins at wild-type levels in N-compartment axons of infected neurons (Fig. 1). Next, we performed live-cell imaging of neurons infected with PRV GS443 (expressing GFP-tagged capsid in a PRV Becker background) or PRV 232 (expressing GFP-tagged capsid in a PRV HF22 background). We observed green puncta moving from the cell bodies into the cognate axons and undergoing subsequent anterograde transport in distal axon segments in samples infected with PRV GS443 (data not shown) or PRV 232 (Fig. 2; see also movie S1 in the supplemental material).
FIG. 1.

gB is not required for axonal sorting of PRV. A diagram of the modified Campenot trichamber system is shown above the figure panels. Dissociated SCG neurons were plated in the S compartment. By 2 weeks postplating, axons penetrate the middle barriers and transit the middle compartment to reach the N compartment. S compartments of neuronal cultures were infected with PRV Becker (A and B) or PRV HF22 (C and D) and processed for immunofluorescence against PRV structural proteins at 24 h postinfection. (E) Mock infection; (F) the accompanying transmitted image. Neuronal somas are shown in panels A, C, and E; N-compartment axons are shown in panels B and D.
FIG. 2.

gB is not required for the axonal sorting of PRV. (A) Dissociated SCG neurons were cultured on glass-bottom MatTek dishes and were infected for 16 h with PRV 232. Color-coded arrows in a time lapse sequence track the movement of individual capsid puncta. (B) PRV 233 was applied to the S compartment; the contents of the N compartment were harvested, and their titers were determined on gB-expressing PK15 cells by inducing fusion using PEG. Shown are plaques produced by PRV 233. Scale bar, 50 μm. (C) PRV 233 titers from N-compartment axons at 24 h postinfection (n = 5). The filled symbol represents the mean.
While our imaging studies imply that the gB-null PRV virions enter axons, we wanted to ascertain whether the fluorescent signal in axons represents fully assembled virions that are defective in cell penetration but are otherwise infectious. We infected the S compartment with PRV 233 (a gB-null PRV strain that expresses diffusible GFP) and collected the axons from the N compartment at 24 h postinfection. The titers of the samples were determined on gB-expressing PK15 cells by inducing fusion between the virions and the cells with PEG. GFP-positive plaques developed on the PK15 monolayer (Fig. 2B). We counted an average of 165 PFU per N compartment (Fig. 2C); without PEG treatment, no plaques formed (data not shown). We conclude that fully assembled PRV virions are sorted to axons in a gB-independent manner.
cis expression of gB is required for neuron-to-cell spread of PRV.
Recent work with human CMV fusion proteins gB, gH, and gL reveals that cells expressing the gH-gL complex undergo fusion with cells expressing gB, indicating that gB complementation can be accomplished via trans expression (47). Therefore, we determined whether gB-null PRV can be transmitted from an infected neuron to an axonally contacted PK15 cell that constitutively expresses PRV gB. To this end, we performed infections of compartmentalized neuronal cultures where gB-expressing PK15 cells were plated onto axons in the N compartment (Fig. 3A). The gB-complementing cells express gB on the cell surface, as determined by immunofluorescence (data not shown). Following replication in the neuronal cell bodies, virions undergo intra-axonal anterograde-directed transport and are transmitted to epithelial cells in the N compartment, which replicate and amplify any transmitted particles. The efficiency of neuron-to-cell viral transmission was determined by harvesting the contents of the N compartment and determining their titers. All titers were determined on gB-expressing PK15 cells. The contents of the S compartment were collected at 24 h postinfection and reflect the input inoculum as well as newly replicated virus. While the initial inoculum of either strain contained 105 PFU, the newly replicated PRV HF22 virions do not contain gB and therefore are not detectable in titering assays. As a result, the S-compartment titers of PRV Becker are 1 log unit higher than those of PRV HF22 (Fig. 3B). PRV Becker N-compartment titers reached 107 PFU by 24 h after infection of somata. However, the gB-null PRV strain HF22 did not produce detectable titers in the N compartment at any time point examined (Fig. 3B and C). The N-compartment titers underscore the gB requirement for neuron-to-cell spread of infection and demonstrate that gB-null PRV cannot be transmitted from a noncomplementing to a gB-complementing cell. We conclude that the function of PRV gB in anterograde spread cannot be complemented via trans expression of the protein. These results are consistent with prior findings that show the requirement for cis expression of transfected PRV or HSV viral fusion proteins for the fusion of epithelial cells in the absence of infection (11, 25).
FIG. 3.

cis expression of gB is required for neuron-to-cell spread of PRV. (A) To study the efficiency of neuron-to-cell viral spread, gB-expressing epithelial cells were plated in the N compartment of the trichamber system, and the inoculum was added to the S compartment. (B) Titers of PRV Becker and PRV HF22 24 h after infection of the S compartment (n = 5). (C) Time course of anterograde infection by PRV HF22 (n = 5). Filled symbols represent means for each data set.
Virions egress along the axonal membrane in a gB-independent fashion and remain cell associated.
We hypothesized that virions may undergo axonal egress in a gB-independent manner and require gB for subsequent entry into the postsynaptic cell. Alternatively, gB may be required for the egress of axonally sorted virions from the axon. Therefore, we sought to determine whether virions undergo axonal egress in the absence of postsynaptic cells. Previous in vitro work demonstrated that no infectious virions are released from axons into the extracellular medium of the N compartment (5). In these experiments, a viral inoculum was applied to the S compartment, and no cells were plated onto N-compartment axons. Extracellular medium from the N compartment was harvested, and titers were determined, revealing an absence of infectious virions at 24 and 36 h postinfection. These data suggest that PRV virions do not undergo axonal egress in the absence of postsynaptic cells.
Another possibility is that virions do undergo release but remain attached to the surface of the axonal membrane and are therefore undetectable in the extracellular medium. To distinguish between these possibilities, we infected the S compartment, and no epithelial cells were plated onto N-compartment axons. We harvested the extracellular medium from the N compartment 24 h postinfection. Next, we treated the N compartment with nonsupplemented DMEM (negative control) or a low-pH citrate solution to inactivate any virions bound to the axonal surface. Virions located within the axons would not be susceptible to inactivation by the citrate wash. After several rinses of the N compartment to remove the solutions, we harvested the axons in fresh medium. While we detected an average of 2 infectious units per chamber in the extracellular medium, the harvested axons yielded almost 100-fold more (an average of 1.3 × 102 PFU). The titers decreased 98% when the axons were treated with the citrate solution prior to harvesting (Fig. 4). We conclude that virions egress from the axon shaft in the absence of any postsynaptic cell but remain tightly associated with the axonal surface.
FIG. 4.
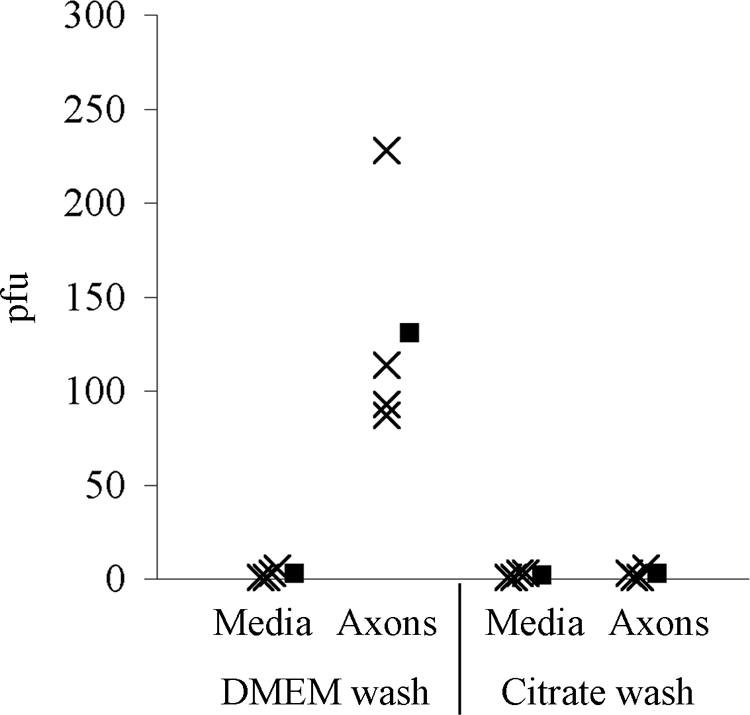
Virions undergo axonal egress and remain cell associated. PRV Becker was used to infect the S compartment of the trichamber system. No epithelial cells were plated in the N compartment. Twenty-four hours postinfection, the extracellular medium from the N compartment was harvested, and titers were determined (media). The N-compartment axons were treated with DMEM or a low-pH citrate solution, rinsed, and harvested, and titers were determined (axons) (n = 4). Filled symbols represent means for each data set.
Strains lacking gB cannot be subjected to this assay, because the virions are not infectious. Therefore, we attempted to detect cell surface-associated virions by immunofluorescence. We hypothesized that such particles would exhibit punctate staining in nonpermeabilized neurons and that the signal would be dependent on Us9-mediated axonal sorting of virions and virion components. We applied PRV GS443 (expressing GFP-capsid protein in a PRV Becker background), PRV 232 (a gB-null PRV strain expressing GFP-capsid protein), or PRV 368 (a Us9-null PRV strain expressing GFP-capsid protein) to the S compartment and stained for the viral membrane gE on the axonal surfaces of nonpermeabilized neurons using a red secondary antibody. gE localized in discrete puncta along the axonal membrane in the N compartments of cultures infected with PRV GS443 or PRV 232. Importantly, the presence of a signal was Us9 dependent; no signal was observed in the axons of PRV 368-infected neurons (Fig. 5). This observation is consistent with the hypothesis that the punctate gE staining along the membrane surface originates from axonally sorted vesicles that release cargo to the axon surface.
FIG. 5.

PRV virions egress from axons in a gB-independent fashion. PRV GS443, PRV 232 (gB null), and PRV 368 (Us9 null) each express GFP fused to the capsid protein VP26 and were used to infect the S compartments of neuron cultures. Samples were fixed and stained for surface gE antigen (red). Imaging was performed on N-compartment axons. Arrowheads indicate colocalization. The side panels are magnified merge images. Bar, 4 μm.
We detected red, green, and yellow puncta in PRV GS443- and PRV 232-infected samples (Fig. 5). Red puncta without GFP-capsid fluorescence may represent L particles that have egressed or vesicle-incorporated gE protein delivered to the axonal surface upon fusion of the cellular transport vesicle with the plasma membrane. GFP fluorescence alone may originate from intracellular virions that are inaccessible to the anti-gE antibody. Alternatively, GFP fluorescence alone may indicate heterogeneity in virion incorporation of gE. Colocalized gE and capsid signals most likely represent released virions bound to the axonal surface. In both PRV GS443- and PRV 232-infected samples, capsid fluorescence colocalized with gE in approximately 50% of the 100 capsid puncta counted in two independent experiments. These data suggest that gB is not required for the egress of virions and L particles from axons.
Efficient neuron-to-cell transmission of infection requires virion membrane incorporation of gB.
Previous data revealed that the membrane dye DiI is transferred from an infected neuron to an axonally contacted PK15 cell during infection, indicating that a syncytium may form between the neuron and the postsynaptic cell (5). Additionally, late in infection, neighboring infected neuronal cell bodies become electrically coupled, suggesting cytoplasmic continuity between the cells. Importantly, the electrical coupling is gB dependent (K. McCarthy, D. Tank, and L. Enquist, unpublished data). We therefore hypothesized that the gB protein localized in the axonal membrane mediates neuron-to-cell spread of PRV by establishing a fusion pore between the axon terminus and the epithelial cell. In this model, the virion-incorporated gB would not be required for efficient neuron-to-cell spread of infection.
To test this hypothesis, we performed infections of compartmentalized neuronal cultures using the well-characterized PRV gB-007 mutant. This strain, derived from the wild-type Kaplan strain of PRV, carries a truncation of the cytoplasmic tail of the gB protein, while the transmembrane region and ectodomain of the protein remain intact (32). The mutation results in a dramatic decrease in virion incorporation of gB. Importantly, PRV gB-007 undergoes cell-to-cell spread, as evidenced by its wild-type kinetics of plaque formation and plaque sizes on rabbit kidney epithelial cells (32, 33). We performed immunofluorescence on compartmentalized neurons to determine whether the truncated gB localizes to the neuronal plasma membrane. Cell surface staining showed strong gB fluorescence in the somatic and axonal membranes (Fig. 6). The presence of gB in the cell membrane was qualitatively greater in PRV gB-007- than in PRV Becker-infected neurons, presumably due to decreased endocytosis of the protein. These findings show that, in PRV gB-007-infected neurons, the gB population in the somatic and axonal membranes is abundant, corroborating the previously published data in epithelial cells (32, 33).
FIG. 6.
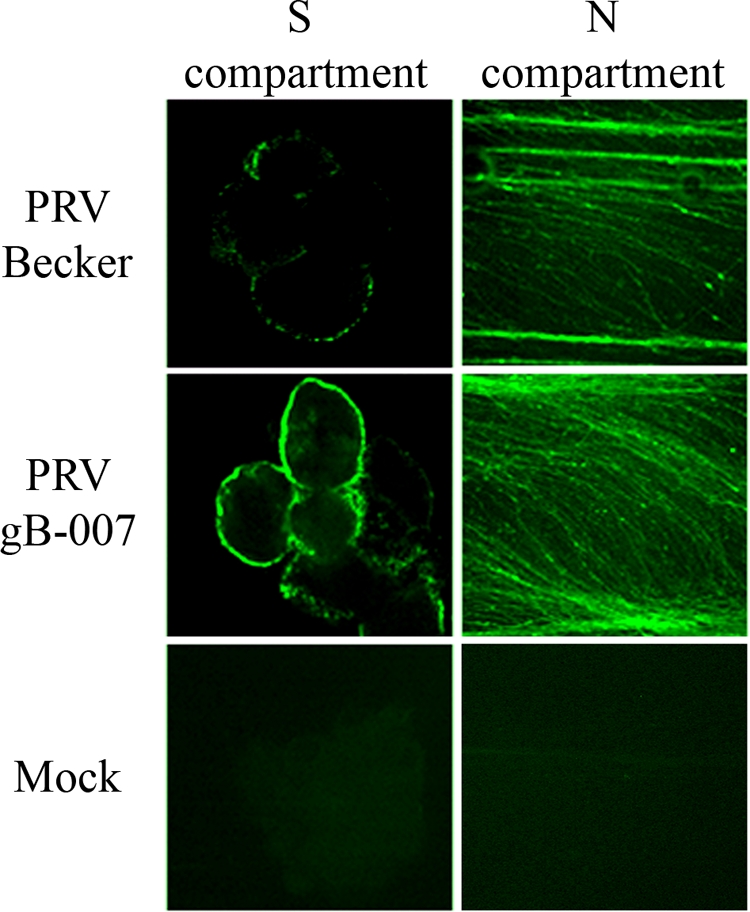
Wild-type gB and truncated gB localize to the plasma membranes of infected neurons. S compartments of neuronal cultures either were infected with PRV Becker or PRV gB-007 or were mock infected, and samples were processed for immunofluorescence against surface gB (green). A single Z-slice through the neuronal soma is shown. N-compartment axons were imaged via epifluorescence microscopy.
We inoculated the soma of compartmentalized neuronal cultures with PRV gB-007 and determined titers in the gB-complementing PK15 cells coplated in the N compartment. The titers of PRV gB-007 were significantly lower than those of PRV Becker. While PRV Becker reached 106 PFU/ml at 12 h postinfection (Fig. 7A), PRV gB-007 reached only 101 PFU/ml (Fig. 7B). This low titer may represent a single infectious unit that underwent neuron-to-cell spread, since one PK15 cell releases approximately 1,000 PFU. At 24 h postinfection, the titers of PRV gB-007 reached 104 PFU (3 log units lower than wild-type titers), likely representing the amplification of infection among epithelial cells in the N compartment. We conclude that neuron-to-cell spread of virions most likely does not proceed through syncytia or fusion pore formation and that virion incorporation of gB is required for efficient transmission of PRV infection.
FIG. 7.

Incorporation of gB into virions is required for efficient neuron-to-cell spread of infection. A PRV Becker (A) or PRV gB-007 (B) inoculum was applied to the S compartments of neuronal cultures. The contents of the S and N compartments were harvested, and titers were determined at 12 or 24 h postinfection (n = 3). Filled symbols represent means for each data set.
DISCUSSION
Infection of laboratory animals with mutant strains of alphaherpesviruses has helped identify virally encoded determinants for pathogenesis and spread in the nervous system. Inoculation of animals with PRV strains lacking the viral membrane gB results in infections that remain limited to the first-order neuron; i.e., transneuronal spread of the virus does not occur (2, 21). In corroboration of these observations, in vitro studies demonstrate that neuron-to-cell spread of PRV requires gB (17, 27). We used compartmented neuronal cultures and imaging techniques to identify the gB-mediated step in transneuronal PRV spread. We found that, unlike the membrane proteins Us9, gE, and gI, gB does not participate in the axonal sorting of virions from the infected cell body to the axonal compartment. Live-cell imaging of fluorescently tagged capsids in SCG cultures enabled us to capture axonal entry and subsequent anterograde trafficking of the gB-null PRV strain. Similarly, Coller and Smith report that gB-null PRV undergoes wild-type anterograde axonal transport in chick dorsal root ganglia (9). Because dissociated SCG neurons form synaptic connections in culture, retrograde viral spread from a postsynaptic to a presynaptic neuron is possible. However, we did not observe any retrograde-trafficking puncta at 17 h postinfection (data not shown). The lack of retrograde-moving puncta further illustrates the requirement for gB in transneuronal spread.
Prior electron microscopy studies of infected neurons have revealed that the fully assembled axonally sorted PRV virions are contained in vesicles, which are most likely derived from the trans-Golgi network during viral membrane acquisition (secondary envelopment) (5, 12, 18, 30). We examined whether gB participates in the fusion of the virion-carrying vesicles with the axonal plasma membrane and therefore in the release of the cargo to the cell surface. We were able to visualize released viral antigens by performing immunofluorescence on nonpermeabilized neurons. Antibodies directed against gE as well as ectodomains of other viral membrane proteins (data not shown) produced punctate staining along axons infected with wild-type and gB-null PRV. The signal was dependent on Us9, a viral protein required for the axonal sorting of virions and L particles. We therefore deduced that the axonally sorted viral cargo is released at the plasma membrane and that the process does not require gB. Importantly, released virions remained tightly cell associated, as evidenced by the fact that they were not detectable in the extracellular medium, and their susceptibility to low-pH inactivation offered further evidence that virions were indeed extracellular. The predicted topology of viral proteins in the vesicles surrounding virions is opposite from that in viral envelopes. Therefore, the fusion of the vesicle with the axonal membrane should not require gB. Similarly, gB is not required for particle release in epithelial cells (24). Given the array of cellular proteins involved in neurosecretion, it is likely that synaptic or dense-core vesicle machinery is recruited to the vesicles carrying viral cargo to mediate the release of virions from the axons. Recent immunoelectron microscopy studies by Miranda-Saksena et al. reveal that the neurosecretion proteins SNAP-25, Rab3A, and GAP-43 are associated with HSV-1 virions in the axons of human fetal dorsal root ganglia (31).
Syncytium formation occurs in various cell types upon infection by alphaherpesviruses and depends on cell surface expression of viral fusion proteins (8, 46). Membrane dye transfer and electrophysiology experiments suggest that transient or permanent membrane and cytoplasmic continuity may exist between an infected neuron and the cell it contacts (5; McCarthy et al., unpublished). We therefore hypothesized that gB may mediate neuron-to-cell spread of PRV by forming syncytia, thereby allowing unimpeded virion traffic between the connected cells. However, the inefficient anterograde spread of PRV strain gB-007, which packages gB into virions poorly and expresses gB abundantly on the cell surface, showed that incorporation of gB is required for the transmission of infection. We conclude that transneuronal spread of PRV most likely does not proceed through syncytial connections. Our data do not discount the possibility that gB-mediated fusion pore formation may occur between neurons and the contacted cells. Such an event may be coupled to viral egress, where fusion of the viral transport vesicle with the axonal plasma membrane would deliver viral proteins found in the vesicle membrane, including gB, to the axonal surface. The accompanying inversion of glycoprotein topology would now enable gB-mediated fusion with the postsynaptic membrane, resulting in the previously observed membrane dye transfer and electrical coupling.
We propose that the axonally sorted PRV virions undergo gB-independent egress by fusion of the cellular transport vesicle with the axonal plasma membrane. Following release to the axonal surface, the virion-incorporated gB mediates the fusion of the viral envelope with a closely apposed epithelial or neuronal cell membrane. In an in vitro study, De Regge et al. showed that the spread of PRV infection from axons to cocultured epithelial cells occurs primarily at axonal varicosities, which form synaptic contacts with the surrounding cells (13). These specialized connections bring membranes of the two cells in close apposition via transmembrane proteins that reach across the 20-nm cleft and impart stability to the synapse. Other in vivo and in vitro studies similarly report that the spread of PRV from an axon to the surrounding cells occurs at scattered sites along the axon and at neuronal termini (5, 45). The synaptic cleft between neurons is not wide enough to accommodate a mature virion, implying that local membrane contortion may accompany transport vesicle fusion with the axonal surface. This event would bring the viral membrane into intimate contact with the postsynaptic cell, enabling the viral fusion machinery to mediate entry, as it would during infection by an extracellular particle. Such proximity between the viral and cell membranes may explain why the viral attachment glycoprotein, gD, though essential for infection by free particles, is dispensable for transneuronal spread of infection (2, 5). Additionally, the close membrane proximity, which would occur only in virion release at specialized connections, may explain the circuit-specific spread of the virus.
It was recently shown that HSV-1 gB and PRV gB engage the paired immunoglobulin-like type 2 receptor α (PILRα) and that the process is required for viral entry into cells transduced with this receptor (1, 40). PILRs are expressed in a variety of tissues, including the nervous system (40). It is exciting to hypothesize that PILRα may participate in the transneuronal spread of infection via interactions with gB.
Supplementary Material
Acknowledgments
We thank Thomas Mettenleiter for providing PRV strain gB-007.
We acknowledge support from the National Institutes of Health (R01 NS33506 and R01 NS60699 to L.W.E.).
Footnotes
Published ahead of print on 3 June 2009.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Arii, J., M. Uema, T. Morimoto, H. Sagara, H. Akashi, E. Ono, H. Arase, and Y. Kawaguchi. 2009. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor α. J. Virol. 834520-4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babic, N., T. C. Mettenleiter, A. Flamand, and G. Ugolini. 1993. Role of essential glycoproteins gII and gp50 in transneuronal transfer of pseudorabies virus from the hypoglossal nerves of mice. J. Virol. 674421-4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brideau, A. D., J. P. Card, and L. W. Enquist. 2000. Role of pseudorabies virus Us9, a type II membrane protein, in infection of tissue culture cells and the rat nervous system. J. Virol. 74834-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Card, J. P., L. Rinaman, J. S. Schwaber, R. R. Miselis, M. E. Whealy, A. K. Robbins, and L. W. Enquist. 1990. Neurotropic properties of pseudorabies virus: uptake and transneuronal passage in the rat central nervous system. J. Neurosci. 101974-1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ch'ng, T. H., and L. W. Enquist. 2005. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J. Virol. 7910875-10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ch'ng, T. H., and L. W. Enquist. 2005. Efficient axonal localization of alphaherpesvirus structural proteins in cultured sympathetic neurons requires viral glycoprotein E. J. Virol. 798835-8846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ch'ng, T. H., E. A. Flood, and L. W. Enquist. 2005. Culturing primary and transformed neuronal cells for studying pseudorabies virus infection. Methods Mol. Biol. 292299-316. [DOI] [PubMed] [Google Scholar]
- 8.Cole, N. L., and C. Grose. 2003. Membrane fusion mediated by herpesvirus glycoproteins: the paradigm of varicella-zoster virus. Rev. Med. Virol. 13207-222. [DOI] [PubMed] [Google Scholar]
- 9.Coller, K. E., and G. A. Smith. 2008. Two viral kinases are required for sustained long distance axon transport of a neuroinvasive herpesvirus. Traffic 91458-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curanović, D., M. G. Lyman, C. Bou-Abboud, J. P. Card, and L. W. Enquist. 2009. Repair of the UL21 locus in pseudorabies virus Bartha enhances the kinetics of retrograde, transneuronal infection in vitro and in vivo. J. Virol. 831173-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis-Poynter, N., S. Bell, T. Minson, and H. Browne. 1994. Analysis of the contributions of herpes simplex virus type 1 membrane proteins to the induction of cell-cell fusion. J. Virol. 687586-7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.del Rio, T., T. H. Ch'ng, E. A. Flood, S. P. Gross, and L. W. Enquist. 2005. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J. Virol. 793903-3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Regge, N., H. J. Nauwynck, K. Geenen, C. Krummenacher, G. H. Cohen, R. J. Eisenberg, T. C. Mettenleiter, and H. W. Favoreel. 2006. Alpha-herpesvirus glycoprotein D interaction with sensory neurons triggers formation of varicosities that serve as virus exit sites. J. Cell Biol. 174267-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ekstrand, M. I., L. W. Enquist, and L. E. Pomeranz. 2008. The alpha-herpesviruses: molecular pathfinders in nervous system circuits. Trends Mol. Med. 14134-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Enquist, L. W., P. J. Husak, B. W. Banfield, and G. A. Smith. 1998. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus Res. 51237-347. [DOI] [PubMed] [Google Scholar]
- 16.Favoreel, H. W., G. Van Minnebruggen, H. J. Nauwynck, L. W. Enquist, and M. B. Pensaert. 2002. A tyrosine-based motif in the cytoplasmic tail of pseudorabies virus glycoprotein B is important for both antibody-induced internalization of viral glycoproteins and efficient cell-to-cell spread. J. Virol. 766845-6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feierbach, B., M. Bisher, J. Goodhouse, and L. W. Enquist. 2007. In vitro analysis of transneuronal spread of an alphaherpesvirus infection in peripheral nervous system neurons. J. Virol. 816846-6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Granzow, H., B. G. Klupp, W. Fuchs, J. Veits, N. Osterrieder, and T. C. Mettenleiter. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 753675-3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hampl, H., T. Ben-Porat, L. Ehrlicher, K. O. Habermehl, and A. S. Kaplan. 1984. Characterization of the envelope proteins of pseudorabies virus. J. Virol. 52583-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hannah, B. P., E. E. Heldwein, F. C. Bender, G. H. Cohen, and R. J. Eisenberg. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 814858-4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heffner, S., F. Kovacs, B. G. Klupp, and T. C. Mettenleiter. 1993. Glycoprotein gp50-negative pseudorabies virus: a novel approach toward a nonspreading live herpesvirus vaccine. J. Virol. 671529-1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heldwein, E. E., H. Lou, F. C. Bender, G. H. Cohen, R. J. Eisenberg, and S. C. Harrison. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313217-220. [DOI] [PubMed] [Google Scholar]
- 23.Husak, P. J., T. Kuo, and L. W. Enquist. 2000. Pseudorabies virus membrane proteins gI and gE facilitate anterograde spread of infection in projection-specific neurons in the rat. J. Virol. 7410975-10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klupp, B., J. Altenschmidt, H. Granzow, W. Fuchs, and T. C. Mettenleiter. 2008. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J. Virol. 826299-6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klupp, B. G., R. Nixdorf, and T. C. Mettenleiter. 2000. Pseudorabies virus glycoprotein M inhibits membrane fusion. J. Virol. 746760-6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LaVail, J. H., A. N. Tauscher, A. Sucher, O. Harrabi, and R. Brandimarti. 2007. Viral regulation of the long distance axonal transport of herpes simplex virus nucleocapsid. Neuroscience 146974-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu, W. W., J. Goodhouse, N. L. Jeon, and L. W. Enquist. 2008. A microfluidic chamber for analysis of neuron-to-cell spread and axonal transport of an alpha-herpesvirus. PLoS ONE 3e2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lyman, M. G., D. Curanovic, and L. W. Enquist. 2008. Targeting of pseudorabies virus structural proteins to axons requires association of the viral Us9 protein with lipid rafts. PLoS Pathog. 4e1000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lyman, M. G., B. Feierbach, D. Curanovic, M. Bisher, and L. W. Enquist. 2007. Pseudorabies virus Us9 directs axonal sorting of viral capsids. J. Virol. 8111363-11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mettenleiter, T. C., B. G. Klupp, and H. Granzow. 2006. Herpesvirus assembly: a tale of two membranes. Curr. Opin. Microbiol. 9423-429. [DOI] [PubMed] [Google Scholar]
- 31.Miranda-Saksena, M., R. A. Boadle, A. Aggarwal, B. Tijono, F. J. Rixon, R. J. Diefenbach, and A. L. Cunningham. 2009. Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J. Virol. 833187-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nixdorf, R., B. G. Klupp, A. Karger, and T. C. Mettenleiter. 2000. Effects of truncation of the carboxy terminus of pseudorabies virus glycoprotein B on infectivity. J. Virol. 747137-7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nixdorf, R., B. G. Klupp, and T. C. Mettenleiter. 2001. Restoration of function of carboxy-terminally truncated pseudorabies virus glycoprotein B by point mutations in the ectodomain. J. Virol. 7511526-11533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nixdorf, R., B. G. Klupp, and T. C. Mettenleiter. 2001. Role of the cytoplasmic tails of pseudorabies virus glycoproteins B, E and M in intracellular localization and virion incorporation. J. Gen. Virol. 82215-226. [DOI] [PubMed] [Google Scholar]
- 35.Peeters, B., N. de Wind, M. Hooisma, F. Wagenaar, A. Gielkens, and R. Moormann. 1992. Pseudorabies virus envelope glycoproteins gp50 and gII are essential for virus penetration, but only gII is involved in membrane fusion. J. Virol. 66894-905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pomeranz, L. E., A. E. Reynolds, and C. J. Hengartner. 2005. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 69462-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rauh, I., and T. C. Mettenleiter. 1991. Pseudorabies virus glycoproteins gII and gp50 are essential for virus penetration. J. Virol. 655348-5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roizman, B., and D. M. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 39.Ruel, N., A. Zago, and P. G. Spear. 2006. Alanine substitution of conserved residues in the cytoplasmic tail of herpes simplex virus gB can enhance or abolish cell fusion activity and viral entry. Virology 346229-237. [DOI] [PubMed] [Google Scholar]
- 40.Satoh, T., J. Arii, T. Suenaga, J. Wang, A. Kogure, J. Uehori, N. Arase, I. Shiratori, S. Tanaka, Y. Kawaguchi, P. G. Spear, L. L. Lanier, and H. Arase. 2008. PILRα is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132935-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith, G., L. Pomeranz, S. P. Gross, and L. W. Enquist. 2004. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. USA 10116034-16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith, G. A., S. P. Gross, and L. W. Enquist. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. USA 983466-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tirabassi, R. S., and L. W. Enquist. 1998. Role of envelope protein gE endocytosis in the pseudorabies virus life cycle. J. Virol. 724571-4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tomishima, M. J., and L. W. Enquist. 2001. A conserved alpha-herpesvirus protein necessary for axonal localization of viral membrane proteins. J. Cell Biol. 154741-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomishima, M. J., and L. W. Enquist. 2002. In vivo egress of an alphaherpesvirus from axons. J. Virol. 768310-8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turner, A., B. Bruun, T. Minson, and H. Browne. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72873-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vanarsdall, A. L., B. J. Ryckman, M. C. Chase, and D. C. Johnson. 2008. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 8211837-11850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Minnebruggen, G., H. W. Favoreel, and H. J. Nauwynck. 2004. Internalization of pseudorabies virus glycoprotein B is mediated by an interaction between the YQRL motif in its cytoplasmic domain and the clathrin-associated AP-2 adaptor complex. J. Virol. 788852-8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang, F., W. Tang, H. M. McGraw, J. Bennett, L. W. Enquist, and H. M. Friedman. 2005. Herpes simplex virus type 1 glycoprotein E is required for axonal localization of capsid, tegument, and membrane glycoproteins. J. Virol. 7913362-13372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whitley, R. J., D. W. Kimberlin, and B. Roizman. 1998. Herpes simplex viruses. Clin. Infect. Dis. 26541-553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.