

Abstract
All herpesviruses have a layer of protein called the tegument that lies between the virion membrane and the capsid. The tegument consists of multiple, virus-encoded protein species that together can account for nearly half the total virus protein. To clarify the structure of the tegument and its attachment to the capsid, we used electron microscopy and protein analysis to examine the tegument of herpes simplex virus type 1 (HSV-1). Electron microscopic examination of intact virions revealed that whereas the tegument was asymmetrically distributed around the capsid in extracellular virions, it was symmetrically arranged in cell-associated virus. Examination of virions after treatment with nonionic detergent demonstrated that: (i) in extracellular virus the tegument was resistant to removal with Triton X-100 (TX-100), whereas it was lost nearly completely when cell-associated virus was treated in the same way; (ii) the tegument in TX-100-treated extracellular virions was asymmetrically distributed around the capsid as it is in unextracted virus; and (iii) in some images, tegument was seen to be linked to the capsid by short, regularly spaced connectors. Further analysis was carried out with extracellular virus harvested from cells at different times after infection. It was observed that while the amount of tegument present in virions was not affected by time of harvest, the amount remaining after TX-100 treatment increased markedly as the time of harvest was increased from 24 h to 64 h postinfection. The results support the view that HSV-1 virions undergo a time-dependent change in which the tegument is transformed from a state in which it is symmetrically organized around the capsid and extractable with TX-100 to a state where it is asymmetrically arranged and resistant to extraction.
All herpesviruses have a tegument, a layer of protein located between the virus membrane and the capsid. Depending on the virus species, the tegument can be 20 to 40 nm in thickness, and it may be uniformly or asymmetrically distributed about the capsid (7, 17, 24, 33). The tegument is composed predominantly of virus-encoded proteins that together can account for up to half or more of the total virion protein mass. Tegument proteins are thought to be those involved in the early stages of infection before progeny virus proteins are synthesized.
The tegument has been most thoroughly studied in herpes simplex virus type 1 (HSV-1). Examination of virions by electron microscopy has demonstrated that the tegument is not highly structured. Its morphology is described as predominantly granular with fibrous elements also present (7, 19). Analysis by cryo-electron microscopy, followed by icosahedral reconstruction has shown that the tegument is not icosahedrally ordered, although a small amount of tegument density is observed close to the capsid surface at the pentons (3, 47).
The HSV-1 tegument is composed of approximately 20 distinct, virus-encoded protein species whose amounts vary considerably. The predominant components are UL47, UL48, and UL49, each of which occurs in more than 800 copies per virion (8, 46). In contrast, others, such as RL2 (ICP0), RS1 (ICP4), UL36, and UL37, occur in ∼100 copies or less. Trace amounts of host cell-encoded proteins are also present (15). Many of the tegument proteins are required for virus replication (34), and functions have been defined for most (9, 12, 31, 40).
Biochemical studies have demonstrated that the tegument makes noncovalent contacts with both the virus capsid and the membrane. Studies of capsid-tegument contacts have emphasized binding of UL36, a tegument protein, to UL25, a capsid protein located near the vertices and involved in DNA encapsidation (5, 20, 29). Other tegument proteins such as UL48 (VP16), UL37, and UL49 (VP22) are found to associate with UL36 and may be bound to the capsid indirectly by way of UL36 (13, 44). UL16 binds reversibly to the capsid while UL46 (VP11/12) has been shown to bind to both the membrane and the capsid (21, 22, 26). Binding of tegument proteins to the membrane has been shown to occur by way of attachment to UL11 (45) and also to the internal domains of membrane glycoproteins, including glycoprotein D (gD), gH, and gE (4, 6, 11).
We describe here the results of a study in which electron microscopy and protein analysis were used to clarify the structure of the HSV-1 tegument and its attachment to the capsid. The study was designed to extend the observation that most of the HSV-1 tegument remains attached to the capsid when the membrane is removed from the virus by treatment with nonionic detergent (19). Cell-associated and extracellular virions were compared after treatment with Triton X-100 (TX-100).
MATERIALS AND METHODS
Cell and virus growth.
All experiments were carried out with the KOS strain of HSV-1 which was grown on monolayer cultures of Vero cells as described previously (35). Cells were grown and infected in 150-cm2 tissue culture flasks or 800-cm2 roller bottles.
Virus and capsid purification.
Extracellular virus was prepared from the infected cell supernatant which was harvested in 15-ml aliquots and centrifuged for 5 min at 1,000 × g to remove cells. The time of harvest varied in individual experiments and is given in the text. Virus was pelleted from the supernatant by centrifugation at 23,000 rpm for 45 min in a Beckman SW41 rotor operated at 4°C. Pellets were resuspended in 300 μl of TNE (0.01 M Tris-HCl, 0.5 M NaCl, 1 mM EDTA [pH 7.5]), and virus was further purified by centrifugation (23,000 rpm, 40 min; SW50.1 rotor; 4°C) on a gradient of 20%-50% sucrose in TNE. The virus band was identified by scattered light and harvested from the gradient with a Pasteur pipette. Preparations yielded ∼300 μl virus at a concentration of ∼0.3 mg of protein/ml from 15 ml of culture supernatant.
Capsids were isolated from extracellular virus by TX-100 treatment of virions purified as described above. Aliquots of 100 μl of virus in TNE were adjusted to 0.5% TX-100 and incubated for 5 min at 4°C. Capsids were isolated by centrifugation (23,000 rpm for 40 min in an SW50.1 rotor at 4°C) on a 600-μl gradient of 20 to 50% sucrose prepared in TNE containing 0.5% TX-100. After centrifugation, gradients were photographed and fractionated to recover capsids (∼100 μl; 0.25 mg of protein/ml).
Capsids from cell-associated virus were isolated beginning with infected cells (∼5 × 108 cells) harvested 18 to 24 h postinfection in batches of three 800-cm2 roller bottles. Cells were harvested by scraping, suspended in 30 ml of water, and frozen at −80°C. After thawing, cells were lysed by adjusting them to 0.5% TX-100 containing 1 mM EDTA, followed by incubation for 5 min at 4°C. Nuclei were then removed from the lysate by centrifugation for 5 min at 800 × g, and capsids were pelleted from the postnuclear supernatant by centrifugation (SW28 rotor; 23,000 rpm for 45 min at 4°C) through a 5-ml cushion containing 35% sucrose, 0.5% TX-100, and 1 mM EDTA. Capsids were then resuspended in 1 ml of TNE containing 2 mM dithiothreitol and purified by two steps of sucrose density gradient ultracentrifugation as described previously (29). Preparations yielded ∼200 μl of capsids at a concentration of 0.25 to 0.5 mg of protein/ml.
Trypsin treatment of tegument-containing capsids.
Studies on the trypsin treatment of tegument-containing capsids were carried out beginning with extracellular HSV-1 harvested 30 to 40 h postinfection and purified as described above. 100-μl aliquots were adjusted to 0.5% TX-100 and trypsin at a concentration of 0.1 or 0.5 μg/ml. Mixtures were incubated for 30 min at 37°C and the capsids were then isolated by ultracentrifugation on a 600-μl gradient of 20 to 50% sucrose in TNE containing 0.5% TX-100 and protease inhibitors as described previously (27). Centrifugation was for 40 min at 23,000 rpm in a Beckman SW55Ti rotor operated at 4°C. After centrifugation, gradients were photographed and fractionated with capsid-containing fractions used for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Comparable experiments were not carried out with cell-associated virus since TX-100 was expected to remove the tegument from these virions.
Electron microscopy.
Infected cells, purified virus and capsids to be examined by thin-section electron microscopy were centrifuged into a small (∼0.3 ml) pellet and fixed overnight with 2% glutaraldehyde. Further steps of fixation, embedding and sectioning were carried out as previously described (16). Sections were stained with 1% uranyl acetate (5 min at room temperature), followed by 0.25% lead citrate (2 min at room temperature). Capsids to be examined by negative staining were adsorbed to carbon-Formvar-coated copper electron microscope grids, stained for 1 min with 1% uranyl acetate, and air dried as described previously (28). All electron microscopy was carried out on a Philips EM400T transmission electron microscope operated at 80 keV. Images were recorded on film and converted to digital form by scanning in a flatbed scanner. Measurement of images was performed with ImageJ and Photoshop CS3. The spacing of the connectors linking capsid and tegument was measured directly from images and also after densitometric scanning of the region between capsid and tegument with ImageJ. The results obtained with the two methods did not differ significantly.
Other methods.
Sucrose gradients were photographed with top illumination from a high intensity lamp so virus and capsid bands could be observed by scattered light. Images were recorded with a digital camera. DNA-containing capsids were identified by spotting 2 μl of capsid suspension onto a polyvinylidene difluoride membrane, staining for 2 min with GelRed (1:10,000 dilution), washing with phosphate-buffered saline, and visualization with UV illumination. Previously described procedures were used for SDS-PAGE, followed by Coomassie blue staining (30). Stained gels were recorded by scanning in a flatbed scanner and the bands were measured quantitatively with UN-SCAN-IT (version 5.2; Silk Scientific).
RESULTS
Intact virus.
Tegument analyses were carried out beginning with two distinct virus populations: cell-associated HSV-1 and virions harvested from the extracellular medium. Examination by electron microscopy showed that most cell-associated virus was located near the outer surface of the cell attached to the surface or to projections from it (Fig. 1a and b). As expected, extracellular virions were found to be free of cell-derived material (Fig. 1d to f). Micrographs showed that the structure of the tegument was quite different in the two virus populations. Whereas it was uniformly distributed around the capsid in cell-associated virus, it was markedly asymmetric in extracellular virions as described previously (7, 17) (compare Fig. 1a- to c with Fig. 1d to f). Counts showed 40 of 40 (100%) and 88 of 488 (18%) virions with a symmetric tegument morphology in cell associated and extracellular virions, respectively. Cell-associated virions were interpreted to be progeny virions as parental virus, derived from an infected cell supernatant, is expected to have an asymmetric tegument.
FIG. 1.

Electron micrographs showing cell-associated (a to c) and extracellular HSV-1 (d to f). Note that the tegument is symmetrically arranged around the capsid in cell-associated virus and asymmetric in extracellular virions. Arrows indicate representative virions with symmetric teguments in panels a and b, while in panels d and e arrows indicate virions where the tegument is asymmetric.
TX-100 extraction.
To examine the tegument in capsids from the two virus populations, virions were treated with 0.5% TX-100 as described in Materials and Methods, and the capsids were purified by sucrose density gradient centrifugation. Examination of the gradients showed that a band of DNA-containing capsids sedimented coincidently with nuclear C capsids in both TX-100-treated cell-associated and extracellular virus populations (Fig. 2a) . Further analysis was done with capsids from the two bands (starred in Fig. 2a) and also with nuclear C capsids (marked as asterisks in the figure) as a control. The two virus-derived capsid populations are identified as TX-100-treated cell-associated viruses (TCAV) and TX-100-treated extracellular viruses (TEV), respectively. In addition to the DNA-containing capsids, each virus population was found to yield small amounts of capsids sedimenting coincidently with nuclear A and B capsids (Fig. 2a).
FIG. 2.

Analysis of HSV-1 capsids by sucrose density gradient centrifugation (a), electron microscopy (b), and SDS-PAGE (c). Analysis was performed with capsids isolated by TX-100 treatment of cell-associated virus (lane 1), TX-100 extraction of extracellular virus (lane 2), and capsids isolated from the nuclei of HSV-1-infected cells (lane 3). Bands marked by asterisks in panel a were the ones used for analysis in panels b and c. Note that tegument remains attached to capsids derived from extracellular, but not cell-associated virus.
Electron microscopic examination showed that whereas TEV capsids were coated with abundant tegument as expected, little or none was observed with TCAV (Fig. 2b; compare lanes 1 and 2). Although the TEV capsid was largely obscured by components of the tegument, in TCAV the capsid was visible with a diameter comparable to that of nuclear C capsids (Fig. 2b; compare lanes 1 and 3). Capsomers were apparent in most TCAV images. Measurement of the particle diameters of TCAV, TEV, and nuclear C capsids yielded values of 111.5 ± 4.2 nm (n = 58), 164.7 ± 9.6 nm (n = 33), and 112.2 ± 1.9 nm (n = 31), respectively (Fig. 3).
FIG. 3.

Capsid diameter as determined in TX-100-extracted cell-associated virus (top), TX-100-extracted extracellular virus (middle), and C capsids isolated from the nuclei of HSV-1-infected cells (bottom). Measurements were made from electron micrographs of specimens after negative staining as described in Materials and Methods. Note the larger diameter of tegument-containing capsids (middle) compared to those lacking tegument (top and bottom).
SDS-polyacrylamide gel analysis supported the view that TCAV contain little tegument. While TEV were found to be composed of capsid proteins UL19, UL38, and UL18 plus additional protein species presumptively identified as tegument proteins UL36, UL46, UL47, UL48, and UL49, TCAV showed capsid proteins with only trace amounts of protein that could correspond to tegument components (compare Fig. 2c, lanes 1 and 2). The TCAV protein composition was quite similar to that of nuclear C capsids (Fig. 2c; compare lanes 1 and 3).
Since it was unexpected to observe that the tegument was removed by TX-100 treatment of cell-associated HSV-1, we considered three alternative possibilities: (i) TCAV were actually C capsids derived from the infected cell nucleus; (ii) TCAV were produced from tegument-containing capsids by the action of host cell enzymes or other factors that would not be present to affect capsids derived from extracellular virus; and (iii) TCAV were produced only at the low salt concentration routinely used for extraction of cell-associated virus.
In response to the first possibility, we note that the distribution of capsid types produced by TX-100 treatment of cell-associated virus (mostly C-like capsids with only a trace of A and B capsids) was quite different from the capsids isolated from infected cell nuclei (predominantly B capsids; Fig. 2a [compare lanes 1 and 3]). Further, the nuclei produced by TX-100 treatment of infected cells were not damaged in a way that would allow capsids to escape. In fact, upon deliberate disruption, these nuclei yielded normal amounts of capsids, including C capsids (data not shown).
The second possibility was addressed by adding extracellular virions to infected cells prior to TX-100 treatment. Such mixtures yielded both TEV and TCAV in the proportion present in the original preparation (data not shown), indicating that host cell factors are not capable of removing the tegument from TEV during the TX-100 extraction process. We conclude that TCAV are most likely to be derived from cell-associated virus as suggested above.
The effect of NaCl concentration was tested by exchanging the salt concentrations used for TX-100 extraction; cell-associated virus was extracted in TNE (0.5 M NaCl), while low-salt buffer was used for extracellular virus. No effect was observed in either case (data not shown). At both salt concentrations tegument was removed from cell-associated virus and was resistant to extraction in virus harvested from the extracellular medium.
Tegument structure.
The structure of the tegument in TEV was further examined by electron microscopy of thin-sectioned material. Images showed DNA-containing capsids surrounded to a variable extent by tegument (Fig. 4). In most images the tegument was seen to be asymmetrically distributed about the capsid as it is in the extracellular virions from which the TEV were prepared. Three images of this type are arrowed in Fig. 4, but others can be identified. In some images the tegument was found to have gaps or irregularities, suggesting that some tegument may have been lost or not incorporated as the virus was formed (Fig. 4). DNA was seen to project from the capsid in some cases, an effect we suggest may be due to capsid or tegument damage during preparation (see boxed image in Fig. 4).
FIG. 4.

Electron micrograph showing extracellular HSV-1 virions after treatment with 0.5% TX-100. Note that the tegument is asymmetrically distributed around the capsid as it is in intact virions (arrows). DNA can be seen to be in the process of being extruded from some capsids; one such capsid is boxed. Gaps or irregularities can be observed in the tegument of many TEV, indicating that it was eroded or was never firmly attached to the capsid.
In some images the tegument could be seen to be attached to the capsid by short connectors (Fig. 5a and b). These were variable in diameter, and most were 6.0 to 10.0 nm in length. Visual examination indicated a degree of regularity to the connector spacing. Measurement of the spacing in 13 capsids yielded a mean value of 16.8 ± 4.8 nm (n = 54; Fig. 5c).
FIG. 5.

Electron microscopic analysis of TEV exhibiting connections between the capsid and the tegument. A representative image of a TEV with connectors is shown at low (a; boxed image) and high (b) magnifications. Connectors are indicated by arrows in panel b. The spacing between connectors was measured in 13 TEV images, and the results are shown in panel c.
Linkage between the capsid and tegument was also probed by trypsin treatment of TEV. It was expected that the degree of trypsin sensitivity in the linkage would be revealing about its overall strength. Experiments were performed by treating extracellular HSV-1 simultaneously with 0.5% TX-100 and trypsin. After incubation, the resulting capsids were examined by sucrose density gradient centrifugation and SDS-PAGE to determine whether the tegument had been removed (Fig. 6).
FIG. 6.

Sucrose density gradient (a) and SDS-PAGE (b) analysis of TEV before (lane 1) and after (lanes 2 and 3) treatment with trypsin. SDS-PAGE analyses were performed with TEV or capsids recovered from gradient bands marked by asterisks in panel a. Note that the effect of trypsin depended on the dose used. At 0.1 μg/ml, trypsin caused removal of the tegument, but DNA was retained in the capsid. In contrast, a concentration of 0.5 μg of trypsin/ml caused the loss of both tegument and DNA.
Sucrose gradient analysis of control reactions incubated without trypsin yielded two bands of DNA containing capsids (Fig. 6a, lane 1). Of these, the predominant band (starred in Fig. 6a) corresponded in migration to TEV, and this identification was confirmed by SDS-polyacrylamide gel analysis, which revealed the presence of tegument proteins, as expected of TEV (Fig. 6b, lane 1). The minor band migrated coincidentally with C capsids (Fig. 6a, lane 1).
When reactions were performed in the presence of trypsin, the results of sucrose gradient analyses were found to depend on the trypsin concentration. At a low concentration (0.1 μg/ml), gradients revealed two bands of capsids: a predominant, DNA-containing band migrating coincidentally with C capsids and a minor band of capsids lacking DNA and migrating with A capsids (Fig. 6a, lane 2). No band of TEV was observed. Only A-like capsids were seen when the trypsin concentration was high (0.5 μg/ml; Fig. 6a, lane 3). SDS-polyacrylamide gel analysis of the C-like and A-like capsids revealed capsid proteins with only negligible amounts of tegument present (Fig. 6b, lanes 2 and 3).
The results of these experiments are interpreted to indicate that tegument proteins were removed from TEV by both low and high trypsin doses. This interpretation is supported by SDS-polyacrylamide gel analyses showing an absence of tegument proteins in the capsids recovered from trypsin-containing incubations (Fig. 6b, lanes 2 and 3). The high trypsin dose additionally caused release of the capsid DNA, as described previously (27). DNA release resulted in the creation of A capsids, as observed by sucrose gradient analysis (Fig. 6a, lane 3).
Effects of extracellular incubation.
Experiments with extracellular virus were carried out to determine when the tegument first becomes resistant to extraction with TX-100. Extracellular virus was harvested at progressively longer times after infection and divided into two aliquots. One was treated with TX-100, while the other was held as a control, and SDS-polyacrylamide gel analysis was used to determine the protein composition of each.
The results demonstrated that the tegument content of virions was not greatly affected by the time of harvest over the range of 24 to 64 h postinfection (Fig. 7a). The three major tegument protein species (UL47, UL48, and UL49) were all found to be present, and the amount did not vary systematically during the harvest period examined. Similarly, the content of a control capsid protein (UL38) did not vary, indicating that analysis was performed with approximately the same amount of virus at each time point (Fig. 7b).
FIG. 7.
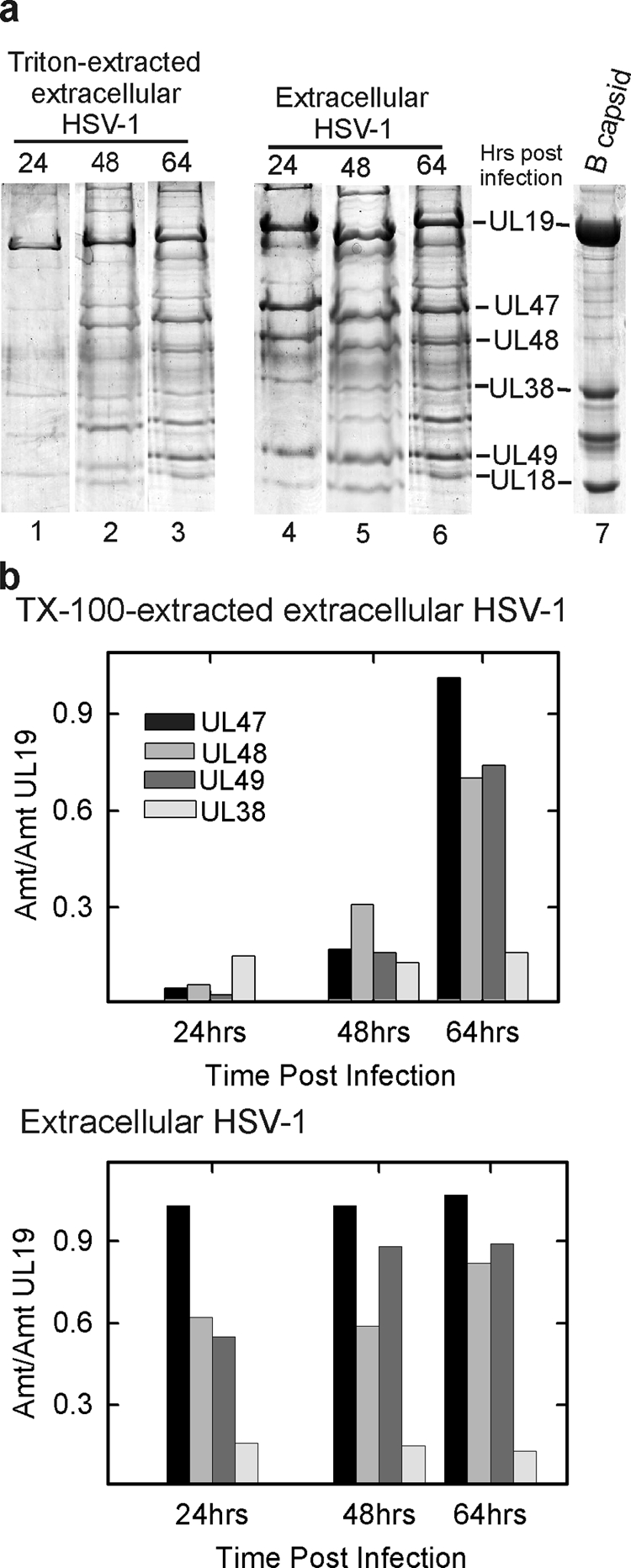
(a) SDS-PAGE analysis of TEV and extracellular HSV-1. Extracellular virus was harvested at different times after infection (24, 48, and 64 h) and divided into two equal aliquots. TEV was prepared from one, while the other served as a control. Gel analysis is shown for TEV (lanes 1 to 3) and for virus (lanes 4 to 6). HSV-1 B capsids were examined as a control (lane 7). (b) Quantitative determination of selected gel bands. Note that while the amount of tegument protein present in TEV increased with time of virus harvest, the amount in virions was not affected.
In contrast, a substantial increase in tegument was observed after TX-100 treatment in 64-h compared to 24-h TEV. As in the case of virus, UL47, UL48, and UL49 were the predominant tegument protein components of TEV, but the amount of each relative to a control protein (UL38) increased substantially with time after infection (Fig. 7a, lanes 1 to 3, and Fig. 7b, top panel). Control experiments demonstrated that the presence of living cells was not required for the observed time-dependent increase in the amount of TX-100-resistant tegument. The same increase was observed when extracellular virus was removed from cells prior to incubation (data not shown). Since the amount of tegument present in virions was not affected by time after infection, it is concluded that the tegument becomes progressively more resistant to TX-100 extraction at increasing times postinfection.
DISCUSSION
Tegument function.
Herpesvirus tegument proteins are thought to be those whose function is required promptly after initiation of infection, before expression of virus genes can take place. The HSV-1 alpha-gene transactivating factor (UL48) is an example of this. UL48 is a tegument protein essential for HSV-1 replication (1, 2, 32). It is released from the tegument as the parental capsid enters the host cell cytoplasm. UL48 then binds to a cellular protein, Oct-1 (18), and in a complex with Oct-1 it activates transcription of immediate-early (alpha) genes such as ICP0, ICP4, and ICP22 (2, 32). Other HSV-1 tegument proteins thought to function soon after virus entry include UL36, UL41, ICP34.5, and ICP22 (12, 34, 39, 40).
In order for a tegument protein to function prior to virus gene expression it must be assembled into the virion tegument as a progeny virion is formed and then released from the capsid when a new host cell is infected. In HSV-1, most tegument proteins become associated with the capsid during secondary envelopment in the cytoplasm. A progeny capsid buds into a vesicle containing a region of tegument on its outer surface, creating a doubly enveloped, tegument-containing capsid. The outer of the two membranes then fuses with the host cell cytoplasmic membrane, exposing a virion to the extracellular environment (23). The tegument is shed completely or nearly so when parental capsids enter the peripheral cytoplasm in the subsequent cycle of infection (23).
Symmetric and asymmetric tegument.
Although individual HSV-1 virions have been observed with either a symmetric or an asymmetric tegument (7, 17, 24, 33), it was novel in the present study to note the correlation of symmetric tegument with cell-associated virus (Fig. 1). It was similarly novel and unexpected to observe that the tegument of cell-associated virus was removed by TX-100 treatment (Fig. 2 and 3). Previous studies with extracellular virus emphasized the resistance of the tegument to TX-100 extraction (19). Since cell-associated virus is a precursor of extracellular virions, the different morphology and TX-100 extractability suggests that the release of progeny virus is accompanied by a global structural transformation or maturation in the tegument. The tegument becomes more tightly associated with the capsid, and much of it is changed in location.
Tegument transformation.
Studies of the time course have demonstrated a time-dependent progression linking virions with extractable and unextractable teguments. The first virions able to be recovered from the extracellular medium (∼24 h postinfection) were found to resemble cell-associated virus in that much of the tegument was removed after treatment with 0.5% TX-100 (Fig. 7). At later times postinfection, an increasing proportion of the tegument became unextractable. This observation indicates that resistance to TX-100 does not occur immediately as progeny virions detach from the host cell but rather it develops gradually over a period of 40 h or more. Resistance to TX-100 extraction appeared to involve all or most of the predominant tegument proteins (Fig. 7).
The most apparent difference between cell-associated and extracellular virus has to do with the host cell in the subsequent cycle of infection. Cell-associated virus is well suited to infect adjacent cells, while extracellular virus has the potential to infect more remote ones. We suggest that the progressively stronger tegument-capsid association observed in extracellular virus may be an adaptation to maintain virus infectivity as it transits through the external environment. Removal of the tegument once a suitable host cell is infected may be caused by host cell factors such as protease activity (10) or possibly by reversibility in the tegument-capsid interaction, as suggested by studies of tegument components UL16 and UL46 (21, 22, 26). Both cell-associated and extracellular virions were found to be infectious (data not shown).
Alternatively, the important feature of extracellular virus may be the asymmetric arrangement of the tegument. An asymmetric tegument may expose the site of DNA exit from the capsid, making uncoating more facile as recently suggested by Maurer et al. (17). It is also possible that resistance of the tegument to TX-100 extraction is simply a manifestation of virion decay. Protein oxidation or denaturation may account for tegument aggregation to one side of the virion and to a strengthened association with the capsid. Either could eventually attenuate the infectivity of extracellular virus.
Tegument-capsid attachment.
Investigations into the nature of capsid-tegument association have focused on UL36, a large tegument protein (3139 amino acids in length) present in ∼120 copies/virion (8). UL36 is attached to the capsid by way of a noncovalent interaction with UL25, a capsid protein located near the vertices and involved in DNA encapsidation (5, 20, 29, 41, 43). Yeast two-hybrid and immunological analyses have demonstrated that UL36 binds directly to other tegument proteins including UL37, UL41, and UL48, suggesting the tegument may be organized around UL36; other tegument proteins would be bound directly or indirectly to UL36 (13, 44). It is consistent with this idea that UL48 binds to tegument proteins UL41, UL46, UL47, and UL49 (14, 25, 36, 44). Two tegument proteins, UL46 and UL16, have been found to attach reversibly to the capsid, indicating that these may also be involved in linking the tegument to the capsid (21, 22, 26).
Electron microscopic examination of TEV as reported here demonstrated the presence of short connections between capsid and tegument with a spacing of 16.8 ± 4.8 nm (Fig. 5). For comparison, the spacing between adjacent capsomers, adjacent triplexes and adjacent pentons is 15.6, 9.4, and 65.6 nm, respectively (48). The observed connector spacing, therefore, correlates best with attachment involving capsomers. The observed spacing is too small to support the view that connections are exclusively at pentons, as suggested by structural analyses and by attachment of UL36 to UL25 sites near the pentons (3, 5, 42, 47).
The sensitivity of the tegument to trypsin digestion in TEV was determined as an overall measure of the strength of the capsid-tegument contact (Fig. 6). In the past, measurement of trypsin sensitivity has been most useful in demonstrating that whereas the major capsid protein is quite resistant to digestion with a high trypsin concentration (1 mg/ml [37]), much lower concentrations were sufficient to detach pentons or to promote DNA release from C capsids (27, 38).
The results described here indicate that the capsid-tegument connection is quite sensitive to trypsin treatment. All or nearly all of the tegument was removed from TEV after a 30-min digestion with 0.1 μg of trypsin/ml at 37°C, a treatment that was too mild to cause DNA release from the same capsids (Fig. 6). We suggest that protease digestion may be a part of the way the tegument is removed from capsids in infected cells shortly after capsids enter the peripheral cytoplasm. Such digestion could account for the protease activity shown to be required for migration of capsids to the nucleus and DNA uncoating (10).
In the future it will be of interest to continue to probe the interaction between the capsid and tegument with the goal of accounting for how the tegument acquires its resistance to TX-100 extraction. Although many possibilities suggest themselves, it is clear that association must be noncovalent in nature. In the present study it was noted that all capsid-tegument contacts were broken when specimens were prepared for SDS-PAGE, even when analysis was performed with the most TX-100 resistant virions.
Acknowledgments
We thank Anna Maria Copeland and Fred Homa for thoughtful comments about the manuscript. We also thank Rebecca Mingo and Jennifer Thompson for suggestions regarding experimental design and interpretation.
This study was supported by NIH award AI041644.
Footnotes
Published ahead of print on 3 June 2009.
REFERENCES
- 1.Batterson, W., and B. Roizman. 1983. Characterization of the herpes simplex virion-associated factor responsible for the induction of alpha genes. J. Virol. 46371-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell, M. E., J. W. Palfreyman, and C. M. Preston. 1984. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate-early transcription. J. Mol. Biol. 1801-19. [DOI] [PubMed] [Google Scholar]
- 3.Chen, D. H., J. Jakana, D. McNab, J. Mitchell, Z. H. Zhou, M. Dougherty, W. Chiu, and F. J. Rixon. 2001. The pattern of tegument-capsid interaction in the herpes simplex virus type 1 virion is not influenced by the small hexon-associated protein VP26. J. Virol. 7511863-11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chi, J. H., C. A. Harley, A. Mukhopadhyay, and D. W. Wilson. 2005. The cytoplasmic tail of herpes simplex virus envelope glycoprotein D binds to the tegument protein VP22 and to capsids. J. Gen. Virol. 86253-261. [DOI] [PubMed] [Google Scholar]
- 5.Coller, K. E., J. I. Lee, A. Ueda, and G. A. Smith. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 8111790-11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farnsworth, A., T. W. Wisner, and D. C. Johnson. 2007. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J. Virol. 81319-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grunewald, K., P. Desai, D. C. Winkler, J. B. Heymann, D. M. Belnap, W. Baumeister, and A. C. Steven. 2003. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 3021396-1398. [DOI] [PubMed] [Google Scholar]
- 8.Heine, J. W., R. W. Honess, E. Cassai, and B. Roizman. 1974. Proteins specified by herpes simplex virus XII. The virion polypeptides of type 1 strains. J. Virol. 14640-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Homa, F. L., and J. C. Brown. 1997. Capsid assembly and DNA packaging in herpes simplex virus. Rev. Med. Virol. 7107-122. [DOI] [PubMed] [Google Scholar]
- 10.Jovasevic, V., L. Liang, and B. Roizman. 2008. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 823311-3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamen, D. E., S. T. Gross, M. E. Girvin, and D. W. Wilson. 2005. Structural basis for the physiological temperature dependence of the association of VP16 with the cytoplasmic tail of herpes simplex virus glycoprotein H. J. Virol. 796134-6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kattenhorn, L. M., G. A. Korbel, B. M. Kessler, E. Spooner, and H. L. Ploegh. 2005. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 19547-557. [DOI] [PubMed] [Google Scholar]
- 13.Klupp, B. G., W. Fuchs, H. Granzow, R. Nixdorf, and T. C. Mettenleiter. 2002. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 763065-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knez, J., P. T. Bilan, and J. P. Capone. 2003. A single amino acid substitution in herpes simplex virus type 1 VP16 inhibits binding to the virion host shutoff protein and is incompatible with virus growth. J. Virol. 772892-2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loret, S., G. Guay, and R. Lippe. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 828605-8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matusick-Kumar, L., W. W. Newcomb, J. C. Brown, P. J. McCann, W. Hurlburt, S. P. Weinheimer, and M. Gao. 1995. The C-terminal 25 amino acids of the protease and its substrate ICP35 of herpes simplex virus type 1 are involved in formation of sealed capsids. J. Virol. 694347-4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maurer, U. E., B. Sodeik, and K. Grunewald. 2008. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc. Natl. Acad. Sci. USA 10510559-10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKnight, J. L., T. M. Kristie, and B. Roizman. 1987. Binding of the virion protein mediating alpha gene induction in herpes simplex virus 1-infected cells to its cis site requires cellular proteins. Proc. Natl. Acad. Sci. USA 847061-7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLauchlan, J., and F. J. Rixon. 1992. Characterization of enveloped tegument structures (L particles) produced by alphaherpesviruses: integrity of the tegument does not depend on the presence of capsid or envelope. J. Gen. Virol. 73269-276. [DOI] [PubMed] [Google Scholar]
- 20.McNab, A. R., P. Desai, S. Person, L. L. Roof, D. R. Thomsen, W. W. Newcomb, J. C. Brown, and F. L. Homa. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J. Virol. 721060-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meckes, D. G., Jr., and J. W. Wills. 2007. Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J. Virol. 8113028-13036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meckes, D. G., Jr., and J. W. Wills. 2008. Structural rearrangement within an enveloped virus upon binding to the host cell. J. Virol. 8210429-10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 761537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgan, C., H. M. Rose, and B. Mednis. 1968. Electron microscopy of herpes simplex virus. I. Entry. J. Virol. 2507-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukhopadhyay, A., G. E. Lee, and D. W. Wilson. 2006. The amino terminus of the herpes simplex virus 1 protein Vhs mediates membrane association and tegument incorporation. J. Virol. 8010117-10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy, M. A., M. A. Bucks, K. J. O'Regan, and R. J. Courtney. 2008. The HSV-1 tegument protein pUL46 associates with cellular membranes and viral capsids. Virol. 376279-289. [DOI] [PubMed] [Google Scholar]
- 27.Newcomb, W. W., F. P. Booy, and J. C. Brown. 2007. Uncoating the herpes simplex virus genome. J. Mol. Biol. 370633-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newcomb, W. W., F. L. Homa, and J. C. Brown. 2005. Involvement of the portal at an early step in herpes simplex virus capsid assembly. J. Virol. 7910540-10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newcomb, W. W., F. L. Homa, and J. C. Brown. 2006. Herpes simplex virus capsid structure: DNA packaging protein UL25 is located on the external surface of the capsid near the vertices. J. Virol. 806286-6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newcomb, W. W., B. L. Trus, F. P. Booy, A. C. Steven, J. S. Wall, and J. C. Brown. 1993. Structure of the herpes simplex virus capsid: molecular composition of the pentons and the triplexes. J. Mol. Biol. 232499-511. [DOI] [PubMed] [Google Scholar]
- 31.Pellett, P. E., J. L. McKnight, F. J. Jenkins, and B. Roizman. 1985. Nucleotide sequence and predicted amino acid sequence of a protein encoded in a small herpes simplex virus DNA fragment capable of trans-inducing alpha genes. Proc. Natl. Acad. Sci. USA 825870-5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Post, L. E., S. Mackem, and B. Roizman. 1981. Regulation of alpha genes of herpes simplex virus: expression of chimeric genes produced by fusion of thymidine kinase with alpha gene promoters. Cell 24555-565. [DOI] [PubMed] [Google Scholar]
- 33.Rixon, F. J. 1993. Structure and assembly of herpesviruses. Semin. Virol. 4135-144. [Google Scholar]
- 34.Roizman, B. 1996. The function of herpes simplex virus genes: a primer for genetic engineering of novel vectors. Proc. Natl. Acad. Sci. USA 9311307-11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheaffer, A. K., W. W. Newcomb, M. Gao, D. Yu, S. K. Weller, J. C. Brown, and D. J. Tenney. 2001. Herpes simplex virus DNA cleavage and packaging proteins associate with the procapsid prior to its maturation. J. Virol. 75687-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smibert, C. A., B. Popova, P. Xiao, J. P. Capone, and J. R. Smiley. 1994. Herpes simplex virus VP16 forms a complex with the virion host shutoff protein vhs. J. Virol. 682339-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spencer, J. V., B. L. Trus, F. P. Booy, A. C. Steven, W. W. Newcomb, and J. C. Brown. 1997. Structure of the herpes simplex virus capsid: peptide A862-H880 of the major capsid protein is displayed on the rim of the capsomer protrusions. Virol. 228229-235. [DOI] [PubMed] [Google Scholar]
- 38.Steven, A. C., C. R. Roberts, J. Hay, M. E. Bisher, T. Pun, and B. L. Trus. 1986. Hexavalent capsomers of herpes simplex virus type 2: symmetry, shape, dimensions and oligomeric status. J. Virol. 57578-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taddeo, B., M. T. Sciortino, W. Zhang, and B. Roizman. 2007. Interaction of herpes simplex virus RNase with VP16 and VP22 is required for the accumulation of the protein but not for accumulation of mRNA. Proc. Natl. Acad. Sci. USA 10412163-12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taddeo, B., W. Zhang, and B. Roizman. 2006. The U(L)41 protein of herpes simplex virus 1 degrades RNA by endonucleolytic cleavage in absence of other cellular or viral proteins. Proc. Natl. Acad. Sci. USA 1032827-2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thurlow, J. K., M. Murphy, N. D. Stow, and V. G. Preston. 2006. Herpes simplex virus type 1 DNA-packaging protein UL17 is required for efficient binding of UL25 to capsids. J. Virol. 802118-2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trus, B. L., W. Gibson, N. Cheng, and A. C. Steven. 1999. Capsid structure of simian cytomegalovirus from cryoelectron microscopy: evidence for tegument attachment sites. J. Virol. 732181-2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trus, B. L., W. W. Newcomb, N. Cheng, G. Cardone, L. Marekov, F. L. Homa, J. C. Brown, and A. C. Steven. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol. Cell 26479-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vittone, V., E. Diefenbach, D. Triffett, M. W. Douglas, A. L. Cunningham, and R. J. Diefenbach. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 799566-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yeh, P. C., D. G. Meckes, Jr., and J. W. Wills. 2008. Analysis of the interaction between the UL11 and UL16 tegument proteins of herpes simplex virus. J. Virol. 8210693-10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang, Y., and J. L. McKnight. 1993. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 671482-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou, Z. H., D. H. Chen, J. Jakana, F. J. Rixon, and W. Chiu. 1999. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J. Virol. 733210-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou, Z. H., M. Dougherty, J. Jakana, J. He, F. J. Rixon, and W. Chiu. 2000. Seeing the herpesvirus capsid at 8.5 Å. Science 288877-880. [DOI] [PubMed] [Google Scholar]