

Abstract
The precise mechanisms regulating hepatitis C virus (HCV) entry into hepatic cells remain unknown. However, several cell surface proteins have been identified as entry factors for this virus. Of these molecules, claudin-1, a tight junction (TJ) component, is considered a coreceptor required for HCV entry. Recently, we have demonstrated that HCV envelope glycoproteins (HCVgp) promote structural and functional TJ alterations. Additionally, we have shown that the intracellular interaction between viral E2 glycoprotein and occludin, another TJ-associated protein, could be the cause of the mislocalization of TJ proteins. Herein we demonstrated, by using cell culture-derived HCV particles (HCVcc), that interference of occludin expression markedly reduced HCV infection. Furthermore, our results with HCV pseudotyped particles indicated that occludin, but not other TJ-associated proteins, such as junctional adhesion molecule A or zonula occludens protein 1, was required for HCV entry. Using HCVcc, we demonstrated that occludin did not play an essential role in the initial attachment of HCV to target cells. Surface protein labeling experiments showed that both expression levels and cell surface localization of HCV (co)receptors CD81, scavenger receptor class B type I, and claudin-1 were not affected upon occludin knockdown. In addition, immunofluorescence confocal analysis showed that occludin interference did not affect subcellular distribution of the HCV (co)receptors analyzed. However, HCVgp fusion-associated events were altered after occludin silencing. In summary, we propose that occludin plays an essential role in HCV infection and probably affects late entry events. This observation may provide new insights into HCV infection and related pathogenesis.
Hepatitis C virus (HCV) is a small enveloped positive-strand RNA virus that belongs to the Flaviviridae family (20). More than 80% of acute infections become chronic, which eventually progress to cirrhosis and hepatocellular carcinoma (28). HCV infects mainly hepatocytes, but the precise mechanisms of infection are largely unknown (11). The HCV particle consists of a nucleocapsid surrounded by a lipid bilayer in which the two envelope glycoproteins (HCVgp), E1 and E2, are anchored as a heterodimer and play a major role in HCV entry (20). The development of an infectious cell culture model based on the production of infective HCV particles (cell culture-derived HCV particles [HCVcc]) (34) and the generation of HCV pseudotyped retroviral particles (HCVpp) (4) have provided powerful tools to study the HCV cycle. HCV entry is a complex multistep process that requires the presence of several factors. There are multiple pieces of evidence for the involvement of host cell proteins in HCV entry, including glycosaminoglycans, the low-density lipoprotein receptor, scavenger receptor class B type I (SR-BI), and the tetraspanin CD81 (11). Recently, claudin-1, a tight junction (TJ) component, has been identified as a coreceptor required for a late step in HCV entry (13).
TJs are major components of cell-cell adhesion complexes and are composed of integral membrane proteins, including occludin and claudins, which associate with actin cytoskeleton-interacting proteins, such as zonula occludens protein 1 (ZO-1) (2). These structures maintain cell polarity, separating apical from basolateral membrane domains, and form a paracellular barrier that allows the selective passage of certain solutes (2). In hepatocytes, TJs seal the bile canaliculi and form the intercellular barrier between bile and blood (12). Recently, we have shown that TJ-associated proteins occludin and claudin-1 disappeared from their normal localization in both HCV-infected and genomic HCV replicon-containing Huh7 cells. Furthermore, TJ function was also altered in these cells (5). In this matter, we have reported an intracellular interaction between E2 and occludin (5). Moreover, it has been reported that claudin-1 and several TJ-associated proteins, such as coxsackievirus and adenovirus receptor (35) and junctional adhesion molecule (JAM) (3), act as virus (co)receptors. Since coxsackievirus entry across epithelial TJs requires occludin (10), we have explored the role of occludin in HCV infection.
MATERIALS AND METHODS
Cell culture, generation of HCV replicon-containing clones, and HCVcc infection.
Huh7, PLC/PRF/5, and 293T cells and their derivatives were grown at 37°C in a 5% CO2 atmosphere in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 2 mM l-glutamine, 50 μg/ml gentamicin, 100 U/ml penicillin, and 100 μg/ml streptomycin. Huh7 cells expressing full-length or subgenomic genotype 1b (Con1; EMBL database accession number AJ238799) HCV replicons were established as previously described (5). For HCVcc infection assays, cells were grown on 96-well plates (3 × 104 cells/cm2) for 24 h, spin infected with HCV isolate JFH1 (multiplicity of infection of 0.1) (34) at 1,200 × g for 2 h (36) and processed 4 days postinfection for real-time reverse transcription-PCR (RT-PCR) or immunofluorescence (see below).
Transgene and short hairpin RNA (shRNA) retroviral transfer.
Full-length human claudin-1 cDNA (GenBank accession number NM_021101) was cloned into the SwaI site of the pRV-IRES-Neo retroviral expression vector (Genetrix S. L., Madrid, Spain). This construct was employed to generate the 293T-claudin-1 cell line.
cDNA oligonucleotides containing the sequence corresponding to nucleotides 1412 to 1430 of human occludin cDNA (GenBank accession NM_002538) were annealed and inserted between the BglII and HindIII sites of pSUPER.retro.puro and pSUPER.retro.neo+GFP (GFP stands for green fluorescent protein) (Oligoengine, Seattle, WA) according to the manufacturer's instructions. This sequence is identical to nucleotides 1480 to 1498 of canine occludin, which has been reported to be an efficient target for occludin knockdown in MDCK cells (37). Generation of the control shRNA retroviral vector, production of retroviral particles, infection, and puromycin selection of target cells were performed as previously described (5). Occludin silencing in HCV replicon-containing clones was performed by transient transfection with the pSUPER.retro.neo+GFP versions of control and occludin shRNA due to the fact that HCV replication was reduced two- to fourfold after retroviral infection and puromycin selection (our unpublished observations). For this purpose, transfections were performed with 2 μg of the indicated plasmids employing Lipofectin (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. After 24 h of culture, GFP-expressing cells were purified by flow cytometry with a FACSAria cell sorter (BD Biosciences, Franklin Lakes, NJ), and 24 h later, RNA was extracted for real-time RT-PCR analysis (see below).
siRNAs and transfections.
Cells were transfected using the Dharmafect 4 protocol with a mix of two On-Targetplus small interfering RNAs (siRNAs) (100 nM each) directed against human occludin (5′-GAAGAAAGAUGGACAGGUA-3′ and 5′-GUACUGGGGUUCAUGAUUA-3′), JAM-A (5′-GGAUAGUGAUGCCUACGAA-3′ and 5′-CGAGUAAGAAGGUGAUUUA-3′), ZO-1 (5′-GAGAAGAAGUGACCAUAUU-3′ and 5′-CUACACUGAUCAAGAACUA-3′), or a nonspecific control siRNA (Dharmacon, Lafayette, CO). After 48 h, cells were reseeded (3 × 104 cells/cm2) and 24 h later infected with luciferase-based pseudoparticles or lysed to check knockdown efficiency by Western blotting (see below).
HCVcc binding assays.
Cells were grown on 24-well plates (3 × 104 cells/cm2) for 24 h. After the cells were washed with cold culture medium, they were incubated with HCVcc (multiplicity of infection of 0.1) supplemented with 20 mM HEPES for 1 h at 4°C with gentle rocking. Where indicated, heparin (250 μg/ml) was added. Cells were washed three times with ice-cold phosphate-buffered saline (PBS) and processed for RT-PCR (see below).
RT-PCR.
RNA extraction, RT, and quantitative PCR were performed as previously described (5). Occludin-specific primers used were 5′-TGCATGTTCGACCAATGC-3′ and 5′-AAGCCACTTCCTCCATAAGG-3′ (1).
Western blots.
Cells were grown on six-well plates (3 × 104 cells/cm2) for 24 h, lysed on the plate with 100 μl of Laemmli buffer, and boiled for 5 min. Western blotting was carried out as previously described (5) with the following antibodies: polyclonal antibodies antioccludin, anti-claudin-1, anti-ZO-1, anti-JAM-A (Zymed, San Francisco, CA) and anti-SR-BI (Novus Biologicals, Inc., Littleton, CO) and monoclonal antibodies anti-CD81 (clone 5A6) and anti-p53 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA).
Plasmid constructs.
The MLV-GFP, CMV-Gag-Pol, and phCMV-RD (encoding the feline endogenous virus RD114 glycoproteins) vectors and the plasmids coding for HCV E1E2 glycoproteins (genotypes 1a and 1b) have been previously described (4). The pVSV-G construct (encoding the vesicular stomatitis virus [VSV] G protein) was purchased from Clontech Laboratories Inc., Mountain View, CA. The pNL4-3.Luc.R-E- plasmid was obtained through the NIH AIDS Research and Reference Reagent Program (Division of AIDS, NIAID, NIH) from Nathaniel Landau (8, 15). The pXP1LTRwt construct (human immunodeficiency virus type 1 [HIV-1] long terminal repeat [LTR]-luciferase reporter plasmid) has been previously described (14). The pRL-null reporter plasmid was purchased from Promega, Madison, WI.
Production of pseudoparticles and infection assays.
GFP-coding pseudoparticles were generated as previously described (4). Production of luciferase-based pseudoparticles was performed by transfection of 293T cells with equal amounts (10 μg) of pNL4-3.Luc.R-E- and either HCV E1E2, phCMV-RD, pVSV-G, or empty vector by the calcium phosphate method. The medium was replaced 16 h after transfection. Supernatants containing the pseudoparticles were harvested 24 h later, filtered through 0.45-μm-pore-size membranes, and used in infection assays. Target cells were grown on 96-well plates (3 × 104 cells/cm2) for 24 h and infected as previously described (4). 293T-derived cells were seeded on plates coated with poly-l-lysine (Sigma, St. Louis, MO). For GFP-based pseudoparticles, infection was enhanced by spinoculation (centrifugal inoculation) (see above), and the percentage of GFP-positive cells was determined using a FACSCalibur flow cytometer (BD Biosciences). Luciferase activity was measured with the luciferase assay system (Promega) according to the manufacturer's instructions and determined in a Sirius single tube luminometer (Berthold Detection Systems GmbH, Pforzheim, Germany).
Immunofluorescence analysis and confocal microscopy.
Cells were grown on coverslips (3 × 104 cells/cm2) for 24 h and processed for immunofluorescence as previously described (5). 293T-derived cells were seeded on coverslips coated with poly-l-lysine. The antibodies used were the same as for Western blots (see above), monoclonal anti-HCV core (clone C7-50; Affinity BioReagents, Goleen, CO) and polyclonal anti-ZO-1 (Zymed). A monoclonal antioccludin antibody (Zymed) was used for double-label immunofluorescence when needed.
Cell surface protein biotinylation.
A previously described protocol for cell surface protein biotinylation (26) was followed with minor modifications. After 24 h of culture (3 × 104 cells/cm2), cells were washed three times with ice-cold calcium- and magnesium-supplemented phosphate-buffered saline (PBS-CM) (MidiMed, Boussens, France) and incubated with 0.5 mg/ml Sulfo-NHS-SS-Biotin [sulfosuccinimidyl 2-(biotinamido)-ethyl-1, 3-dithiopropionate] (Pierce, Rockford, IL) in PBS-CM for 45 min at 4°C. Cells were washed three times with ice-cold Tris-buffered saline and lysed in buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 1% NP-40, and EDTA-free Halt protease inhibitor cocktail (Pierce) for 30 min on ice. After sonication for 1 min in a Soniprep 150 (MSE Ltd., Crawley, United Kingdom), lysates were cleared by centrifugation (23,440 × g) for 15 min at 4°C, and the protein concentration was measured with the Bio-Rad protein assay (Bio-Rad, Hercules, CA). Cleared lysates (1 mg) were incubated with 100 μl of 50% immobilized NeutrAvidin protein (Pierce) overnight at 4°C with mixing. Samples were centrifuged (1 min, 500 × g), and precipitates were washed three times with ice-cold lysis buffer, resuspended in Laemmli buffer, and boiled for 10 min. Bound proteins were separated on a sodium dodecyl sulfate-polyacrylamide gel and analyzed by Western blotting using the indicated antibodies.
Cell-cell fusion assays.
Cell-cell fusion experiments were performed essentially as previously described (19). 293T cells were transduced with occludin shRNA-coding retroviral particles and selected with puromycin (see above). 293T-occludin shRNA “donor” cells (5 × 105 cells/well seeded into the wells of six-well plates [wells coated with poly-l-lysine 24 h prior to transfection]) were cotransfected using Lipofectamine (Invitrogen) with 100 ng of pXP1LTRwt plasmid and either 2 μg of empty vector, 2 μg of phCMV-H77, or 0.4 μg of pVSV-G plus 1.6 μg of empty vector. After 24 h, transfected cells were reseeded (2 × 104 cells/well) in 24-well plates. Huh7-Tat indicator cells (19) were transfected with control or occludin siRNAs (see above); after 48 h, cells were detached and added (8 × 104 cells/well) to the transfected 293T-occludin shRNA “donor” cells. After 24 h of cocultivation, cells were washed with PBS, incubated for 5 min in citric acid buffer (15 mM citric acid, 150 mM NaCl) at pH 5, and washed three times with medium. The luciferase activity was measured 24 h later as described above. In order to test whether occludin knockdown affected Tat-associated transcriptional activity, control and occludin siRNA-transfected Huh7-Tat cells were seeded in the wells of 24-well plates (6 × 104 cells/well) 24 h after siRNA transfection, cultured overnight, and transiently cotransfected with 20 ng of pXP1LTRwt, 4 ng of pRL-null, and 200 ng of carrier plasmid. Parental Huh7 cells (not expressing Tat) were transfected in parallel and used as a control for Tat-dependent HIV-1 LTR induction. After 48 h, luciferase activity was measured with the dual-luciferase reporter assay system (Promega) according to the manufacturer's instructions.
RESULTS
The TJ-associated protein occludin is relevant to HCV infection.
Recently, it has been suggested that claudin-1 is implicated in HCV infection (13). To evaluate the contribution of the TJ-associated protein occludin to HCV infection, we used a retroviral knockdown approach with control or occludin-specific shRNAs in both Huh7 and PLC/PRF/5 human hepatocyte-derived cell lines. Furthermore, we included 293T-claudin-1 cells, derived from a nonhepatic cell line in which ectopically expressed claudin-1 renders 293T cells susceptible to HCVpp infection (13). Western blot analysis showed that occludin shRNA caused a marked downregulation of occludin expression in all cell lines tested (Fig. 1A). Then, we performed infection assays using HCVcc and evaluated productive infection by real-time RT-PCR. Occludin knockdown sharply reduced the susceptibility of Huh7 and PLC/PRF/5 cells to HCVcc infection compared to cells expressing control shRNA (Fig. 1B). The effect of occludin downregulation on HCVcc infection of 293T-claudin-1 cells could not be evaluated because no detectable levels of viral RNA were found after JFH-1 HCVcc infection (data not shown). This observation is consistent with a previous report and probably reflects the low permissiveness for initiation of HCV replication in 293T cells (32). These data were further confirmed by immunofluorescence analysis of viral protein core accumulation in control or occludin shRNA Huh7 cells (Fig. 1C).
FIG. 1.

HCVcc infection is impaired by occludin knockdown. (A) Occludin was silenced using shRNA technology on Huh7, PLC/PRF/5 (PLC), and 293T-Claudin-1 cells. Occludin knockdown was confirmed by Western blotting using antioccludin and anti-p53 (loading control) antibodies. The positions of molecular mass markers (in kilodaltons) are indicated to the right of the blots. Results are representative of two independent experiments. (B) Cells were infected with HCVcc, and 1-μg RNA samples were analyzed by real-time RT-PCR using specific primers to determine HCV RNA levels. Histone H3 mRNA levels were used for sample normalization. Data are expressed as HCV RNA levels relative to control shRNA-transduced cells. Data are represented as the mean values plus standard deviations (SD) (error bars) from three experiments. (C) HCVcc-infected Huh7 cells were processed for immunostaining using an antibody directed specifically against HCV core protein (green). The merged image with the nuclei stained with 4′,6′-diamidino-2-phenylindole (DAPI) (blue) is shown. Results are representative of two separate experiments. Bars, 250 μm. (D) Occludin was silenced using shRNA technology on Huh7 clones harboring the genomic replicon I389/Core-3′/5.1 (HCV-G1) or the subgenomic replicon I377/NS3-3′ (HCV-NSA). Occludin and HCV RNA levels were determined by real-time RT-PCR as described in Materials and Methods. Data are expressed as the mean values plus SD (error bars) of the results obtained with both clones.
In order to test whether occludin downregulation affects viral replication, we analyzed HCV RNA levels in Huh7 cells harboring genomic (HCV-G1) or subgenomic (HCV-NSA) replicons (5) after occludin knockdown. No differences in HCV RNA levels were found between control and occludin shRNA-expressing replicon-containing cells (Fig. 1D), indicating that HCV replication was not affected by occludin silencing. All together, these findings suggest that occludin expression is essential for HCV infection without affecting HCV replication.
Occludin knockdown impairs HCV entry.
HCVpp carrying envelope proteins from different HCV genotypes have been validated for the study of HCV entry (4). HCVpp consist of full-length HCV envelope glycoproteins assembled onto retroviral core particles containing a retrovirus-derived genome harboring different reporter genes such as those encoding GFP or luciferase (4). To evaluate the contribution of occludin to HCV entry, control or occludin shRNA-transduced cells were challenged with GFP-coding HCVpp. Data showed that occludin knockdown resulted in a reduction of infectivity of both genotype 1a- and 1b-derived HCVpp without affecting VSV pseudotyped particles (VSVpp) or RD114 pseudotyped particles (RD114pp) infection levels (Fig. 2A). These data were further confirmed using luciferase-coding HCVpp (Fig. 2B).
FIG. 2.

Occludin knockdown alters HCV entry. (A) Infectivity of murine leukemia virus-derived GFP-coding pseudoparticles. Control or occludin shRNA-transduced cells were infected with HCVpp (genotypes 1a and 1b), VSVpp, or RD114pp. Data are expressed as percentages of GFP-positive cells relative to control shRNA-transduced cells. The percentage of GFP-positive cells observed with pseudoparticles bearing no envelope proteins (background reading) was subtracted from the values obtained with HCVpp, RD114pp, and VSVpp. Data are represented as the mean values plus standard deviations (SD) (error bars) of the results of three experiments performed in duplicate. (B) Infectivity of HIV-derived luciferase-coding pseudoparticles. Data are expressed relative to control shRNA-transduced cells. Background readings were always less than 2% of the values obtained with pseudoparticles containing envelope proteins. Data are represented as the mean values plus SD of the results of at least four experiments performed in triplicate. RLUs, relative luciferase units. (C) Effects of occludin-, JAM-A-, and ZO-1-specific siRNAs on pseudoparticle infectivity. PLC/PRF/5 cells were transiently transfected with control or specific siRNAs against occludin, ZO-1, and JAM-A. After 48 h, cells were reseeded (3 × 104 cells/cm2), and 24 h later, the cells were infected with luciferase-based pseudoparticles or lysed to check knockdown efficiency by Western blotting using antioccludin, anti-JAM-A, anti-ZO-1, and anti-p53 (loading control) antibodies. Expression levels of claudin-1, SR-BI and CD81 were also monitored. The positions of molecular mass markers (in kilodaltons) are indicated to the right of the blots. Data are expressed relative to control siRNA-transfected cells and represented as the mean values plus SD of the results of two independent experiments performed in triplicate.
The TJ multiprotein complex is composed by integral membrane proteins, such as JAM-A, which serves as a receptor for mammalian reovirus, and occludin, which associates with actin cytoskeleton-interacting proteins like ZO-1 (2). To evaluate the contributions of different TJ-associated proteins to HCV infection, Huh7 and PLC/PRF/5 cell lines were transiently transfected with occludin-, ZO-1-, and JAM-A-specific siRNAs. As expected, occludin knockdown after siRNA transfection resulted in a reduction of infectivity of HCVpp without affecting VSVpp infection levels on PLC/PRF/5 cells (Fig. 2C) or Huh7 cells (data not shown). In contrast, reduction of ZO-1 or JAM-A expression did not affect HCVpp infectivity (Fig. 2C). All together these findings suggest that occludin, but not other TJ-associated proteins, such as JAM-A or ZO-1, is essential for HCV entry.
Occludin knockdown does not affect initial HCV attachment or expression levels and subcellular localization of its (co)receptors.
The HCV cell entry process starts with the binding of the viral particles to the surface of target cells via interactions between HCVgp and specific cell surface receptors (11). We carried out HCVcc binding assays in order to examine whether occludin plays a role in initial cell binding events. Total RNA was extracted after incubating HCVcc with cells for 1 hour at 4°C. As shown in Fig. 3, we found no differences in HCVcc attachment between control and occludin shRNA-transduced cells (Huh7 and PLC/PRF/5 cells). Similar results were obtained by transient transfection of Huh7 cells with control or occludin siRNAs (data not shown). The specificity of HCVcc binding to cells was demonstrated by competition with heparin, a homolog of highly sulfated heparan sulfate, as a control to reduce HCV attachment to the cell surface (Fig. 3). These data suggest that occludin is not involved in the initial attachment of virions to target cells.
FIG. 3.
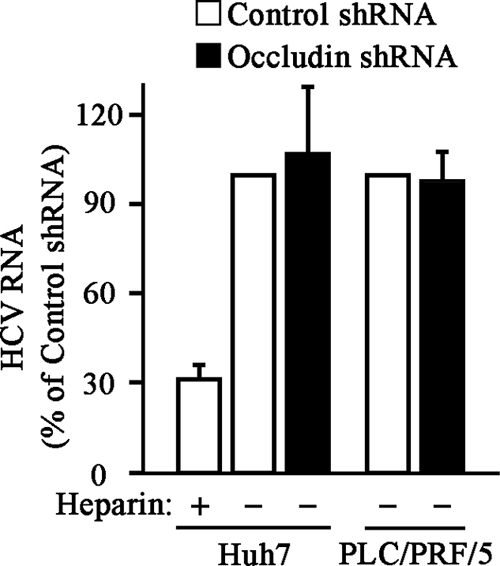
Occludin knockdown does not affect HCV initial attachment to target cells. Control or occludin shRNA-transduced Huh7 or PLC/PRF/5 cells were washed with cold culture medium and incubated with HCVcc for 1 h at 4°C in the presence (+) or absence (−) of heparin (250 μg/ml). Unbound viral particles were removed, and total RNA was extracted. Samples (1 μg) of RNA were analyzed by real-time RT-PCR using specific primers to determine HCV RNA levels. Histone H3 mRNA levels were used for sample normalization. Data are expressed as HCV RNA levels relative to control shRNA-transduced cells in the absence of heparin. Data are represented as the mean values plus standard deviations (error bars) of the results of two independent experiments performed at least in duplicate.
It has been suggested that the precise localization of HCV (co)receptors and their coordinate interactions with different proteins are required for productive HCV infection (11). In this context, we investigated whether occludin silencing affected expression levels and/or cell surface localization of HCV (co)receptors. When Huh7 cells were subjected to biotinylation of cell surface proteins, occludin was efficiently biotinylated and isolated by NeutrAvidin beads, and no occludin was detected when the biotinylation step was omitted (Fig. 4A). As expected, p53 was not detected in the NeutrAvidin precipitates, indicating no intracellular biotinylation (Fig. 4A). Western blot analysis of both total lysates and cell surface biotinylated extracts showed similar levels of HCV (co)receptors SR-BI, CD81, and claudin-1 between control and occludin shRNA-transduced cell lines (Fig. 4B). It is noteworthy that transient transfection of cells with occludin siRNA did not alter expression levels of HCV (co)receptors either (Fig. 2C).
FIG. 4.

Expression levels and cell surface localization of HCV (co)receptors are not altered after occludin knockdown. (A) After Huh7 cells were subjected to biotinylation (+), total lysates and NeutrAvidin-bound proteins obtained from mock-treated or biotin-treated cells were analyzed by Western blotting using antioccludin and anti-p53 antibodies. (B) Control or occludin shRNA-transduced cells were cell surface biotinylated. Western blot analysis of both total lysates and NeutrAvidin-bound proteins was performed using antioccludin, anti-claudin-1, anti-SR-BI, anti-CD81, and anti-p53 antibodies. Data shown are representative of at least two independent experiments. The positions of molecular mass markers (in kilodaltons) are indicated to the right of the blots.
Next we examined by confocal microscopy-based immunofluorescence analysis whether occludin silencing could alter the localization of the different HCV (co)receptors on cell surface, such as CD81 and SR-BI, and TJ-associated proteins claudin-1, occludin, and ZO-1 (Fig. 5). Given that the level of occludin silencing was not the same among all cells, we focused on fields where occludin expression was undetectable. We observed that in both control and occludin shRNA-transduced PLC/PRF/5 cells, claudin-1 and ZO-1 localized at the apical poles of lateral cell membranes, the typical distribution of these proteins in TJ-forming cells. Furthermore, CD81 and SR-BI appeared diffusely distributed across the plasma membrane independently of occludin silencing (Fig. 5). Similar results were obtained with Huh7 and 293T-claudin-1 cells (data not shown). In summary, these data indicate that occludin knockdown did not affect expression levels, cell surface localization, and spatial distribution of the HCV (co)receptors analyzed.
FIG. 5.

Subcellular localization of HCV (co)receptors and TJ-associated protein ZO-1 is not affected by occludin silencing. Control or occludin shRNA-transduced PLC/PRF/5 cells were processed for immunofluorescence with antibodies against occludin, ZO-1, claudin-1, CD81, and SR-BI. Localization of occludin (red) and HCV (co)receptors or ZO-1 (green) was analyzed by immunofluorescence confocal analysis (the merged projection of confocal stacks is shown by the large labeled panel at the top, and the x-z section is shown by the thin panel at the bottom). Nuclei were stained with DAPI (blue). z sections were compiled by taking 0.5-μm steps through each x-y section. In order to clearly show the obtained results, due to the fact that occludin knockdown was not homogeneous among the occludin shRNA-transduced polyclonal cell population, cells with undetectable levels of occludin are shown. Images are representative of two independent experiments. The black arrows indicate the planes corresponding to the x-z sections shown. Bar, 50 μm.
Occludin is implicated in HCV late entry events.
Binding of HCV to target cells is followed by internalization of the viral particle by clathrin-mediated endocytosis (6). It has been established that VSV entry also relies on clathrin-mediated endocytosis (31). Since we observed that occludin shRNA did not induce any changes on VSVpp entry (Fig. 2), it is conceivable that occludin silencing did not affect clathrin-mediated endocytosis. However, in order to further confirm this issue, we examined whether transferrin uptake, which is a clathrin-dependent process (16), was altered after occludin knockdown. Experiments showed that transferrin internalization was not affected in occludin shRNA-transduced Huh7 and 293T-claudin-1 cells (data not shown).
After HCV internalization, fusion has been proposed to occur within early endosomes (25), where the acidic pH triggers the fusion process, probably by inducing conformational changes in the envelope proteins (6, 17, 18, 33). To determine whether occludin is required for HCVgp-mediated membrane fusion, we carried out cell-cell fusion assays. In these experiments, either untransfected Huh7 cells or occludin and control siRNA-transfected Huh7-Tat “indicator” cells (19) were cocultured with 293T-occludin shRNA “donor” cells cotransfected with a HIV-1 LTR-luciferase reporter construct and plasmids coding for either HCV or VSV envelope proteins. Fusion between “donor” and “indicator” cells results in a Tat-mediated transactivation of the HIV-1 LTR measured as luciferase expression. Occludin knockdown in the “donor” cells was performed to avoid cis-interactions with HCVgp and trans-interactions with occludin expressed in the “indicator” cells. For similar reasons, we decided not to express claudin-1 in 293T “donor” cells. As expected, coculture of HCVgp-expressing “donor” cells with control siRNA-transfected Huh7-Tat cells produced almost 100-fold-more luciferase activity than parental Huh7 cells did (Fig. 6A), demonstrating effective cell-cell fusion and Tat-mediated transactivation of the HIV-1 LTR, thus validating the experimental setup. Interestingly, when HCVgp-expressing “donor” cells were used, occludin knockdown of Huh7-Tat cells resulted in a decrease of luciferase activity, indicating an impairment of HCVgp-dependent cell fusion (Fig. 6A). The specificity of occludin effects on HCVgp-mediated fusion was demonstrated by the fact that occludin knockdown of Huh7-Tat cells did not alter the fusion with “donor” cells expressing the VSV envelope protein (Fig. 6A). Occludin knockdown in Huh7-Tat cells was confirmed by Western blotting 48, 72, and 96 h after siRNA transfection, time points corresponding to the beginning of the coculture, the acid wash, and the measurement of luciferase activity, respectively (data not shown and Fig. 6B). Additionally, in order to rule out the possibility that occludin silencing affected Tat-associated transcriptional activity, control and occludin siRNA-transfected Huh7-Tat cells were transiently transfected with the HIV-1 LTR reporter plasmid, showing similar luciferase activities (Fig. 6B). All together, these results indicate that occludin is required for HCVgp-dependent cell fusion.
FIG. 6.

Occludin knockdown affects HCVgp-mediated cell fusion. (A) 293T occludin-shRNA “donor” cells cotransfected with either empty vector, HCVgp, or VSVgp and a HIV-1 LTR-luciferase reporter construct were cocultured with control or occludin siRNA-transfected Huh7-Tat “indicator” cells. Parental Huh7 cells (not expressing Tat) were used as a control for Tat-dependent HIV-1 LTR induction. After 24 h, cells were treated at pH 5 for 5 min, and the luciferase activity induced by the fusion between “donor” and “indicator” cells was measured 24 h later. Unspecific fusion, measured as the luciferase activity obtained when using “donor” cells expressing no envelope proteins, was less than 15% of the values obtained with HCVgp (data not shown). The level of occludin knockdown in 293T-occludin shRNA “donor” cells was similar to that shown in Fig. 1 for shRNA-transduced 293T-claudin-1 cells (data not shown). Data are expressed relative to control siRNA-transfected Huh7-Tat cells and represented as the mean values plus standard deviations (SD) (error bars) of an experiment performed in triplicate. Results are representative of three independent experiments. RLUs, relative luciferase units. (B) Forty-eight hours after transfection of Huh7-Tat cells with control or occludin siRNA, cells were cotransfected with the HIV-1 LTR-luciferase reporter construct and the reporter plasmid pRL-null, which bears a promoterless Renilla luciferase gene and serves as control for transfection efficiency. Parental Huh7 cells were also included in the experiment. After 48 h of further culture, knockdown efficiency was checked by Western blotting and luciferase activity was measured. The positions of molecular mass markers (in kilodaltons) are shown to the right of the blots. Renilla-normalized data are expressed relative to control siRNA-transfected Huh7-Tat cells and represented as the mean values plus SD of an experiment performed in triplicate. Results are representative of three independent experiments.
DISCUSSION
The most striking finding of this study is that the TJ-associated protein occludin is essential for HCV infection of several cell lines using both HCVcc and HCVpp. These data, along with the fact that occludin knockdown did not affect HCV replication, strongly suggest that occludin plays an essential role in HCV entry.
HCV entry is initiated by the binding of the particle to an attachment factor, which helps to concentrate viruses on the cell surface. After the initial attachment to the target cell, the virus binds to specific entry factors which are responsible for initiating a series of events that eventually lead to viral entry (11). Our data suggest that occludin is not involved in the initial attachment of virions to target cells, an infection step which probably depends on other virus (co)receptors, such as glycosaminoglycans and lipoprotein receptors.
Currently, it seems that HCV entry requires an unexpectedly large group of cellular (co)receptors, including SR-BI, CD81, and claudin-1 implicated at the postbinding stage (11). Our experiments suggest that the impairment of HCV entry after occludin knockdown was not due to a reduction of cell surface expression or spatial redistribution of the HCV (co)receptors analyzed. Furthermore, since occludin shRNA did not induce any changes in VSVpp infection and transferrin uptake, our data suggested that occludin knockdown did not affect clathrin-mediated endocytosis. Taken together, these findings suggest a rather direct and specific role of occludin in HCV entry.
HCV internalization is followed by membrane fusion, which likely occurs in early endosomes (25). HCVgp are resistant to inactivation by low pH (33), suggesting that HCVgp pH sensitivity occurs during cell entry probably mediated by cell surface molecules which could trigger conformational rearrangements and/or promote acid pH sensitivity of HCVgp. Occludin knockdown promoted a significant reduction of HCVgp-dependent cell-cell fusion, indicating its possible role in the fusion process or at an earlier step required to render the virus competent for low-pH-triggered entry. Interestingly, it has been shown that claudin-1 is required for HCVgp-dependent cell fusion (13), suggesting that the entry steps mediated by occludin and claudin-1 might involve similar mechanisms which could take place at the TJ. However, the relevance of TJs on HCV infection is still controversial and a matter of debate. After TJ disruption by calcium depletion, a significantly increased viral entry in Caco-2 cells (24) and an impaired HCV infection in Huh7 cells (7) have been observed. Our experiments showed that localization of TJ-associated proteins ZO-1 and claudin-1 was not significantly affected by occludin silencing. Furthermore, HCVpp infectivity was not altered by ZO-1 and JAM-A knockdown, suggesting that TJ function itself may not be required for HCV cell entry. However, further functional experiments will be required to determine whether TJs are necessary for HCV infection and whether occludin knockdown alters hepatocyte TJ function.
A number of studies suggest that viruses move laterally on the plasma membrane before being internalized (22). These movements take place until the viruses have engaged sufficient receptors to initiate signaling events required for internalization. In the case of HCV, it has been suggested that the virus exploits CD81-mediated lateral migration to move to the TJs where the (co)receptor claudin-1 specifically localizes (7). A similar event has been demonstrated for the human coxsackievirus, whose binding on the apical surface of polarized cells triggers intracellular signals that permit virus to move to the TJ where it interacts with its primary receptor, coxsackievirus and adenovirus receptor, to enter target cells (9). Emerging evidence indicates that TJs are not an absolute and static barrier but rather a very dynamic cellular structure (29). In particular, occludin is recycled continuously even in epithelial cells with intact cell-cell contacts (30). Interestingly, the basolateral membrane is an obligatory intermediate in the transport of occludin to TJs (23). After HCV attachment, which presumably takes place on the basolateral membrane of hepatocytes in contact with the sinusoidal blood, occludin could be implicated in viral movement to the site where internalization occurs. Our previous recent results demonstrated an intracellular interaction between E2 and occludin (5). Thereby, it is plausible that the E2/occludin association could also take place extracellularly during a postbinding step probably to promote the subsequent viral internalization. Further studies are necessary to confirm this hypothesis.
It is important to point out that Liu and colleagues have also recently demonstrated that occludin participates in HCV infection (21). Furthermore, it has recently been shown that human occludin is an essential HCV cell entry factor, providing an important advance toward developing mouse models for HCV infection (27). Our data contribute important mechanistic details of the role of occludin in late steps of HCV entry. All together, these observations may provide new insights into HCV tropism, infection, and spreading that could help in understanding HCV-related pathogenesis.
Acknowledgments
This work was supported in part by the following grants: (i) a grant from CIBER-ehd to R. Moreno-Otero, M. López-Cabrera, and P. Majano; (ii) grant SAF2007-61201 from the Ministerio de Educación y Ciencia to M. López-Cabrera; (iii) grant CP 03/0020 from Instituto Salud Carlos III; (iv) grant SAF2007-60667 from the Ministerio de Educación y Ciencia to P. Majano; and (v) a grant from the European Research Council (ERC-2008-AdG-233130 “HEPCENT”) to F.-L. Cosset. I. Benedicto was financially supported by CIBER-ehd, and F. Molina-Jiménez was supported by ISCIII and FIB Hospital Universitario de la Princesa.
We express our gratitude to R. Bartenschlager, E. Muñoz, and T. Wakita for providing us with critical reagents. We also thank R. Samaniego for his technical assistance in confocal microscopy experiments.
Footnotes
Published ahead of print on 10 June 2009.
REFERENCES
- 1.Abe, T., E. Sugano, Y. Saigo, and M. Tamai. 2003. Interleukin-1beta and barrier function of retinal pigment epithelial cells (ARPE-19): aberrant expression of junctional complex molecules. Investig. Ophthalmol. Vis. Sci. 444097-4104. [DOI] [PubMed] [Google Scholar]
- 2.Aijaz, S., M. S. Balda, and K. Matter. 2006. Tight junctions: molecular architecture and function. Int. Rev. Cytol. 248261-298. [DOI] [PubMed] [Google Scholar]
- 3.Barton, E. S., J. C. Forrest, J. L. Connolly, J. D. Chappell, Y. Liu, F. J. Schnell, A. Nusrat, C. A. Parkos, and T. S. Dermody. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104441-451. [DOI] [PubMed] [Google Scholar]
- 4.Bartosch, B., J. Dubuisson, and F. L. Cosset. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197633-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benedicto, I., F. Molina-Jimenez, O. Barreiro, A. Maldonado-Rodriguez, J. Prieto, R. Moreno-Otero, R. Aldabe, M. Lopez-Cabrera, and P. L. Majano. 2008. Hepatitis C virus envelope components alter localization of hepatocyte tight junction-associated proteins and promote occludin retention in the endoplasmic reticulum. Hepatology 481044-1053. [DOI] [PubMed] [Google Scholar]
- 6.Blanchard, E., S. Belouzard, L. Goueslain, T. Wakita, J. Dubuisson, C. Wychowski, and Y. Rouille. 2006. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 806964-6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brazzoli, M., A. Bianchi, S. Filippini, A. Weiner, Q. Zhu, M. Pizza, and S. Crotta. 2008. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J. Virol. 828316-8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connor, R. I., B. K. Chen, S. Choe, and N. R. Landau. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206935-944. [DOI] [PubMed] [Google Scholar]
- 9.Coyne, C. B., and J. M. Bergelson. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124119-131. [DOI] [PubMed] [Google Scholar]
- 10.Coyne, C. B., L. Shen, J. R. Turner, and J. M. Bergelson. 2007. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2181-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubuisson, J., F. Helle, and L. Cocquerel. 2008. Early steps of the hepatitis C virus life cycle. Cell. Microbiol. 10821-827. [DOI] [PubMed] [Google Scholar]
- 12.Easter, D. W., J. B. Wade, and J. L. Boyer. 1983. Structural integrity of hepatocyte tight junctions. J. Cell Biol. 96745-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans, M. J., T. von Hahn, D. M. Tscherne, A. J. Syder, M. Panis, B. Wolk, T. Hatziioannou, J. A. McKeating, P. D. Bieniasz, and C. M. Rice. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446801-805. [DOI] [PubMed] [Google Scholar]
- 14.Gomez-Gonzalo, M., M. Carretero, J. Rullas, E. Lara-Pezzi, J. Aramburu, B. Berkhout, J. Alcami, and M. Lopez-Cabrera. 2001. The hepatitis B virus X protein induces HIV-1 replication and transcription in synergy with T-cell activation signals: functional roles of NF-kappaB/NF-AT and SP1-binding sites in the HIV-1 long terminal repeat promoter. J. Biol. Chem. 27635435-35443. [DOI] [PubMed] [Google Scholar]
- 15.He, J., S. Choe, R. Walker, P. Di Marzio, D. O. Morgan, and N. R. Landau. 1995. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 696705-6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirst, J., and M. S. Robinson. 1998. Clathrin and adaptors. Biochim. Biophys. Acta 1404173-193. [DOI] [PubMed] [Google Scholar]
- 17.Hsu, M., J. Zhang, M. Flint, C. Logvinoff, C. Cheng-Mayer, C. M. Rice, and J. A. McKeating. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 1007271-7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koutsoudakis, G., A. Kaul, E. Steinmann, S. Kallis, V. Lohmann, T. Pietschmann, and R. Bartenschlager. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 805308-5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lavillette, D., E. I. Pecheur, P. Donot, J. Fresquet, J. Molle, R. Corbau, M. Dreux, F. Penin, and F. L. Cosset. 2007. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J. Virol. 818752-8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindenbach, B. D., and C. M. Rice. 2005. Unravelling hepatitis C virus replication from genome to function. Nature 436933-938. [DOI] [PubMed] [Google Scholar]
- 21.Liu, S., W. Yang, L. Shen, J. R. Turner, C. B. Coyne, and T. Wang. 2009. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J. Virol. 832011-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marsh, M., and A. Helenius. 2006. Virus entry: open sesame. Cell 124729-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matter, K., and M. S. Balda. 1998. Biogenesis of tight junctions: the C-terminal domain of occludin mediates basolateral targeting. J. Cell Sci. 111511-519. [DOI] [PubMed] [Google Scholar]
- 24.Mee, C. J., J. Grove, H. J. Harris, K. Hu, P. Balfe, and J. A. McKeating. 2008. Effect of cell polarization on hepatitis C virus entry. J. Virol. 82461-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meertens, L., C. Bertaux, and T. Dragic. 2006. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J. Virol. 8011571-11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morimoto, S., N. Nishimura, T. Terai, S. Manabe, Y. Yamamoto, W. Shinahara, H. Miyake, S. Tashiro, M. Shimada, and T. Sasaki. 2005. Rab13 mediates the continuous endocytic recycling of occludin to the cell surface. J. Biol. Chem. 2802220-2228. [DOI] [PubMed] [Google Scholar]
- 27.Ploss, A., M. J. Evans, V. A. Gaysinskaya, M. Panis, H. You, Y. P. de Jong, and C. M. Rice. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457882-886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poynard, T., M. F. Yuen, V. Ratziu, and C. L. Lai. 2003. Viral hepatitis C. Lancet 3622095-2100. [DOI] [PubMed] [Google Scholar]
- 29.Shen, L., C. R. Weber, and J. R. Turner. 2008. The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. J. Cell Biol. 181683-695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian, V. S., J. S. Marchant, D. Ye, T. Y. Ma, and H. M. Said. 2007. Tight junction targeting and intracellular trafficking of occludin in polarized epithelial cells. Am. J. Physiol. Cell Physiol. 293C1717-C1726. [DOI] [PubMed] [Google Scholar]
- 31.Sun, X., V. K. Yau, B. J. Briggs, and G. R. Whittaker. 2005. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology 33853-60. [DOI] [PubMed] [Google Scholar]
- 32.Timpe, J. M., Z. Stamataki, A. Jennings, K. Hu, M. J. Farquhar, H. J. Harris, A. Schwarz, I. Desombere, G. L. Roels, P. Balfe, and J. A. McKeating. 2008. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 4717-24. [DOI] [PubMed] [Google Scholar]
- 33.Tscherne, D. M., C. T. Jones, M. J. Evans, B. D. Lindenbach, J. A. McKeating, and C. M. Rice. 2006. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 801734-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walters, R. W., P. Freimuth, T. O. Moninger, I. Ganske, J. Zabner, and M. J. Welsh. 2002. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 110789-799. [DOI] [PubMed] [Google Scholar]
- 36.Ye, L., X. Wang, S. Wang, G. Luo, Y. Wang, H. Liang, and W. Ho. 2008. Centrifugal enhancement of hepatitis C virus infection of human hepatocytes. J. Virol. Methods 148161-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu, A. S., K. M. McCarthy, S. A. Francis, J. M. McCormack, J. Lai, R. A. Rogers, R. D. Lynch, and E. E. Schneeberger. 2005. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. Am. J. Physiol. Cell Physiol. 288C1231-C1241. [DOI] [PubMed] [Google Scholar]