

Abstract
Although enveloped virus assembly in the host cell is a crucial step in the virus life cycle, it remains poorly understood. One issue is how viruses include lipids in their membranes during budding from infected host cells. To analyze this issue, we took advantage of the fact that baby hamster kidney cells can be infected by two different viruses, namely, vesicular stomatitis virus and Semliki Forest virus, from the Rhabdoviridae and Togaviridae families, respectively. We purified the host plasma membrane and the two different viruses after exit from the host cells and analyzed the lipid compositions of the membranes by quantitative shotgun mass spectrometry. We observed that the lipid compositions of these otherwise structurally different viruses are virtually indistinguishable, and only slight differences were detected between the viral lipid composition and that of the plasma membrane. Taken together, the facts that the lipid compositions of the two viruses are so similar and that they strongly resemble the composition of the plasma membrane suggest that these viruses exert little selection in including lipids in their envelopes.
Enveloped viruses acquire their lipid envelope from the membranes of host cells (43). In this process, the nucleocapsid or the nucleocapsid-matrix complex of the viruses buds out of the cell and becomes enveloped by a segment of the host membrane. This membrane segment is modified during the budding process, such that virally encoded membrane proteins are included in the viral envelope, while most host proteins are excluded. Since viruses usually do not carry lipid-synthesizing enzymes, the lipids in the viral envelope are derived from the host membrane. The lipid compositions of enveloped viruses have been studied for years (2, 15, 17, 18, 23, 25, 34, 36, 38, 40). One question that remains to be answered is whether the lipids are included passively, and thus the lipid composition of the envelope reflects the lipid composition of the host membrane, or whether lipid sorting occurs, leading to selective inclusion of some lipids and exclusion of others. This issue has been complicated by the fact that the lipid bilayer is no longer considered a homogenous liquid but contains fluctuating nanoscale assemblies of sphingolipids, saturated phospholipids, cholesterol (Chol), and proteins, called lipid rafts (13, 44). Lipid rafts can be induced to coalesce—usually by protein-protein interactions—into larger, dynamic platforms that function in signal transduction, intracellular membrane transport, and other membrane functions (45). It was also proposed that viruses make use of these membrane domains during their exit from cells (29, 32).
A major complication in comparing viral envelopes with host cell membranes is the difficulty in obtaining host cell membranes of purity similar to that of the easily purified viruses. Many studies are faulted by the impurity of the cell membranes analyzed. Moreover, the early work in this field employed conventional analytical methods (such as thin-layer chromatography) that provide only semiquantitative estimates of the total abundance of the major lipid classes. Most importantly, lipid species diversity could not be analyzed. Recent developments in mass spectrometry (MS) have enabled comprehensive and quantitative analyses of lipidomes at the level of individual molecular species. The lipidomes of human immunodeficiency virus (HIV), murine leukemia virus (6, 7), and several bacteriophages (20, 21) were recently analyzed by these new methods.
This paper focuses on two well-characterized enveloped viruses, Semliki Forest virus (SFV) and vesicular stomatitis virus (VSV). SFV is an RNA virus belonging to the Togaviridae family of the Alphaviridae that acquires its envelope by budding from the host cell plasma membrane (PM) (46). Early studies analyzed the lipid composition of the viral envelope and also that of the host cell PM (39, 40). These studies revealed strong similarity between the envelope of SFV and the host PM, but one important discrepancy was the higher Chol-to-phospholipid ratio in the virus.
VSV is an RNA virus belonging to the Rhabdoviridae family and also hijacks its envelope from the host cell PM (35), but the lipid specificity of the budding process remains controversial. The most recent studies claim that VSV buds from localized regions that do not reflect the average composition of the PM (23, 36). It has also been claimed that lipid rafts are involved in VSV envelope assembly during budding (37).
We used BHK-21 cells as host cells to purify SFV and VSV. The purposes of this study were (i) to establish a robust, comprehensive, and quantitative method to analyze lipidomes, including the full complement of glycerolipid, glycerophospholipid, and sphingolipid species as well as Chol; (ii) to establish a protocol for purification of PM suitable for MS analysis; and (iii) to analyze and compare the lipidomes of SFV, VSV, and the BHK-21 PM.
We found that the lipidomes of SFV and VSV are similar in molecular composition and are closely related to that of the BHK-21 PM. The small differences observed could be explained by the high degrees of curvature generated during the viral budding process.
MATERIALS AND METHODS
Media, buffers, and organic solvents.
All common chemicals were purchased from Sigma Chemicals (St. Louis, MO) and were of the best analytical grade. Minimal essential medium was from PAA, Pashing, Austria. Penicillin-streptomycin, glutamate, and fetal calf serum were obtained from Gibco, and 100× nonessential amino acids were obtained from Sigma. All buffer components (HEPES, Tris, and inorganic salts) were proanalytica grade. Water for MS, methanol (both LiChrosolv grade), and chloroform (liquid chromatography [LC] grade) were from Merck (Darmstadt, Germany). Synthetic lipid standards were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). The GM3 bovine mixture was purchased from Matreya Biochemicals, LLC (Pleasant Gap, PA). Phosphatidylinositol (PI) 17:0-17:0 was provided by Christoph Thiele (Max Planck Institute of Molecular Cell Biology and Genetics).
Beads, antibodies, and other reagents.
Dynabeads M-500 subcellular beads were purchased from Invitrogen and conjugated to neutravidin protein from Pierce (Rockford, IL) according to the manufacturer's instructions, except that gelatin was used for blocking rather than bovine serum albumin, which would bind lipids. Conjugated beads were stored in 0.1% gelatin at 4°C. Ultralink monomeric avidin Sepharose and EZ-Link sulfo-NHS-LC-biotin were purchased from Pierce. Anti-calnexin antibody (rabbit polyclonal) was from Sigma and was used at a 1:2,000 dilution. Horseradish peroxidase (HRP)-conjugated ExtrAvidin was also from Sigma and was used at 1:500. OptiPrep density medium was from AxisShield Laboratories, latrunculin A was from Calbiochem, and molecular size markers for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were purchased from Invitrogen.
Cells and viruses.
The BHK-21 cells used in this study are indexed as BHK-21 CCL10 in the ATCC collection. The cells were maintained in minimal essential medium (Rochester, NY) supplied with 5% fetal calf serum, penicillin-streptomycin, glutamate, and nonessential amino acids and were passaged every 2 to 3 days. The VSV and SFV stocks were produced as described previously (16, 24).
Cell infection and virus production and purification.
The optimal amount of virus and duration of virus production were determined by immunofluorescence using anti-SFV and anti-VSV-glycoprotein antibodies. Our aim was to use the smallest possible amount of virus to have about 80% of cells infected in the first round.
For large-scale virus production for lipid analysis, we used five T175 flasks for the production of SFV and VSV.
Virus was diluted in virus production medium (minimal essential medium supplied with glutamate and 20 mM HEPES), and 5 ml was used for each T175 flask of phosphate-buffered saline (PBS)-washed cells. Infection was carried out for 1 h at 37°C and 5% CO2. The virus-containing medium was then removed, and virus production medium plus nonessential amino acids was added (15 ml/T175 flask). Cells were incubated for 17 h at 37°C and 5% CO2.
The medium was collected and subjected to 5 min of centrifugation at 300 × g. The supernatant was transferred to a fresh tube and centrifuged for 5 min at 1,000 × g, and the supernatant obtained thereafter was centrifuged for 30 min at 10,000 × g. The resulting supernatant was transferred to an ultraclear SW28 tube (Beckman, Fullerton, CA) and underlayered, using a syringe with a long needle, with 3 to 5 ml 20% (wt/vol) sucrose in HEPES buffer. Centrifugation was carried out for 3 h in an SW28 rotor at 27.5 krpm at 4°C. The supernatant, including the sucrose cushion, was decanted, and all liquid was carefully removed by drying the walls of the tubes with a tissue. The pellets were then resuspended overnight at 4°C after adding 80 μl HEPES-buffered saline (HBS) buffer and sealing the tubes with Parafilm. The next day, the resuspended pellets from the same samples were combined (the total volume to be loaded on one gradient must not exceed 250 μl) and loaded onto a velocity gradient. The gradients were prepared in ultraclear tubes by the underlayering technique, starting from the top. The layers were as follows: 650 μl of 7.5% and 650 μl 10% Optiprep in HBS buffer, followed by 500-μl layers in 1.25% Optiprep steps, i.e., 11.25, 12.5, …27.5%. The gradients were sealed and linearized by overnight incubation at 4°C.
The gradients were centrifuged for 25 to 30 min at 26.5 krpm in an SW40 rotor at 4°C. After the spin, the virus bands were collected by fractionating the gradient in 1-ml fractions from the top; the virus band was collected in the smallest possible volume, typically 800 to 1,000 μl, and subsequently diluted approximately 10× with HBS and pelleted for 3 h at 27 krpm and 4°C (SW40 rotor). The resulting pellets were washed again in 1 ml HBS, using a TLA55 rotor in a tabletop centrifuge, for 45 min at 38 krpm and 4°C. The final pellets were washed (TLA55 rotor) with 150 mM ammonium bicarbonate buffer and finally resuspended in 100 μl ammonium bicarbonate buffer and subjected to lipid extraction and analysis.
Cell surface biotinylation and purification of PM.
Cells were grown for 2 to 3 days to ∼80 to 90% confluence; cells from 350 to 700 cm2 were used for each preparation and analysis by MS. Culture flasks were moved to 4°C and allowed to cool for ∼10 min prior to biotinylation to prevent internalization of surface components. Cells were washed twice with ice-cold PBS, and 1 mg/ml NHS-sulfo-LC-biotin in PBS, pH 8, was added (∼0.02 ml/cm2). Biotinylation was allowed to proceed for 20 min with gentle rocking at 4°C. The biotin solution was then removed, and cells were washed twice with PBS, once with 0.1 M glycine to quench any remaining unreacted biotinylation reagent, and finally once with PBS. Cells were then scraped into 20 mM Tris, pH 8.35-200 mM sucrose-0.2 mM MgCl2 (scraping buffer; used at ∼0.01 ml/cm2) and either directly processed or frozen at −80°C.
Cell clumps were disrupted by being pipetted up and down against the flask or tube walls, and neutravidin-conjugated Dynabeads M-500 subcellular beads (in PBS plus 0.1% gelatin from fish skin) were added (∼0.5 μl at 4 × 108 beads/ml per cm2 of cells). The suspension was mixed, and binding was allowed to proceed at 4°C with mixing for 20 to 30 min. Binding can be assessed by inspecting a small amount of sample under a microscope. Ideally, all cells should have a few beads bound to the surface. Excess beads must be avoided (cells completely covered with beads appear as “grapes”; these armored structures do not break easily and would contaminate the preparation). Beads with bound cells were then removed from the suspension by means of a magnet or magnetic rack (it is essential to work with small volumes to be able to perform this separation quickly to prevent sedimentation of unbound cells). The beads with bound cells were washed twice with scraping buffer before the addition of hypoosmotic buffer (“shocking buffer”; 20 mM Tris pH 9.1-25 mM sucrose-0.2 mM MgCl2). The samples were incubated for ∼10 min at 4°C with mixing; this treatment leads to osmotic swelling, eventually causing the cells to burst. Shocking buffer was exchanged and supplied with 250 nM latrunculin A and allowed to work during a short (∼2 to 5 min) incubation at room temperature with mixing. After this treatment, almost no whole cells should be visible under a microscope. The beads were then washed with 0.2 M NaBr-0.2 M KCl-50 mM Tris, pH 6.8-250 nM latrunculin A and transferred to a new tube, followed by a quick wash with 0.1 M Na2CO3, pH 11; this treatment reveals any contamination with whole cells and nuclei—the alkaline carbonate disrupts the endoplasmic reticulum (ER) and causes the nuclei to form “slimy” clumps that can be removed with a pipette or by transferring the rest of the suspension to a new tube. The beads were then washed once with 0.1 M NaBr-0.1 M KCl-25 mM Tris, pH 6.8, and once with freshly prepared 150 mM NH4HCO3, pH 8, and finally resuspended in 200 μl 150 mM NH4HCO3 buffer and analyzed by Western blotting and MS.
The quality and yield of PM preparations depend mostly on the efficiency of binding of the beads to the cell surface. When too few beads bind, the yield is too low, and then intracellular membranes may contaminate the resulting sample material. In contrast, when too many beads bind to the cells, the cells are more difficult to lyse. Thus, the resulting preparations may be contaminated with whole cells. The quality of the beads (affinity and blocking) and the amount of beads used are therefore crucial parameters influencing the quality of the obtained PM. We tried different kinds of magnetic beads for conjugation of the biotin-binding proteins. The best results were obtained with Dynabeads M-500 subcellular neutravidin-conjugated beads (other beads that we tried included MyOne T1 streptavidin beads and Dynabeads M450 beads). Freshly conjugated beads suspended in 0.1% gelatin were found to yield better results than those obtained with beads that had been stored. It is critically important to cool the cells to 4°C before starting with surface biotinylation to prevent endocytosis of the biotinylated PM and to strictly carry out all subsequent steps on intact cells at 4°C. It is also important to transfer the beads with bound PM to a fresh tube for the final washing steps. The walls of standard polypropylene microtubes tend to become coated with cell debris, which would later contaminate the lipid extracts. The resulting material is irreversibly bound to the magnetic beads via the biotin-neutravidin interaction and hence is useful only for analysis that makes use of solubilization, harsh elution, or extraction of the bound material (solubilization by SDS-PAGE loading buffer or extraction with organic solvents). However, for our purpose, to analyze the lipidome after organic solvent extraction, the method was optimal.
Lipid extraction and MS.
Purified viruses and PM were subjected to a quantitative shotgun lipidomic analysis. In short, samples were spiked with 10 μl of internal standard lipid mixture, providing a total spike of 20 pmol diacylglycerol (DAG) 17:0-17:0, 24 pmol phosphatidic acid (PA) 17:0-17:0, 52 pmol phosphatidylethanolamine (PE) 17:0-17:0, 7.5 pmol phosphatidylglycerol (PG) 17:0-17:0, 43 pmol phosphatidylserine (PS) 17:0-17:0, 40 pmol phosphatidylcholine (PC) 18:3-18:3, 54 pmol PI 17:0-17:0, 10 pmol ceramide (Cer) 18:0;3/18:0;0, 40 pmol sphingomyelin (SM) 18:1;2/17:0, 53 pmol GM3 bovine mixture, 20 pmol GalCer 18:1;2/12:0, 20 pmol LacCer 18:1;2/12:0, and 50 pmol Chol-d7. Samples were subsequently subjected to lipid extraction executed at 4°C (9). Briefly, approximately 10 μg of protein (as determined by Bradford assay) from each sample was dissolved in 200 μl of 155 mM ammonium bicarbonate and then extracted with 1 ml chloroform-methanol (10:1) for 2 h. The lower, organic phase was collected, and the aqueous phase was reextracted with 1 ml chloroform-methanol (2:1) for 1 h. The lower, organic phase was collected and evaporated in a vacuum desiccator at 4°C. Lipid extracts were dissolved in 100 μl chloroform-methanol (1:2 [vol/vol]) and subjected to quantitative lipid analysis on both a hybrid QSTAR Pulsar i quadrupole time-of-flight mass spectrometer (MDS Sciex, Concord, Canada) and an LTQ Orbitrap instrument equipped with a TriVersa NanoMate robotic nanoflow ion source (Advion Biosciences, Inc., Ithaca, NJ). DAG, PA, PS, PE, PI, and PG species were quantified by negative-ion-mode multiple-precursor ion scanning analysis (8); PC and SM species were quantified by positive-ion-mode precursor ion scan m/z 184.1. Cer, glucosylceramide-plus-galactosylceramide (HexCer), and lactosylceramide-plus-digalactosylceramide (diHexCer) species were quantified by positive-ion-mode Fourier transform MS analysis on an LTQ Orbitrap instrument. GM3 species were quantified by negative-ion-mode Fourier transform MS analysis on an LTQ Orbitrap instrument. Chol was quantified after chemical acetylation in positive ion mode by multiple-reaction monitoring (22). Automated processing of acquired mass spectra and identification and quantification of detected molecular lipid species were performed by Lipid Profiler software (MDS Sciex, Concord, Canada) (8) and Pecoder software, developed in-house (42).
Lipid species were annotated by their molecular composition. Glycerophospholipid and DAG species are annotated as follows: lipid class followed by the number of carbon atoms in the first FA moiety:number of double bonds in the first FA moiety-number of carbon atoms in the second FA moiety:number of double bonds in the second FA moiety (e.g., PI 16:0-18:1). PC species are annotated as follows: lipid class followed by the number of carbon atoms in the first FA moiety plus number of carbon atoms in the second FA moiety:number of double bonds in the first FA moiety plus number of double bonds in the second FA moiety (e.g., PC 34:1). Sphingolipid species are annotated as follows: lipid class followed by the number of carbon atoms in the long-chain base moiety plus number of carbon atoms in the fatty acid moiety:number of double bonds in the long-chain base moiety plus number of double bonds in the fatty acid moiety;number of hydroxyl groups in the long-chain base moiety plus number of hydroxyl groups in the fatty acid moiety (e.g., SM 34:1;2).
RESULTS
VSV and SFV purification and lipid composition.
The procedure described in this paper reliably yielded pure, infectious virus particles suitable for further analysis. Only viral proteins were detected in the preparations, as judged by silver-stained SDS-PAGE gels (Fig. 1). The preparations were also devoid of cell-derived lipids. We prelabeled BHK cells with [1,2-14C]acetate, mixed the media from these cells and cells infected with SFV, and then purified the virus as described in Materials and Methods. Almost all (>95%) of the radioactivity was removed during the step of pelleting virus through a sucrose cushion. Practically no radioactivity was detected in the final step of virus purification, employing velocity gradient centrifugation (see Fig. S1 in the supplemental material). We also tried an additional density gradient centrifugation step in the virus purification procedure and found no differences in the virus lipid compositions (data not shown).
FIG. 1.
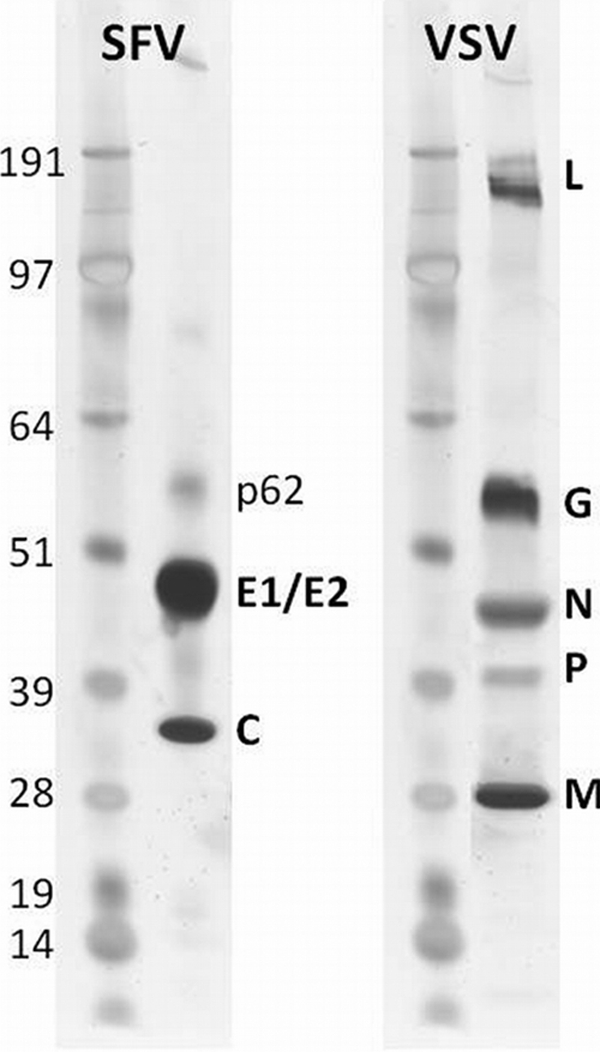
SDS-PAGE analysis of purified viruses. Approximately 1 μg of final virus preparation was loaded onto a NuPAGE 4 to 12% BisTris gel, run in morpholinepropanesulfonic acid (MOPS) buffer, and stained with silver nitrate.
Next, we characterized and quantified the main lipid classes of the purified viruses. These comprise the glycerophospholipids, i.e., PA, PC, PE, PG, PS, and PI; the sphingolipids, i.e., Cer, SM, HexCer, diHexCer, and the ganglioside GM3; DAG; and Chol. All of these lipid classes can be extracted efficiently by conventional extraction, with the exception of GM3 (3, 10). In order to enhance the recovery of GM3, we employed a two-step extraction procedure (9) in which samples are first extracted with the apolar solvent mixture chloroform-methanol (10:1 [vol/vol]). This extraction recovers PC, PE, PG, DAG, Cer, SM, HexCer, diHexCer, and Chol. Subsequently, the remaining aqueous sample was extracted with the polar solvent mixture chloroform-methanol (2:1 [vol/vol]), which efficiently recovers PA, PS, PI, and GM3. This two-step lipid extraction procedure allowed sensitive analysis of GM3, prompted by increased recovery of GM3 and improved ionization efficiency due to the separation of the lipid classes. For lipid analysis, we employed a lipidomic platform based on nanoelectrospray ionization combined with quantitative high-resolution MS analysis of the lipid extracts (9, 42). The comparative lipidomic analysis achieved the absolute quantification of 159 individual molecular lipid species encompassing 13 lipid classes. Absolute quantification was achieved by the addition of a set of internal standards, comprising all lipid classes analyzed, prior to the lipid extraction (8) (see Materials and Methods). Lipid content is presented as mol% of individual lipid species normalized to the total amount of lipid quantified.
The lipid compositions of these structurally very different viruses are remarkably similar. We did not observe statistically significant differences at the lipid class level (Table 1; see Fig. 3). At the lipid species level, the PC compositions of both viruses revealed that saturated and monounsaturated PC species were slightly enriched in SFV compared to VSV (see Fig. 5 and Fig. S9 in the supplemental material). This trend became more evident when we analyzed the fatty acid saturation profile of all glycerophospholipids (see Fig. 6A). At the fatty acid chain length level, there was also a slight increase of glycerophospholipid species with 30 and 32 carbon atoms, while lipid species with 38 carbon atoms in their fatty acid moiety were depleted (see Fig. 6B) in SFV compared to VSV. The state of unsaturation and chain length profiles of the sphingolipids were the same for both viruses.
TABLE 1.
Lipid class composition of BHK cells, their PMs, and VSV and SFV produced in BHK cells
| Lipid | Avg mol% ± SEa
|
|||
|---|---|---|---|---|
| BHK cells | PM | VSV | SFV | |
| PC | 23.1 ± 1.6 | 10.5 ± 2.4 | 8.6 ± 1.0 | 10.4 ± 1.2 |
| PC O- | 8.2 ± 0.2 | 3.9 ± 0.7 | 2.8 ± 0.4 | 3.7 ± 0.6 |
| PE | 5.3 ± 1.1 | 2.7 ± 0.9 | 5.0 ± 0.7 | 3.9 ± 0.2 |
| PE O- | 7.2 ± 2.1 | 7.7 ± 1.5 | 9.6 ± 0.6 | 7.3 ± 0.2 |
| PG | 1.2 ± 0.4 | 0.2 ± 0.08 | 0.06 ± 0.00 | 0.08 ± 0.01 |
| PI | 5.7 ± 1.7 | 1.9 ± 0.2 | 1.0 ± 0.3 | 0.8 ± 0.1 |
| PS | 4.7 ± 1.2 | 8.1 ± 0.9 | 9.8 ± 1.8 | 10.6 ± 1.7 |
| SM | 5.7 ± 1.1 | 8.6 ± 1.1 | 11.5 ± 0.7 | 12.9 ± 0.6 |
| DAG | 0.4 ± 0.2 | 1.3 ± 0.9 | 0.2 ± 0.01 | 0.3 ± 0.2 |
| PA | 0.8 ± 0.5 | 0.3 ± 0.1 | 0.1 ± 0.05 | 0.2 ± 0.03 |
| Cer | 0.3 ± 0.16 | 0.09 ± 0.03 | 0.09 ± 0.01 | 0.1 ± 0.02 |
| HexCer | 0.2 ± 0.02 | 0.07 ± 0.05 | ||
| diHexCer | 0.04 ± 0.01 | 0.2 ± 0.09 | 0.1 ± 0.06 | |
| GM3 | 3.7 ± 1.0 | 11.6 ± 0.8 | 7.7 ± 2.4 | 6.8 ± 1.2 |
| Chol | 28.8 ± 1.5 | 42.1 ± 2.6 | 43.6 ± 1.8 | 42.4 ± 1.9 |
For four independent experiments.
FIG. 3.

Lipid class compositions of BHK cells, PM, VSV, and SFV. Lipid class average compositions were determined by adding the abundances of individual lipid species from each class (Fig. 5; see Fig. S2 to S8 in the supplemental material). Error bars are the correspondent standard deviations (n = 4 for each sample).
FIG. 5.

Molecular composition of major lipid classes. The abundance of each lipid class is represented in mol% (absolute amount of lipid species/absolute amount of total identified lipid species).
FIG. 6.

Profiling of glycerophospholipid chain length (A) and saturation (B). The relative abundances of each chain length and unsaturation state were normalized to the total glycerophospholipid in each sample.
PM purification and lipid composition.
The PM comprises only a few percentage points of the total membranes of a cell (approximately 10% seems to be a reasonable estimate for BHK cells [11]), while the ER makes up more than 80% of the total cell membranes. Thus, the ER is the major contaminant of PM preparations. We used the ER marker calnexin as a measure of purity for our PM preparations. We tried different methods to purify the BHK cell PM. We finally ended up with a method in which surface biotinylation and neutravidin-coupled magnetic beads were used to achieve a highly purified PM. With this method, only negligible ER contamination was seen, often with no calnexin detectable (Fig. 2). From 350 cm2 of ∼80% confluent cells, about 3 to 4 nmol (∼3 μg) of lipid was recovered by using the two-step lipid extraction procedure described above.
FIG. 2.
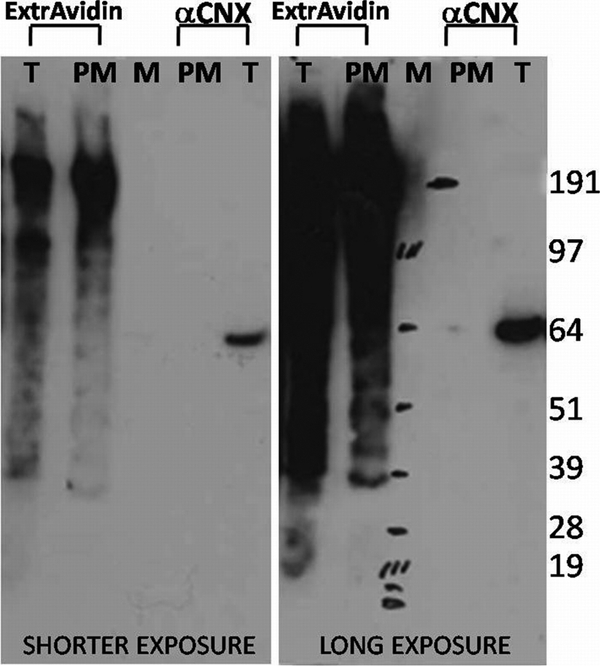
Western blot analysis of total cells and purified PM. About 500 ng of protein (total cells; lanes T) and 5% purified PM (lanes PM) were loaded onto 4 to 12% BisTris NuPAGE gels, run in MOPS buffer, and blotted onto a nitrocellulose membrane. The membrane was cut along the marker (lane M), and the halves were developed separately, using polyclonal anti-calnexin (αCNX) antibody produced in rabbits with HRP-conjugated anti-rabbit antibody and HRP-conjugated ExtrAvidin biotin-binding protein.
The lipid composition of the PM was very different from that of the whole BHK cell. We observed overall increases in sphingolipids (from 9.9 mol% ± 2.2 mol% to 20.4 mol% ± 2.0 mol%) and Chol (from 28.8 mol% ± 1.5 mol% to 42.1 mol% ± 2.6 mol%) and a decrease in glycerophospholipids (from 56.2 mol% ± 8.7 mol% to 35.2 mol% ± 6.8 mol%) in the PM compared to whole BHK lipids. This is in good agreement with previous reports (7, 39). We found significant reductions in PG, PE, PI, and PC, while SM, Chol, PS, and GM3 were increased in the PM compared to the BHK cell (Fig. 3 and Fig. 4A). The most depleted lipid classes in the PM were previously shown to be located predominantly in internal organelles. On the other hand, the enriched lipid classes (sphingolipids and Chol) were previously shown to be increased in the PM (49). This observation testifies to the high purity of the obtained PM fractions. Besides the reduction of glycerophospholipids in the PM, we also found that these lipid species are mainly diunsaturated in BHK cells, while in the PM they are monounsaturated (Fig. 5). This is due mostly to a decrease in the amount of the PC 36:2 species, from 6.9 mol% ± 0.7 mol% in the whole cell to 2.1 mol% ± 1.0 mol% in the PM (Fig. 6).
FIG. 4.

(A) Enrichment plot of PM versus BHK cells. (B) Enrichment plot of VSV versus PM and SFV versus PM. Enrichment is defined as the ratio of the average quantity of each lipid class in a given sample to the average quantity of each lipid class in another sample. Only the statistically significant (by ANOVA plus Tuckes test) changes are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The lipid compositions of VSV and SFV are similar but not identical to that of the BHK PM.
One aim of this work was to try to understand the principles of lipid incorporation of viruses during budding from the host cell PM. From a comparison of the lipidomes of the PM and both purified viruses, we could conclude that the lipid compositions of the viruses are similar to the PM lipidome, with some noteworthy differences. SM was enriched in both viruses, while PG and PI were depleted (Fig. 3). Interestingly, while the SM lipid class was enriched only 1.3- and 1.5-fold in VSV and SFV, respectively, relative to the PM, the SM 34:0;2 species (dihydrosphingomyelin) was enriched 2.7- and 3.5-fold, respectively. Moreover, SM species with long fatty acids (C24:0 and C24:1) were also enriched in the viruses, by 2.5- and 5-fold, respectively. This result is in good agreement with previous reports suggesting the involvement of dihydrosphingomyelin in virus budding (6, 7). The most significant difference within the glycerophospholipids was the elevation in the viruses of diunsaturated PS species, mainly PS 36:2 (18:1-18:1) and PS 34:2 (18:1-16:1), by 1.6- and 2.1-fold, respectively (Fig. 5). In summary, we observed little specificity in lipid uptake from the PM into the viral envelope. Nevertheless, the viruses studied showed some selectivity for SM (especially long-chain SM species and dihydrosphingomyelin), while GM3 was depleted.
DISCUSSION
Advanced MS combined with optimized lipid recovery recipes enabled absolute quantification of 159 individual molecular species from 13 major lipid classes in the lipidomes of VSV and SFV, as well as in the PM of BHK cells. For the first time, this quantitative analysis included gangliosides. We found that the simplest ganglioside, GM3, was a major constituent of the PM and the viral envelopes, and thus lipidomic analysis excluding these lipid classes will be incomplete. Moreover, the versatility and sensitivity of the method will also enable us to extract lipids and to determine their composition quantitatively from biological samples purified in very small amounts.
The most startling result of the present study is that the lipid compositions of SFV and VSV are so similar. This is the case for the lipid class distribution as well as for the individual species within each lipid class. Also important is the high content of Chol, at 43.6 mol% ± 1.8 mol% for VSV and 42.4 mol% ± 1.9 mol% for SFV. Early studies of the lipid composition of SFV by Renkonen et al. (40) had similarly high values, while those reported for VSV were lower (25, 34).
To compare the lipidome of the host membrane with those of the viruses, we developed methodology to purify the PM from BHK-21 cells. The major contaminant of membranes is the ER, and this is also the most troublesome impurity because the lipid composition of this organelle is quite different from that of the PM (49). Only by introducing an affinity step based on cell surface-specific biotinylation were we able to obtain PM preparations devoid of ER contamination, as judged by Western blotting with calnexin antibodies. Another criterion from the lipid composition results is the high Chol content. The concentrations of Chol and sphingolipids are known to increase along the biosynthetic pathway from the ER to the cell surface (49). In our analysis, the Chol content of the PM was 42.1 mol% ± 2.6 mol%. The lower values reported previously (15, 25, 34, 36) for Chol and sphingolipids can probably be attributed to insufficient purity of the isolated PMs. An additional measure of the purity of our PM preparation is the threefold reduction in the level of Cer, which is synthesized in the ER and predominantly enriched there. Also, the sphingolipid concentrations, with the major classes being SM and the ganglioside GM3, were much higher in the PM than in the whole-cell lipidome.
Thus, we can conclude that our PM preparation from BHK cells was quite pure and therefore that the lipidome of the PM can be compared usefully to the lipidomes of the two viruses. Based on our analyses, it is obvious that all three lipidomes are quite similar. The Chol concentration is higher than 40%, and also, the sphingolipid concentration is around 20% in all of the lipidomes. However, concerning the sphingolipid composition, the SM content is slightly higher in the viral envelopes, but this is compensated by a similar decrease in GM3 content in the viral envelopes. In comparing the lipid compositions of the viruses and the PM, it is also important to keep membrane geometry in mind. While both SFV and VSV are highly curved, the PM is probably less curved. The difference between the areas of the inner and outer leaflets of membrane increases with curvature. Thus, in SFV (the diameter of the lipid envelope without spikes is approximately 50 nm), the inner leaflet will cover only about 65% of the area presented by the outer leaflet, and for the less-curved, bullet-shaped VSV (180 by 70 nm), the coverage will be 80% (47). The differences between the areas of the outer and inner leaflets could account to some extent for the differences—albeit small—in the lipid compositions of the PM and the viruses. For instance, considering that PS species should populate the inner leaflet of the BHK PM, PS species with bulkier fatty acid moieties (e.g., diunsaturated species) could be sorted to the viral bud regions (1, 28, 33, 50, 51). Curvature and lipid asymmetry were previously proposed to play a role in the formation of bacteriophage envelopes (19-21). However, since we do not have information on lipid asymmetry of the viral envelopes extended to individual lipid species, these remain issues for future work. The existing methods for determining lipid asymmetry are based on low-resolution thin-layer chromatographic methods and therefore do not include lipid species (33). The time is now ripe to revisit this area of research by developing new methodology based on the high sensitivity and high resolution of MS methods available today.
One important outcome of this work is the comprehensive lipidomic analysis of the highly purified PM. It is worthwhile to notice the features that seem to be the hallmarks of PM composition, i.e., the remarkably high Chol content (∼40 mol%) and the low PC content (10.5% ± 2.4% ester and 3.9% ± 0.7% ether). The successful purification and analysis of the PM have made it possible to convincingly relate the compositions of the viral envelopes to that of the PM.
The main conclusion of this paper is that SFV and VSV acquire their lipid bilayers from the host PM, such that the lipid composition of the bilayer undergoes only minor changes during the budding process. Moreover, both SFV and VSV organize their budding sites in the same way with respect to the lipid bilayer because their lipidomes are so similar. Thus, the viral membrane glycoproteins and the inner core proteins, the capsid protein for SFV and the M protein for VSV, do not seem to influence the budding process with respect to the lipid bilayer.
How does this conclusion fit with earlier studies on phenotypic mixing by VSV? This virus can incorporate glycoproteins from other viruses (such as retrovirus spike proteins or influenza virus hemagglutinin) as well as host membrane proteins into its envelope (5, 52). SFV, in contrast, rigorously excludes other virus glycoproteins and PM proteins from the host cell during the budding process (5, 43, 52). A hypothesis employing lipid rafts as platforms mediating phenotypic mixing has been put forward (37). During the assembly process, lipid rafts would accumulate at the budding site, bringing together both viral proteins and raft-associated host proteins. Thus, raft affinity could explain why foreign proteins are included in the viral envelope. However, if this were the case for VSV, then one would expect the viral envelope to be enriched in raft lipids, such as sphingolipids, saturated PCs, and Chol. In the present study, we show that this is not the case. Also, the analysis of the saturation level of the glycerophospholipids, including PC, demonstrated that there were no striking differences between VSV, SFV, and the PM in this respect. Moreover, the VSV glycoprotein does not have the typical characteristics of raft-associated proteins (41). This singly spanning transmembrane protein is not insoluble in Triton X-100 at 4°C. In lateral diffusion studies using fluorescence recovery after photobleaching of the apical membranes of MDCK cells, the VSV glycoprotein behaved as a non-raft-partitioning protein (26). In copatching studies, a very minor fraction colocalized with raft proteins (14). The bulk of the protein behaves like a nonraft protein, such as the transferrin receptor. On the basis of these studies, the VSV glycoprotein does not fit into the category of raft proteins exemplified by the influenza virus hemagglutinin. Thus, the hypothesis that lipid rafts mediate VSV assembly and pseudotyping should be reconsidered. An alternative model has been proposed by Metsikko and Garoff (27), who demonstrated that about 15% of the protein in the VSV envelope can be occupied by non-VSV proteins and that the non-VSV integral proteins might be included more or less passively, depending mostly on the concentration of the proteins in the PM and not on the basis of raft affinity. Considering our results that the lipid compositions of SFV, VSV, and the host PM are very similar, we favor the latter hypothesis. However, further work will be necessary to sort out how phenotypic mixing works mechanistically. Although SFV and VSV do not seem to fall into the category of enveloped viruses that employ lipid rafts during budding, there are viruses that do so. One representative of such a virus class is HIV (30, 31). Two recent studies have employed MS methods to analyze the lipidome of HIV (6, 7). In contrast to our results for SFV and VSV, the HIV envelope was strongly enriched in Cer and GM3 (4, 5) compared to the concentrations in the host PM (5). The enrichment of Cer was an interesting feature of the HIV envelope and may reflect the involvement of Cer-induced raft domains in HIV assembly (12, 48). In the SFV and VSV envelopes, Cer was starkly depleted. These studies indeed suggest that the viruses may acquire their lipid envelopes by different mechanisms. Blom et al. did study the lipid class compositions of VSV and influenza virus budding from fibroblasts (4). They observed an increase in glycosphingolipids in influenza virus compared to those in VSV. These studies should be repeated as a full-scale lipidome analysis. Only by demonstrating that one virus selects specific lipids while another does not will it be possible to prove that there are viruses that employ raft coalescence as a mechanism to assemble their membranes. Enveloped viruses can thus be employed as tools to understand how protein-lipid and lipid-lipid interactions contribute to bilayer organization and its plasticity.
Supplementary Material
Acknowledgments
We thank the Simons lab for both critical and positive feedback. We thank Christoph Thiele for providing the synthetic internal standard PI 17:0-17:0. We thank Vineeth Srendranath and Dominik Schwudke for developing the Pecoder software to analyze the LTQ-Orbitrap spectra. We thank Roger Sandhoff for the quantification of GM3 standard extract.
This work was supported by grant DFG SFB/TR13-04 and by EU FP6 PRISM contract LSHB-CT 2007-037740. S.C. had an EMBO postdoctoral fellowship.
Footnotes
Published ahead of print on 27 May 2009.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Allan, D., and P. Quinn. 1989. Membrane phospholipid asymmetry in Semliki Forest virus grown in BHK cells. Biochim. Biophys. Acta 987199-204. [Google Scholar]
- 2.Aloia, R. C., F. C. Jensen, C. C. Curtain, P. W. Mobley, and L. M. Gordon. 1988. Lipid composition and fluidity of the human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 85900-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bligh, E. G., and W. J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37911-917. [DOI] [PubMed] [Google Scholar]
- 4.Blom, T. S., M. Koivusalo, E. Kuismanen, R. Kostiainen, P. Somerharju, and E. Ikonen. 2001. Mass spectrometric analysis reveals an increase in plasma membrane polyunsaturated phospholipid species upon cellular cholesterol loading. Biochemistry 4014635-14644. [DOI] [PubMed] [Google Scholar]
- 5.Briggs, J. A. G., T. Wilk, and S. D. Fuller. 2003. Do lipid rafts mediate virus assembly and pseudotyping? J. Gen. Virol. 84757-768. [DOI] [PubMed] [Google Scholar]
- 6.Brugger, B., B. Glass, P. Haberkant, I. Leibrecht, F. T. Wieland, and H. G. Krausslich. 2006. The HIV lipidome: a raft with an unusual composition. Proc. Natl. Acad. Sci. USA 1032641-2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan, R., P. D. Uchil, J. Jin, G. Shui, D. E. Ott, W. Mothes, and M. R. Wenk. 2008. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J. Virol. 8211228-11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ejsing, C. S., E. Duchoslav, J. Sampaio, K. Simons, R. Bonner, C. Thiele, K. Ekroos, and A. Shevchenko. 2006. Automated identification and quantification of glycerophospholipid molecular species by multiple precursor ion scanning. Anal. Chem. 786202-6214. [DOI] [PubMed] [Google Scholar]
- 9.Ejsing, C. S., J. L. Sampaio, V. Surendranath, E. Duchoslav, K. Ekroos, R. W. Klemm, K. Simons, and A. Shevchenko. 2009. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. 1062136-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Folch, J., M. Lees, and G. H. S. Stanley. 1957. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 226497-509. [PubMed] [Google Scholar]
- 11.Griffiths, G., G. Warren, P. Quinn, O. Mathieu-Costello, and H. Hoppeler. 1984. Density of newly synthesized plasma membrane proteins in intracellular membranes. I. Stereological studies. J. Cell Biol. 982133-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gulbins, E., and R. Kolesnick. 2003. Raft ceramide in molecular medicine. Oncogene 227070-7077. [DOI] [PubMed] [Google Scholar]
- 13.Hancock, J. F. 2006. Lipid rafts: contentious only from simplistic standpoints. Nat. Rev. Mol. Cell Biol. 7456-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harder, T., P. Scheiffele, P. Verkade, and K. Simons. 1998. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141929-942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirschberg, C. B., and P. W. Robbins. 1974. The glycolipids and phospholipids of Sindbis virus and their relation to the lipids of the host cell plasma membrane. Virology 61602-608. [DOI] [PubMed] [Google Scholar]
- 16.Kaariainen, L., K. Simons, and C. H. von Bonsdorf. 1969. Studies in subviral components of Semliki Forest virus. Ann. Med. Exp. Biol. Fenn. 47235-248. [PubMed] [Google Scholar]
- 17.Klenk, H. D., and P. W. Choppin. 1971. Glycolipid content of vesicular stomatitis virus grown in baby hamster kidney cells. J. Virol. 7416-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klenk, H. D., and P. W. Choppin. 1970. Glycosphingolipids of plasma membranes of cultured cells and an enveloped virus (SV5) grown in these cells. Proc. Natl. Acad. Sci. USA 6657-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laurinavicius, S., D. H. Bamford, and P. Somerharju. 2007. Transbilayer distribution of phospholipids in bacteriophage membranes. Biochim. Biophys. Acta 17682568-2577. [DOI] [PubMed] [Google Scholar]
- 20.Laurinavicius, S., R. Käkelä, D. H. Bamford, and P. Somerharju. 2004. The origin of phospholipids of the enveloped bacteriophage phi6. Virology 326182-190. [DOI] [PubMed] [Google Scholar]
- 21.Laurinavicius, S., R. Käkelä, P. Somerharju, and D. H. Bamford. 2004. Phospholipid molecular species profiles of tectiviruses infecting gram-negative and gram-positive hosts. Virology 322328-336. [DOI] [PubMed] [Google Scholar]
- 22.Liebisch, G., M. Binder, R. Schifferer, T. Langmann, B. Schulz, and G. Schmitz. 2005. High throughput quantification of cholesterol and cholesteryl ester by electrospray ionization tandem mass spectrometry (ESI-MS/MS). Biochim. Biophys. 1761121-128. [DOI] [PubMed] [Google Scholar]
- 23.Luan, P., L. Yang, and M. Glaser. 1995. Formation of membrane domains created during the budding of vesicular stomatitis virus. A model for selective lipid and protein sorting in biological membranes. Biochemistry 349874-9883. [DOI] [PubMed] [Google Scholar]
- 24.Matlin, K. S., H. Reggio, A. Helenius, and K. Simons. 1982. Pathway of vesicular stomatitis virus entry leading to infection. J. Mol. Biol. 156609-631. [DOI] [PubMed] [Google Scholar]
- 25.McSharry, J. J., and R. R. Wagner. 1971. Lipid composition of purified vesicular stomatitis viruses. J. Virol. 759-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meder, D., M. J. Moreno, P. Verkade, W. L. C. Vaz, and K. Simons. 2006. Phase coexistence and connectivity in the apical membrane of polarized epithelial cells. Proc. Natl. Acad. Sci. USA 103329-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Metsikko, K., and H. Garoff. 1989. Role of heterologous and homologous glycoproteins in phenotypic mixing between Sendai virus and vesicular stomatitis virus. J. Virol. 635111-5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mouritsen, O. G. 2005. Life—as a matter of fat. The emerging science of lipidomics. Springer-Verlag, Heidelberg, Germany.
- 29.Nayak, D. P., and E. K. Hui. 2004. The role of lipid domains in virus biology. Subcell. Biochem. 37443-491. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen, D. H., and J. E. K. Hildreth. 2000. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J. Virol. 743264-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ono, A., and E. O. Freed. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA 9813925-13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ono, A., and E. O. Freed. 2005. Role of lipid rafts in virus replication. Adv. Virus Res. 64331-358. [DOI] [PubMed] [Google Scholar]
- 33.Op den Kamp, J. A. F. 1979. Lipid asymmetry in membranes. Annu. Rev. Biochem. 4847-71. [DOI] [PubMed] [Google Scholar]
- 34.Patzer, E. J., N. F. Moore, Y. Barenholz, J. M. Shaw, and R. R. Wagner. 1978. Lipid organization of the membrane of vesicular stomatitis virus. J. Biol. Chem. 2534544-4550. [PubMed] [Google Scholar]
- 35.Patzer, E. J., R. R. Wagner, and E. J. Dubovi. 1979. Viral membranes: model systems for studying biological membranes. CRC Crit. Rev. Biochem. 6165-217. [DOI] [PubMed] [Google Scholar]
- 36.Pessin, J. E., and M. Glaser. 1980. Budding of Rous sarcoma virus and vesicular stomatitis virus from localized lipid regions in the plasma membrane of chicken embryo fibroblasts. J. Biol. Chem. 2559044-9050. [PubMed] [Google Scholar]
- 37.Pickl, W. F., F. X. Pimentel-Muinos, and B. Seed. 2001. Lipid rafts and pseudotyping. J. Virol. 757175-7183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quigley, J. P., D. B. Rifkin, and E. Reich. 1971. Phospholipid composition of Rous sarcoma virus, host cell membranes and other enveloped RNA viruses. Virology 46106-116. [DOI] [PubMed] [Google Scholar]
- 39.Renkonen, O., C. G. Gahmberg, K. Simons, and L. Kaariainen. 1972. The lipids of the plasma membranes and endoplasmic reticulum from cultured baby hamster kidney cells (BHK21). Biochim. Biophys. Acta 25566-78. [DOI] [PubMed] [Google Scholar]
- 40.Renkonen, O., L. Kaarainen, K. Simons, and C. G. Gahmberg. 1971. The lipid class composition of Semliki Forest virus and plasma membranes of the host cells. Virology 46318-326. [DOI] [PubMed] [Google Scholar]
- 41.Scheiffele, P., A. Rietveld, T. Wilk, and K. Simons. 1999. Influenza viruses select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem. 2742038-2044. [DOI] [PubMed] [Google Scholar]
- 42.Schwudke, D., J. T. Hannich, V. Surendranath, V. Grimard, T. Moehring, L. Burton, T. Kurzchalia, and A. Shevchenko. 2007. Top-down lipidomic screens by multivariate analysis of high-resolution survey mass spectra. Anal. Chem. 794083-4093. [DOI] [PubMed] [Google Scholar]
- 43.Simons, K., and H. Garoff. 1980. The budding mechanisms of enveloped animal viruses. J. Gen. Virol. 501-21. [DOI] [PubMed] [Google Scholar]
- 44.Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature 387569-572. [DOI] [PubMed] [Google Scholar]
- 45.Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 131-39. [DOI] [PubMed] [Google Scholar]
- 46.Simons, K., and G. Warren. 1984. Semliki Forest virus: a probe for membrane traffic in the animal cell. Adv. Protein Chem. 3679-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas, D., W. W. Newcomb, J. C. Brown, J. S. Wall, J. F. Hainfeld, B. L. Trus, and A. C. Steven. 1985. Mass and molecular composition of vesicular stomatitis virus: a scanning transmission electron microscopy analysis. J. Virol. 54598-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trajkovic, K., C. Hsu, S. Chiantia, L. Rajendran, D. Wenzel, F. Wieland, P. Schwille, B. Brugger, and M. Simons. 2008. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 3191244-1247. [DOI] [PubMed] [Google Scholar]
- 49.van Meer, G. 1989. Lipid traffic in animal cells. Annu. Rev. Cell Biol. 5247-275. [DOI] [PubMed] [Google Scholar]
- 50.van Meer, G., K. Simons, J. A. F. Op den Kamp, and L. L. M. Van Deenen. 1981. Phospholipid asymmetry in Semliki Forest virus grown on baby hamster kidney (BHK-21) cells. Biochemistry 201974-1981. [DOI] [PubMed] [Google Scholar]
- 51.Whatmore, J. L., and D. Allan. 1994. Phospholipid asymmetry in plasma membrane vesicles derived from BHK cells. Biochim. Biophys. Acta 119288-94. [DOI] [PubMed] [Google Scholar]
- 52.Zavadova, A., J. Zavada, and R. A. Weiss. 1977. Unilateral phenotypic mixing of envelope antigens between togaviruses and vesicular stomatitis virus or avian RNA tumour virus. J. Gen. Virol. 37557-567. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.