

Abstract
Influenza A virus segment 2 is known to encode two polypeptides in overlapping open reading frames: PB1, the polymerase, and PB1-F2, a proapoptotic virulence factor. We show that a third major polypeptide is synthesized from PB1 mRNA via differential AUG codon usage. PB1 codon 40 directs translation of an N-terminally truncated version of the polypeptide (N40) that lacks transcriptase function but nevertheless interacts with PB2 and the polymerase complex in the cellular environment. Importantly, the expression of N40, PB1-F2, and PB1 are interdependent, and certain mutations previously used to ablate PB1-F2 production affected N40 accumulation. Removal of the PB1-F2 AUG upregulated N40 synthesis, while truncating PB1-F2 after codon 8 (with a concomitant M40I change in PB1) abolished N40 expression. A virus lacking both N40 and PB1-F2 replicated normally. However, viruses that did not express N40 but retained an intact PB1-F2 gene overexpressed PB1 early in infection and replicated slowly in tissue culture. Thus, the influenza A virus proteome includes a 12th primary translation product that (similarly to PB1-F2) is nonessential for virus viability but whose loss, in particular genetic backgrounds, is detrimental to virus replication.
Influenza A virus (IAV) is a major pathogen that infects a broad variety of homeothermic vertebrates, including humans and domesticated animal species such as chickens, swine, and horses (27). Individual strains of IAV tend to be specific to a single host species and, although they may infect alternative hosts, this is generally a sporadic, inefficient process until adaptation to the new host occurs (26). IAVs also vary widely in pathogenicity, with high virulence often being associated with cross-species transmission events. An extreme example of this in humans is the contrasting mortality rates resulting from infection with human-adapted H1N1 viruses (<0.1% case fatality) and current avian H5N1 strains (>60%) (36, 40; http://www.who.int/csr/disease/avian_influenza/country/en/).
The IAV genome is carried on eight segments of negative-sense RNA. Host range and pathogenicity are polygenic traits, but it is well established that segment 2 plays an important role (3, 4). In part, this reflects the essential functions of PB1, the primary protein product of the segment. PB1 is the core subunit of the viral RNA-dependent RNA polymerase, which is responsible for transcription and replication of the virus genome (14). However, nearly 20 years after the first complete nucleotide sequence of segment 2 was published (6), a second polypeptide made from segment 2 mRNA was discovered (9). Most, but not all strains of IAV express a small (up to 90-amino-acid) polypeptide (designated PB1-F2) from an overlapping open reading frame (ORF) embedded entirely within the 5′ end of the PB1 cistron (8, 9, 51). PB1-F2 localizes to mitochondria through its C-terminal region and induces apoptosis in certain cell types (9, 17, 47, 49). The proapoptotic effects of overexpressed PB1-F2 suggested the hypothesis that it is a virulence factor (9), and several studies have correlated its loss of expression and/or sequence polymorphisms with alterations to virus pathogenicity (10, 31, 50). However, no evidence has been presented to date to show that the increased pathogenicity observed in vivo is caused by PB1-F2 induced apoptosis. It has also been suggested that PB1-F2 influences the activity and intracellular localization of PB1 itself through an interaction occurring in the nuclei of cells (30).
The expression of PB1-F2 is thought to occur by leaky ribosomal scanning past three upstream AUG codons (9). Although not formally tested, this is plausible since neither the PB1 AUG codon nor those of two intervening short ORFs are set in strong Kozak translation initiation contexts (24), whereas the AUG of PB1-F2 is (Fig. 1A). It has also been proposed that shorter PB1-F2 polypeptides are synthesized from one or more AUG codons further downstream of the PB1-F2 start codon (50), but for this to occur by leaky scanning would require the unlikely scenario of ribosomes bypassing a minimum of three further AUG codons with strong initiation contexts to initiate on AUGs with moderate contexts (Fig. 1A). Thus, there are interesting and (given the significance of PB1 and PB1-F2 to IAV pathogenicity) important unresolved questions regarding the regulation of gene expression from segment 2. Toward this, we show here that a third major polypeptide species is translated from segment 2 mRNA; PB1 N40, an N-terminally truncated and functionally distinct variant of PB1. Furthermore, we find that, whereas N40 is not essential for virus replication in cultured cells, the expression of PB1, PB1 N40, and PB1-F2 is interdependent, and the absence of N40 in the presence of an intact PB1-F2 ORF leads to slower virus replication kinetics.
FIG. 1.

Expression of multiple protein species from IAV segment 2. (A) Diagram of the 5′ end of segment 2 mRNA. ORFs in the three reading frames are indicated by boxes. AUG codons are color coded according to the relative strength of their Kozak consensus (green = strong, −3A/G, +4G; amber = intermediate, −3A/G or + 4G; red = weak, −3U, +4U), and those that initiate identified polypeptides are indicated. (B) Detection of a minor PB1 species from infected cells. [35S]methionine-labeled lysate from MDCK cells infected (lanes I) or mock infected (lanes M) with Cambridge PR8 virus were immunoprecipitated with anti-PB1 V19, anti-PA V35, or anti-PB2 V28 as indicated and analyzed by SDS-PAGE and autoradiography in parallel with a radiolabeled in vitro translation reaction of WT segment 2 (in lane R, a lower exposure of the same track was used). (C) Lysates from cells infected (or mock infected) with the indicated panel of viruses, as well as in vitro-translated N40 were analyzed by Western blotting for PB1 with anti-PB1 V19 serum. (D) Aliquots of in vitro translation reactions programmed with the indicated plasmids (lane VOC [vector only control]) were analyzed by SDS-PAGE and autoradiography. (E) Lysates from 293T cells transfected with the indicated plasmids (pDUAL; empty vector control) were analyzed by SDS-PAGE and Western blotting with anti-PB1 MAb 10.4 (top panel) or anti-PB1-F2 C-terminal specific serum (bottom panel). The migration of molecular mass markers (in kilodaltons), as well as specified polypeptides, is indicated.
MATERIALS AND METHODS
Plasmids, antisera, and viruses.
Plasmids pcDNA-PB2, -PA, and -NP, containing cDNA copies of the influenza A/PR/8/34 (PR8) genes have been previously described (32). Plasmids pPolI-Flu-ffLuc containing an influenza virus-based luciferase minireplicon vRNA under the control of the human RNA polymerase I promoter and pcDNA mMx1 containing the murine Mx1 gene were provided by Laurence Tiley (5). Plasmid pcDNA-N40, containing a cDNA copy of PR8 segment 2 (accession no. EF467819) lacking start codons upstream of AUG 5 was constructed by PCR to insert bp 121 to 2307 of segment 2 into the XhoI/XbaI sites of pcDNA3. Plasmids pcDNA PB1-, PB2-, and PA-GFP were provided by Ervin Fodor (16). pEGFP-TAP, encoding a green fluorescent protein (GFP) fusion with the human mRNA export Tip-associated protein was supplied by Adrian Whitehouse (45). Dual promoter reverse genetics plasmids for PR8 segments 1 and 3 to 8 were given by Ron Fouchier (11). A similar construct for segment 2 was generated by subcloning the human polymerase I promoter, viral sequences (originally cloned from the NIBSC strain of PR8), and hepatitis delta ribozyme sequence from a previously described plasmid (44) into the pcDNA3 multicloning site. Sequence analysis of this plasmid showed the influenza gene to be identical to the clone reported by (11) with the exception of two nucleotide changes (T1204C and T1434C). Other mutants had additional substitutions introduced by site-directed mutagenesis as follows: F2-8, G144A; F2-57, G291A and T309A; ΔAUG, A115T, G116C, C117A, and T120C; M40L, A142T; and M40I, G144T. The predicted phenotypic effects of these mutations on the segment 2 ORFs are summarized in Table 1. All plasmids were sequence verified.
TABLE 1.
Phenotypic summary of segment 2 mutants
| Virus | Predicted structure of ORFs
|
Major phenotype(s) | Replication kinetics | ||
|---|---|---|---|---|---|
| PB1 | PB1-F2 | N40 | |||
| F2-8 | M40I | Δ8 | Null | WT | |
| F2-57 | WT | Δ57 | WT | WT | |
| ΔAUG | WT | Null | WT | Overexpresses N40; delayed PB1 expression; increased cytoplasmic PB1 | WT |
| M40L | M40L | WT | Null | Early increase in PB1 | Delayed |
| M40I | M40I | W9L | Null | Early increase in PB1 | Delayed |
Mouse monoclonal anti-PB1 clone 10.4 (directed against an epitope contained between residues 44 and 69) and a rabbit polyclonal serum raised against a peptide corresponding to amino acids 740 to 757 of PB1 were generously provided by Mark Krystal. Rabbit polyclonal anti-PB1 serum V19 raised against amino acids 50 to 370 of PR8 PB1, as well as similar anti-PA, anti-PB2, and anti-NP (2915) sera, have been previously described (13, 22). A rabbit antiserum to the C terminus of the PB1-F2 protein was kindly provided by Jonathan Yewdell. Rat monoclonal anti-tubulin YL1/2 was purchased from Serotec. IR800 and IR680 dye-conjugated anti-rabbit immunoglobulin G (IgG) and anti-mouse IgG sera were purchased from LiCor, while IR680 dye conjugated anti-rat IgG was purchased from Invitrogen. Alexa 594-conjugated anti-mouse IgG and fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG were purchased from Invitrogen and Dako, respectively.
Nonrecombinant viruses PR8 (H1N1, Cambridge lineage) A/Udorn/72 (Udorn; H3N2) A/USSR/77 (USSR; H1N1) were sourced from the Division of Virology's collection of influenza viruses (15, 41). A/Eq/Newmarket/11/03 (Eq/Nkt; H3N8) was kindly provided by Janet Daly, while A/Dk/Singapore/5/97 (Dk/Sing; H5N3) was originally obtained from the Health Protection Agency collection held at Colindale (42). Recombinant PR8 viruses were produced by transfection of plasmids into 293T cells as previously described (22) and passaged once in MDCKs and then once in 8-day-old embryonated eggs. Segment 2 was sequenced from each virus stock to ensure the presence of the correct mutations. Virus titers were determined by plaque assay in MDCK cells, and plaque sizes were measured as previously described (22).
Protein analyses.
Coupled in vitro transcription-translation reactions were carried out in rabbit reticulocyte lysate using the Promega TNT system according to the manufacturer's instructions. Metabolically labeled infected cell lysates were generated and subjected to immunoprecipitation as previously described (13, 15). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blots were performed according to standard procedures. Blots were imaged by using infrared fluorescence of appropriately tagged secondary antibodies and quantified by using a LiCOR Odyssey scanner and software. Influenza RNP reconstitution assays were carried out as previously described (32), except that a synthetic vRNA encoding luciferase was used as the minigenome.
Microscopy.
Fluorescence microscopy of cells fixed with 4% formaldehyde was carried out as previously described (15, 41). Samples were imaged by using a Leica TCS SP confocal microscope. Postcapture processing of images to allow daylight visualization and reproduction was applied equally to each experiment by using Adobe Photoshop. For FRAP analysis, cells were maintained at 37°C in CO2 independent Leibovitz L-15 medium (Gibco). Images were captured on a Zeiss LSM 510 confocal microscope using a ×63 objective and a digital zoom factor of 5. GFP was excited using the 488-nm laser line of a 30-mW Ar laser running at 6.1 A and 1% output. Photobleaching was performed on a 1.4-μm2 bleach window at 100% laser output. Five prebleach and seventy postbleach images were collected at 0.39-s intervals. Fluorescence intensities of regions of interest were obtained using the LSM510 software. After correction for background fluorescence and acquisition photobleaching, diffusion coefficient (DC) values were calculated by using an adaptation of classical FRAP analysis (2) as described elsewhere (25; E. M. Loucaides et al., unpublished data).
RESULTS
Polypeptide expression patterns from IAV segment 2.
PB1-F2 expression has been postulated to occur by translation initiation at AUG4 on the PB1 mRNA (9), after scanning ribosomes have bypassed the suboptimal context AUGs 1 to 3 (Fig. 1A). Use of one or more of AUGs 7 to 9 to give rise to C-terminal PB1-F2 polypeptides has also been suggested (50). Inspection of the influenza PR8 virus segment 2 sequence shows two intervening AUG codons that are both in strong Kozak initiation consensus sequences (Fig. 1A; AUGs 5 and 6), which must therefore also be considered candidates for translation initiation. We considered AUG6 to be of less potential significance since it accesses a short ORF in reading frame 3 (Fig. 1A) and it is not well conserved in IAV, being present in only around one-third of virus isolates (18). In contrast, AUG5 is very highly conserved among natural virus isolates, being absent from only 26 of 7411 publicly accessible (GenBank) PB1 sequences (9, 18) (Table 2) . This AUG is 115 nucleotides downstream of AUG1 and 21 nucleotides downstream of AUG4. It is in the same reading frame as AUG1, and its use as an initiation codon would be predicted to give rise to a protein product identical to PB1 except for the lack of the first 39 amino acids (termed here PB1 N40).
TABLE 2.
IAV isolates lacking the PB1-N40 AUG codon
| Virus | Accession no. | Subtype | PB1 codon 40a | Length of PB1-F2 ORFb (codons) |
|---|---|---|---|---|
| A/chicken/NJ/17169/1993 | EU743092 | H5N2 | M40L | 79* |
| A/chicken/Yokohama/aq144/2001 | AB256744 | H9N2 | M40L | 79 |
| A/swine/Alberta/56626/03 | DQ280198 | H1N1 | M40I | 11† |
| A/chicken/Chis/15224/1997 | CY006041 | H5N2 | M40I | 0 |
| A/duck/Germany/1215/1973 | CY014715 | H2N3 | M40I | 8 |
| A/Ontario/1252/2007 | EU399757 | H3N2 | M40I | 8 |
| A/swine/Saitama/1996 | AB434414 | H1N2 | M40I | 8 |
| A/chicken/Laos/P0171/2007 | CY034718 | H5N1 | M40I | 8 |
| A/northern pintail/Alaska/44204-075/2006 | EU557446 | H3N8 | M40I | 90† |
| A/Chicken/Hong Kong/739/94 | AF156422 | H9N2 | M40I | 8 |
| A/partridge/Shantou/1913/2004 | EU050495 | H6N2 | M40I | 8 |
| A/chukkar/Shantou/1530/2005 | EU050533 | H6N1 | M40I | 8 |
| A/silky chicken/Shantou/1826/2004c | CY023302 | H9N2 | M40I | 8 |
| A/chicken/Hunan/774/2002 | CY023766 | H9N2 | M40I | 8 |
| A/turkey/Shantou/1915/2004 | EU050496 | H6N2 | M40I | 8 |
| A/chukkar/Shantou/89/2005 | EU050528 | H6N1 | M40I | 8 |
| A/silky chicken/Korea/S3/03 | AY862694 | H9N2 | M40I | 8 |
Substitution at PB1 codon 40.
*, This ORF has a mutation in the mitochondrial targeting sequence; †, these ORFs also possess the W9L mutation.
Nine related sequences from other H9N2 viruses are not shown (46).
Therefore, we first investigated whether PB1 N40 was produced in cells infected with IAV. Radiolabeled cell lysates from MDCK cells infected (or mock infected) with PR8 virus were treated with SDS to disrupt noncovalent protein-protein interactions and then immunoprecipitated with monospecific serum against the IAV P proteins before analysis by SDS-PAGE and autoradiography. In parallel, a plasmid containing a cDNA clone of wild-type (WT) PR8 segment 2 was in vitro translated in rabbit reticulocyte lysate to provide a marker. Polypeptides of the expected size for full-length PB1, PB2, and PA were apparent in the samples immunoprecipitated from infected cells (Fig. 1B, lanes 2, 4, and 6). However, while single polypeptide species were precipitated by anti-PB2 or anti-PA sera, two proteins were apparent in the PB1 sample (lane 2). Western blotting of the immunoprecipitates confirmed that both species were PB1 related (data not shown). Two major polypeptide species were also produced when segment 2 was in vitro translated, and these comigrated with the products from infected cells (lane 1). To investigate whether production of a shorter form of PB1 was a consistent feature of IAV, MDCK cells were infected with a panel of human, avian, and equine viruses, and the cell lysates were examined by immunoblotting. In all cases, as well as a predominant PB1 polypeptide, a shorter protein product was also detected, which, furthermore, comigrated with an in vitro-translated N40 polypeptide (Fig. 1C, compare lanes 2 to 7 with lane 8).
We hypothesized that, like PB1-F2, N40 is expressed by leaky ribosomal scanning. This hypothesis predicts that some of the mutations used by previous studies to ablate PB1-F2 expression will affect N40 expression. To date, all such studies have used (either wholly or in part) viruses in which the PB1-F2 AUG has been mutated (ΔAUG) (9, 30, 31, 50), which would be predicted to increase use of the downstream AUG5 and hence increase N40 expression levels. In addition, two studies used the strategy of altering codon W9 of PB1-F2 to a stop codon (F2-8), coincidentally introducing a nonsynonymous M40I mutation in PB1 and removing the N40 initiation codon (9, 50). Accordingly, we introduced ΔAUG and F2-8 mutations into a cDNA copy of PR8 segment 2 cloned into a bidirectional reverse genetics plasmid. Three further mutants were created as controls. First, in the F2-57 construct, codons 58 and 64 of PB1-F2 were altered to termination codons through changes that are silent in the PB1 ORF. This construct mimics the truncated PB1-F2 found in many human H1N1 viruses from 1947 onward (8, 51); these mutations were not predicted to alter N40 expression. Finally, two additional mutations were made to examine the interdependence of N40 and PB1-F2 expression. The first of these, M40L, destroys the N40 AUG but is silent in the F2 ORF. The second also alters AUG5 through an M40I change as in the F2-8 mutant. However, rather than introducing a stop codon (as in F2-8), a W9L mutation is introduced into PB1-F2. These changes to segment 2 ORFs are summarized in Table 1.
As a first test of whether AUG5 was used to initiate synthesis of a shorter form of PB1, the WT and mutated plasmids were in vitro translated. As before, WT segment 2 produced two polypeptides, of which the smaller form comigrated with in vitro-translated N40 (Fig. 1D, lanes 2 and 8). The F2-57 mutant directed translation of products similar to those of the WT gene, but plasmids lacking AUG5 (F2-8, M40L, and M40I) produced only a single polypeptide of the expected size for full-length PB1 (lanes 3, 6, and 7). In the case of the ΔAUG construct, the shorter species was translated in greater abundance than full-length PB1 (lane 5). To confirm these results and verify the PB1-F2 status of these mutants, the plasmids were transiently transfected into 293T cells, and cell lysates were examined by immunoblotting with antisera to PB1 and PB1-F2. In agreement with the in vitro results, plasmids containing WT and F2-57 segment 2 produced both full-length and, in lesser abundance, shorter forms of PB1 (Fig. 1E, upper panel, lanes 2 and 4), but only the full-length protein was produced from F2-8, M40L, or M40I plasmids (lanes 3, 6, and 7). Again, the ΔAUG plasmid expressed higher levels of the truncated PB1 polypeptide (lane 5). As predicted, an antiserum raised against the C terminus of PB1-F2 detected synthesis of the protein in cells transfected with WT, M40I, and M40L plasmids (Fig. 1E, lower panel, lanes 2, 6, and 7), although the M40I plasmid expressed somewhat lower levels, perhaps suggesting that the W9L mutation in PB1-F2 may have destabilized the protein. No specific PB1-F2 signal was detected from segment 2 plasmids with the F2-8, F2-57, or ΔAUG mutations (Fig. 1E, lower panel, lanes 3 to 5), although since the truncation of the PB1-F2 ORF in the F2-57 construct would prevent expression of the antibody epitope, we cannot exclude that a short 57-amino-acid PB1-F2 protein is made in this case. Conversely, we did not detect production of N-terminally truncated PB1-F2 polypeptides produced from AUGs 7 to 9, as seen in a previous study (50). Overall, these experiments strongly supported the hypothesis that AUG5 is used to direct translation of a shorter form of PB1 (named here as N40), as well as demonstrating that mutations affecting PB1-F2 expression have the potential to alter its expression.
In order to investigate the function of N40 and to examine the effect of altering the relative levels of segment 2 gene products during infection, reverse genetics was used to introduce the segment 2 mutations described above into PR8 virus. Viruses were rescued by transfection of eight bidirectional plasmids into 293T cells, and stocks were amplified in MDCK cells and embryonated eggs. Initially, expression of segment 2 polypeptides was analyzed at 8 h postinfection (p.i.) in MDCK cells by immunoblotting for PB1 and PB1-F2. Mirroring the plasmid expression data, PB1-F2 was only expressed in cells infected with the M40L, M40I, and WT viruses (Fig. 2A, lower panel). As before (Fig. 1B and C), a monoclonal antibody recognizing an epitope in amino acids 44 to 69 of PB1 detected two forms of the protein from cells infected with WT PR8 virus: a predominant species of the expected size for the full-length protein and a lower-abundance, slightly smaller species (Fig. 2A, top panel, lane 2). Again, in agreement with the plasmid-based data, this pattern was unchanged with the F2-57 virus (lane 4), but viruses F2-8, M40L, and M40I failed to express N40 (lanes 3, 6, and 7), while the ΔAUG virus produced equivalent amounts of short and long forms of PB1 (lane 5). To further probe the origin of the short form of PB1, a duplicate filter was probed with an antibody raised against the C-terminal 18 residues of PB1. The N40 polypeptide was detected in lysates from cells infected with WT, F2-57, and ΔAUG viruses (Fig. 2A, middle panel), indicating that the protein has an intact C terminus.
FIG. 2.

Expression of segment 2 polypeptides in virus-infected cells. MDCK cells were infected (or mock infected) with the panel of viruses as labeled, and cell lysates were obtained at various times p.i. (panel A, 8 h p.i.; panels C to E, as labeled) analyzed by Western blotting for the indicated polypeptides. Representative experiments are shown in panels A and C. (B, D, and E) The accumulation of the indicated polypeptides was quantified. The means ± the standard errors of the mean (SEM) of four independent experiments using two independently rescued virus stocks are plotted in panels B and D, while the data in panel E reflect the means and ranges of two independent experiments.
Overall, therefore, the variation in abundance of the lower-molecular-weight PB1 N40 species according to the presence, absence, or context of AUG5 both in DNA-based and authentic viral expression settings, coupled with its reactivity with antibodies directed against PB1 residues 44 to 69 and residues 740 to 757 provides strong evidence that, in addition to AUGs 1 and 4, AUG5 is also used for translation initiation in segment 2 mRNA. Next, we examined the kinetics of its expression. Low overall amounts of PB1 at 4 h p.i. made detection of the N40 form difficult, except in the case of the ΔAUG virus, where it was present in higher abundance than the full-length protein (Fig. 2C, upper panels, lane 5). At a late time point of 20 h p.i., N40 was visible in all samples from viruses with an intact AUG5 (Fig. 2C, lower panels) but in apparently lower relative amounts to the full-length protein. In confirmation of this, quantification of replicate experiments showed that, whereas N40 represented ca. 6 to 10% of the total PB1 polypeptides in cells infected with WT and F2-57 viruses at 8 h p.i., by 20 h p.i. this level had dropped to ca. 2% (Fig. 2B). Similarly, in the case of the ΔAUG virus, where N40 was more abundant than WT PB1 at 8 h p.i., by 20 h p.i. it had fallen to ca. 20% of the total PB1 protein (Fig. 2B).
During these infections, the accumulation of full-length PB1 in F2-8 and F2-57 virus-infected cells appeared near normal (Fig. 2A, C, and D). However, compared to WT virus, an early delay in PB1 expression in ΔAUG-infected cells and, conversely, increased early expression of PB1 in M40L- and M40I-infected cells was apparent (Fig. 2C, upper panels, compare lane 2 with lanes 5 to 7). Quantification confirmed a reproducible difference, with average PB1 levels in ΔAUG virus-infected cells at 4 h p.i. reduced by nearly threefold compared to WT virus, whereas those in M40L- and M40I -infected cells were raised by more than 1.5- and 3-fold, respectively (Fig. 2D). However, as infection proceeded, the differences became less marked, with PB1 levels for all mutant viruses at 20 h p.i. appearing near normal. When PB1-F2 expression was quantified, no significant accumulation was seen in cells infected with the F2-8, F2-57, and ΔAUG viruses (Fig. 2E), as expected. The levels of the protein were near normal in the case of virus M40L but reduced more than twofold for virus M40I (Fig. 2E).
In summary, these data show that N40 is expressed during infection with WT IAV where, especially at earlier times p.i., it provides an appreciable fraction of the total PB1-related polypeptides. Furthermore, its expression is altered by mutations previously used by others to abolish PB1-F2 production. Importantly, a complex interplay of expression of the three segment 2 gene products was evident, since the ΔAUG virus that expressed elevated levels of N40 in the absence of PB1-F2, displayed reduced accumulation of PB1 at early time points p.i. In contrast, N40 null viruses that also expressed PB1-F2 (M40L and M40I) displayed elevated levels of PB1 early in infection.
Function of the N40 polypeptide.
A recent study suggested that PB1-F2 influences the intracellular localization of PB1, since mutations that abolished PB1-F2 expression rendered PB1 localization more cytoplasmic late in infection (30). Accordingly, we examined PB1 localization at 8 h p.i. by immunofluorescence in cells infected with our panel of viruses, using an antiserum that reacted with both full-length PB1 and N40. As expected, PB1 was concentrated in the nuclei in cells infected with WT virus, although in many cells, some staining was also apparent in the cytoplasm (Fig. 3D). Similar patterns were also seen in cells infected with the F2-8, F2-57, M40I, and M40L viruses. However, after infection with the ΔAUG virus, PB1 staining was more notably more cytoplasmic, with many cells showing equal- or higher-intensity staining in the cytoplasm than in the nucleus. This latter result is similar to the phenotype seen by Mazur et al. (30), who also studied viruses lacking AUG4 and correlated a cytoplasmic shift of PB1 with the lack of PB1-F2 expression. However, PB1 staining remained predominantly nuclear in cells infected with the F2-8 virus (Fig. 3D) that is also PB1-F2 null but does not overexpress N40.
FIG. 3.

Intracellular localization of PB1-related polypeptides. (A and B) HeLa cells were transfected with plasmids encoding N40, PB1, GFP, or PA-GFP as indicated; fixed 24 h later; stained with anti-PB1 MAb 10.4; and imaged by confocal microscopy. (C) MDCK cells transfected with plasmids encoding N40 or (bottom row) empty vector were infected 24 h later with Dk/Sing virus and at 8 h p.i. were fixed and stained with anti-PB1 MAb 10.4 and anti-NP 2915 before imaging by confocal microscopy. (D) MDCK cells were infected with the indicated viruses, fixed, and stained with anti-PB1 MAb 10.4 at 8 h p.i. Confocal settings were kept consistent throughout each experiment.
Next, we examined the intracellular localization of N40. Since N40 lacks the sequences responsible for binding PA (21, 34, 37) and, since PB1 and PA only undergo efficient nuclear import as a dimer (16, 33), it would be predicted to be largely cytoplasmic in the absence or presence of PA. To test this, cells were transfected with plasmids that expressed N40 or the WT PB1 gene in combinations with either GFP or PA-GFP and monitored by fluorescence microscopy. Both N40 and PB1 were predominantly (but not exclusively) cytoplasmic in the absence of other viral proteins, while coexpressed GFP localized throughout the cell (Fig. 3A). PA-GFP was also largely cytoplasmic when expressed alone but, in combination with PB1, both polypeptides were concentrated in the nucleus (Fig. 3B). In contrast, when PA-GFP was coexpressed with N40, both proteins remained mostly cytoplasmic, a finding consistent with their predicted inability to form a stable complex.
PB1 also interacts with PB2 and NP, as well as potentially forming homo-oligomers (7, 13, 23). Therefore, N40 localization in the context of IAV infection was investigated. Since the N40 product does not contain any unique sequences compared to PB1, this was achieved by transfecting cells with a plasmid that expressed the PR8 N40 polypeptide (which reacts with the anti-PB1 MAb 10.4), followed by superinfection of the cells with an avian influenza virus (Dk/Sing) whose PB1 lacks the epitope. Virus-infected cells that had been transfected with empty plasmid vector stained well for NP but not for PB1, demonstrating infection and the lack of reactivity of the Dk/Sing PB1 with the anti-PB1 MAb (Fig. 3C, bottom row). In uninfected cells, N40 was largely cytoplasmic, as before (top row). However, in infected cells it localized throughout both the nucleus and the cytoplasm, indicating partial nuclear uptake in the presence of other viral proteins. This implies that, during the course of infection, N40 interacts with another viral protein(s), resulting in increased nuclear uptake.
Since N40 lacks a PA binding site, it would not be predicted to be functionally equivalent to PB1, since PA increases the affinity of PB1 for the vRNA promoter and contains the endonuclease activity involved in cap-snatching, and most studies agree that a trimeric polymerase complex is required for all forms of viral RNA synthesis (12, 14, 20, 28, 48). Accordingly, we tested the ability of N40 to support viral gene expression in an RNP reconstitution assay. Cells were cotransfected with plasmids encoding PB2, PA, NP, a synthetic vRNA containing a luciferase reporter gene, and various combinations of N40, WT PB1, and (to balance plasmid amounts where necessary) empty vector. In the presence of all four WT RNP proteins, the vRNA molecule was efficiently transcribed to give rise to large amounts of luciferase activity, whereas only background amounts were produced in the absence of PB1 (Fig. 4A). N40 was unable to replace the WT PB1 gene in this system, producing background levels of luciferase similar to those of samples lacking any source of PB1. We also examined whether N40 could act as a trans-dominant inhibitor of the viral polymerase complex by cotransfecting a 10-fold excess of the N40 expression plasmid along with the WT RNP components. Although luciferase accumulation was slightly reduced in the presence of excess N40, the degree of inhibition was less than that seen when a similar amount of plasmid vector was cotransfected and markedly less than the inhibition caused by murine Mx1 (Fig. 4A), used as a known inhibitor of IAV gene expression (5). Thus, N40 did not support transcriptional activity of the virus polymerase complex, but neither did it act as a trans-dominant inhibitor.
FIG. 4.

Ability of segment 2 polypeptides to support IAV gene expression. 293T cells were transfected with plasmids encoding PB2, PA, NP, and a synthetic vRNA analogue containing a luciferase gene along with the indicated combinations of plasmids (WT [WT PB1]) and, 48 h later, the luciferase activity quantified. The data in panel A are the means ± the SEM of three independent experiments plotted relative to the activity seen in the absence of a PB1 gene, whereas the data in panel B are the means ± the SEM of a minimum of six independent experiments plotted as the percent activity seen with WT PB1 for each experiment. **, P < 0.01.
These analyses were extended to examine whether the segment 2 mutations that alter N40 and PB1-F2 expression also affected transcriptional activity of the RNP complex. RNPs were reconstituted by plasmid transfection, and luciferase output was measured as described above. All of the segment 2 mutant plasmids supported substantial levels of influenza gene expression (Fig. 4B). Although the F2-8, F2-57, and ΔAUG mutants were indistinguishable from WT, the M40L and M40I genes consistently supported significantly higher levels of luciferase expression. Thus, in this system, the loss of N40 expression in the presence of an intact PB1-F2 ORF increases viral gene expression, whereas the loss of both gene products leaves it unaltered from WT levels.
To further examine interactions between N40 and the rest of the viral polymerase components, we turned to the sensitive technique of examining protein dynamics in live cells by fluorescence recovery after photobleaching (FRAP). We have recently established this system for analysis of the influenza virus RNP components (25; Loucaides et al., unpublished), based on a previously described set of GFP-tagged constructs (16). When the live cell dynamics of nuclear resident PB2-GFP were examined, the bleached area recovered fluorescence intensity rapidly and to near 100% of the initial value (Fig. 5A). In contrast, recovery of the full 3P complex (tagged with PB1-GFP) was slower and incomplete, indicating the presence of a substantial immobile fraction. Both recovery curves were shifted downward in the presence of additional N40, suggesting a reduction in the mobility of the GFP-tagged components. In confirmation of this, when the DC values were calculated from the recovery curves, both PB2-GFP and the full polymerase complex showed highly significant reductions of ∼2-fold in their mobility (Fig. 5C), strongly indicating an interaction with N40. This interaction was specific, since the recovery curves for an unrelated cellular nuclear protein, GFP-TAP, were essentially superimposable in the presence or absence of N40 (Fig. 5B) and there was no significant change in DC values (Fig. 5C).
FIG. 5.

FRAP analysis of N40. 293T cells were transfected with plasmids encoding PB2-GFP or PB1-GFP, PA, and PB2 (3P) (A) or GFP-TAP (B) in combination with either N40 or an equivalent amount of empty vector, and the dynamics of the GFP-tagged proteins were examined by FRAP. Average (means ± the SEM) recovery curves from at least 32 individual cells are plotted in panel A and from at least 20 cells are plotted in panel B. (C) Mean plus SEM DC values are plotted. **, P < 0.01; ***, P < 0.001.
Growth properties of the segment 2 mutant viruses.
Overall, these data indicate that the N40 polypeptide interacts with other polymerase components and therefore may have an influence on virus replication. Accordingly, the growth properties of the segment 2 mutant viruses were examined. When multicycle replication was examined in MDCK and A549 cells and in eggs, there was no significant difference in output titer, as assessed by plaque assay, between the WT virus and any of the mutants (Fig. 6A and data not shown). However, a minor but statistically significant difference in plaque size was apparent for the viruses M40L and M40I, which produced plaque areas ca. 20 to 25% smaller than WT PR8 at 24 h p.i. (Fig. 6B). This led us to examine the kinetics of virus replication in single cycle infections in MDCK cells. Under these conditions all viruses replicated to similar final titers of ∼108 PFU/ml (Fig. 6C). However, while the kinetics of the F2-8, F2-57, and ΔAUG viruses were very similar to those of the WT virus, the M40I and M40L viruses showed a notable delay in replication, with ∼100-fold lower titers at 6 h p.i. Both of these viruses have a mutated PB1 protein, as well as lacking N40 expression. However, the F2-8 virus, which has the same PB1 mutation as M40I (and the consequent inability to express N40) but is also deficient in PB1-F2 expression, showed normal single-cycle growth kinetics (Fig. 6C). Overall, we conclude that while N40 is clearly not essential for viral growth, its absence, in the context of an intact PB1-F2 ORF, leads to slower replication kinetics in cultured cells.
FIG. 6.
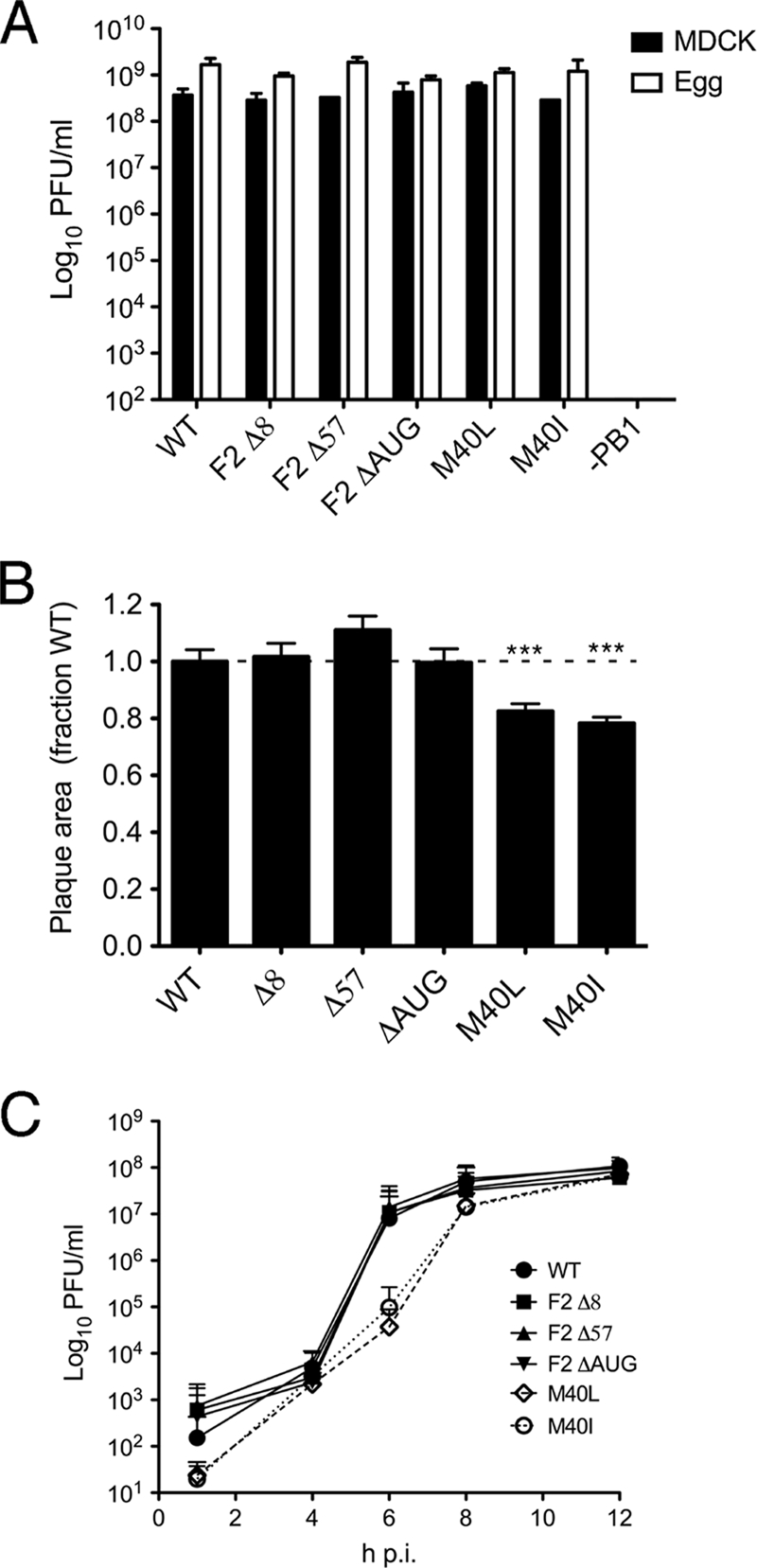
Growth properties of segment 2 mutant viruses. (A) Titers achieved at 48 h p.i. in MDCK cells infected at an MOI of 0.01 or at 48 h p.i. from eggs inoculated with 1,000 PFU are plotted. The means plus the ranges of two independent experiments are shown. (B) Mean plus SEM plaque areas in MDCK cells at 24 h p.i. are plotted. At least 150 plaques were measured from three separate experiments, and the data were normalized with respect to the mean plaque area of a matching WT virus. ***, P < 0.001. (C) Single cycle growth curves from MDCK cells infected at an MOI of 3. The means plus the SEM of four independent experiments using a minimum of two separate virus isolates are plotted.
DISCUSSION
We show here the existence of a third polypeptide species synthesized from IAV segment 2 mRNA. The protein, named here PB1 N40, is an N-terminally truncated version of PB1 that arises from translation initiation at AUG5/PB1 codon 40 in the mRNA, most likely from leaky ribosomal scanning. We base this conclusion on the observations that the polypeptide reacts with PB1 antisera whose epitopes are contained between amino acids 44 to 69 and amino acids 740 to 757 of the full-length polypeptide, its size (predicted molecular mass of 82 kDa versus 87 kDa for PB1), comigration with recombinant N40 and, most importantly, the finding that its expression levels vary predictably according to the presence, absence, or context of AUG5 both in DNA-based and in authentic viral expression settings. Its expression was detected in a variety of IAV strains, including representative human, equine, and avian strains (Fig. 1) and in substantial levels, accounting for 5 to 10% of total PB1 levels in cells infected with WT PR8 virus at 8 h p.i. (Fig. 2). Although (to the best of our knowledge) this is the first report to systematically define the origin and investigate the function of this novel addition to the IAV proteome, our data are compatible with previous studies. Akkina et al. noted that a variety of IAV strains expressed a slightly shorter PB1-related polypeptide that was unlikely to be produced by proteolysis of the full-length protein (1). Perez and Donis also commented on the presence of an N-terminally truncated form of PB1 when expression of the gene in transfected cells was driven by the vaccinia virus/T7 RNA polymerase system (38).
Having identified a 12th primary translation product of IAV, the important question becomes the function of the protein. Although N40 contains 718 of the 757 residues present in WT PB1, the lack of the N-terminal 39 amino acids removes the sequences defined as the primary interaction site with PA, which would be expected to result in profound functional differences between the two proteins (21, 34, 37). In confirmation of this, we found that N40 did not form a complex with PA that was capable of directing efficient nuclear import of the two proteins (Fig. 3) or support IAV gene expression in an RNP reconstitution assay (Fig. 4). Overexpression of N40 in the same system did not lead to trans-dominant inhibition of IAV gene expression by the WT protein (Fig. 4), and neither was expression of the N40 gene product essential for virus replication in tissue culture cells or embryonated eggs (Fig. 6). We have therefore considered the hypothesis that the N40 protein has no function in the virus life cycle and is merely an inconsequential by-product of PB1-F2 expression operating via leaky ribosomal scanning and the PB1 gene containing a downstream AUG codon. However, several arguments support more positive hypotheses. First, microscopy of N40 localization in infected cells showed increased uptake of the protein into the nucleus (Fig. 3), a finding suggestive of an interaction with one or more nuclear resident viral proteins other than PA. In addition to PA, PB1 is known to interact with PB2, NP, PB1-F2, and itself (7, 13, 23, 30), and while the location of homo-oligomerization, PB1-F2- and NP-binding domains are not known, the N40 polypeptide certainly contains the primary PB2 interaction site (19, 35, 39, 43). Consistent with this, FRAP analysis of GFP-tagged versions of PB2 and the polymerase trimer indicated an interaction with N40 in the nucleus of cells (Fig. 5). Second, mutations that affected the production of N40 revealed a complex interdependence of the expression kinetics of the three proteins translated from segment 2 mRNA (Table 1). Although the observation that loss of the PB1-F2 AUG in the ΔAUG virus upregulates translation of the downstream initiating AUG of N40 is readily explainable, the finding that the same mutation leads to decreased PB1 accumulation at early times p.i. (Fig. 2C) was unexpected. Similarly, the >3-fold difference in PB1 levels at 4 h p.i. during infection with the M40I and F2-8 viruses (Fig. 2D), both of which contain the same mutation in PB1 that abolishes N40 expression but which differ in the presence or absence of an intact PB1-F2 ORF, respectively, is further evidence that expression of the three polypeptides is not independent. In all cases however, PB1 accumulation balanced out to near-normal levels later in infection. To what extent this interdependence (and the changing relative proportions of N40 and PB1 accumulation during infection with WT virus) results from translational mechanisms and/or the differential stability of the three proteins remains to be determined. The data indicating a physical interaction between PB1 and PB1-F2 (30) and a study suggesting that PB1 abundance is regulated by additional factors than simply the poor Kozak consensus of AUG1 (29) are consistent with both possibilities. Third, the loss of N40 expression in the presence of an intact PB1-F2 gene leads to a replication defect in MDCK cells (Fig. 6). The overlapping/coincident nature of the PB1, PB1-F2, and N40 ORFs prevents the design of “clean” mutations that affect the structure and expression of only one protein at a time, but this conclusion can safely be drawn from comparing the behavior of the F2-8, M40I, and M40L viruses. While the slow replication kinetics of the M40L virus could perhaps be attributed to its eponymous PB1 mutation, the M40I and F2-8 viruses again provide a matched comparison: they contain the same PB1 mutation, the same deficit in N40 synthesis but differ in their respective ability or inability to express PB1-F2. The former virus that expresses two of the three segment 2 polypeptides, PB1 and PB1-F2, grew slowly, while the latter virus, which expresses only PB1, grew normally. We also note that whereas the vast majority of natural IAV isolates have PB1 M40, the small number that do not, possess either an isoleucine or a leucine residue (Table 2). Multiple IAVs with a PB1 M40I mutation (similar to our F2-8 virus) were isolated from a variety of avian species in the South China area (46), ruling out the possibility of sequencing errors. Furthermore, all known influenza virus B and C PB1 proteins have an isoleucine or leucine residue, respectively, at position 40, overall suggesting that a methionine is not obligatory for polymerase function. Thus, we have two current working hypotheses: first, that N40 expression has no direct functional relevance per se but is indirectly important in maintaining the correct balance between PB1 and PB1-F2 expression. In this regard, it is interesting that of the 26 natural virus isolates lacking the N40 AUG codon, only three possess a long (>78-amino-acid) PB1-F2 ORF that includes the mitochondrial targeting sequence, and of these, one virus has alterations to two amino acid residues shown to be important for mitochondrial localization (47), while a third has the W9L mutation that in PR8 M40I reduced the steady-state abundance of the protein (Table 2 and Fig. 2A and E). A second hypothesis, not incompatible with the first, holds that the N40 protein has a nonessential and as-yet-undetermined function in virus replication. Further work is necessary to test these proposals.
Whether or not the N40 product has a positive function in the virus life cycle, our finding that mutations of the type used in several prior studies to investigate the role of PB1-F2 affect its expression and that of PB1 is important. Elucidation of the function of PB1-F2 has not been straightforward. Although all studies in which segment 2 has been mutated to block PB1-F2 expression agree that the protein is not essential for virus replication in tissue culture, eggs, or mice, various phenotypes have been observed for the PB1-F2 null viruses. These phenotypes range from apparently unaltered replication kinetics in any tested system to both slight increases and decreases in growth parameters in MDCK cells, along with a general but not consistent tendency to reduce pathogenicity in mice (9, 30, 31, 50). However, we note that the original publication on PB1-F2 (9) relied in part for its conclusions on a virus in which the N40 AUG had been mutated (similar to our F2-8 virus), thus blocking expression of N40, as well as full-length PB1-F2. Moreover, all of the studies examining PB1-F2 null viruses that we are aware of used, at least in part, viruses in which the PB1-F2 AUG had been mutated, which is analogous to our ΔAUG virus. Our data predict that these viruses will similarly overexpress the N40 product. If this is indeed the case, some aspects of these data may be open to reinterpretation, at least until the role of N40 is better defined. As a case in point, while our data showing increased cytoplasmic staining with a PB1 antibody after infection with the ΔAUG virus (Fig. 3) recapitulates a previous study (30) with an equivalent virus, the fact that we did not see a similar outcome with other PB1-F2 deletion viruses, coupled with the observation that only the ΔAUG virus expresses much higher levels of the largely cytoplasmic N40 protein, strongly argues that, at least for our virus, the cytoplasmic species is in fact the shorter form of PB1.
To summarize, we have shown that three, not two, of the 5′-proximal AUG codons in segment 2 mRNA direct translation initiation, resulting in the expression of not only the previously defined PB1 and PB1-F2 polypeptides but also a novel PB1 variant, PB1 N40. The function of the N40 polypeptide remains to be defined, but the overlapping nature and apparent translational interdependence of the segment 2 cistrons makes it potentially very difficult to disentangle the relative contributions of the three protein products to viral replication and pathogenicity. Recent studies in which virulence has been correlated with PB1-F2 sequence polymorphisms well downstream of the ribosomal initiation sites in the mRNA provide compelling evidence for the importance of PB1-F2 (10, 31) even in light of the findings reported here. Nevertheless, we would argue that further work is necessary to fully understand the contributions the segment 2 gene products make to IAV pathogenicity.
Acknowledgments
We thank Ervin Fodor, Ron Fouchier, Mark Krystal, Laurence Tiley, Jonathan Yewdell, and Adrian Whitehouse for the gift of reagents.
This study was supported by grants from the Medical Research Council (G0700815 to P.D.) and the Biotechnology and Biological Sciences Research Council (BB/C516495/2 to W.S.B.). A.F. is supported by a Ph.D. studentship from the Wellcome Trust. W.S.B. is a member of the Wellcome Trust funded Centre for Respiratory Infections.
Footnotes
Published ahead of print on 3 June 2009.
REFERENCES
- 1.Akkina, R. K., J. C. Richardson, M. C. Aguilera, and C. M. Yang. 1991. Heterogeneous forms of polymerase proteins exist in influenza A virus-infected cells. Virus Res. 1917-30. [DOI] [PubMed] [Google Scholar]
- 2.Axelrod, D., D. E. Koppel, J. Schlessinger, E. Elson, and W. W. Webb. 1976. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 161055-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baigent, S. J., and J. W. McCauley. 2003. Influenza type A in humans, mammals and birds: determinants of virus virulence, host-range and interspecies transmission. Bioessays 25657-671. [DOI] [PubMed] [Google Scholar]
- 4.Basler, C. F., and P. V. Aguilar. 2008. Progress in identifying virulence determinants of the 1918 H1N1 and the Southeast Asian H5N1 influenza A viruses. Antivir. Res. 79166-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benfield, C. T., J. W. Lyall, G. Kochs, and L. S. Tiley. 2008. Asparagine 631 variants of the chicken Mx protein do not inhibit influenza virus replication in primary chicken embryo fibroblasts or in vitro surrogate assays. J. Virol. 827533-7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bishop, D. H., J. A. Huddleston, and G. G. Brownlee. 1982. The complete sequence of RNA segment 2 of influenza A/NT/60/68 P1 protein. Nucleic Acids Res. 101335-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biswas, S. K., P. L. Boutz, and D. P. Nayak. 1998. Influenza virus nucleoprotein interacts with influenza virus polymerase proteins. J. Virol. 725493-5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, G. W., C. C. Yang, K. C. Tsao, C. G. Huang, L. A. Lee, W. Z. Yang, Y. L. Huang, T. Y. Lin, and S. R. Shih. 2004. Influenza A virus PB1-F2 gene in recent Taiwanese isolates. Emerg. Infect. Dis. 10630-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, W., P. A. Calvo, D. Malide, J. Gibbs, U. Schubert, I. Bacik, S. Basta, R. O'Neill, J. Schickli, P. Palese, P. Henklein, J. R. Bennink, and J. W. Yewdell. 2001. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 71306-1312. [DOI] [PubMed] [Google Scholar]
- 10.Conenello, G. M., D. Zamarin, L. A. Perrone, T. Tumpey, and P. Palese. 2007. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 31414-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Wit, E., M. I. Spronken, T. M. Bestebroer, G. F. Rimmelzwaan, A. D. Osterhaus, and R. A. Fouchier. 2004. Efficient generation and growth of influenza virus A/PR/8/34 from eight cDNA fragments. Virus Res. 103155-161. [DOI] [PubMed] [Google Scholar]
- 12.Dias, A., D. Bouvier, T. Crepin, A. A. McCarthy, D. J. Hart, F. Baudin, S. Cusack, and R. W. Ruigrok. 2009. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458914-918. [DOI] [PubMed] [Google Scholar]
- 13.Digard, P., V. C. Blok, and S. C. Inglis. 1989. Complex formation between influenza virus polymerase proteins expressed in Xenopus oocytes. Virology 171162-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elton, D., P. Digard, L. Tiley, and J. Ortin. 2006. Structure and function of the influenza virus RNP, p. 1-36. In Influenza virology: current topics. Caister Academic Press, Wymondham, United Kingdom.
- 15.Elton, D., M. Simpson-Holley, K. Archer, L. Medcalf, R. Hallam, J. McCauley, and P. Digard. 2001. Interaction of the influenza virus nucleoprotein with the cellular CRM1-mediated nuclear export pathway. J. Virol. 75408-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fodor, E., and M. Smith. 2004. The PA subunit is required for efficient nuclear accumulation of the PB1 subunit of the influenza A virus RNA polymerase complex. J. Virol. 789144-9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibbs, J. S., D. Malide, F. Hornung, J. R. Bennink, and J. W. Yewdell. 2003. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J. Virol. 777214-7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gog, J. R., S. Afonso Edos, R. M. Dalton, I. Leclercq, L. Tiley, D. Elton, J. C. von Kirchbach, N. Naffakh, N. Escriou, and P. Digard. 2007. Codon conservation in the influenza A virus genome defines RNA packaging signals. Nucleic Acids Res. 351897-1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez, S., T. Zurcher, and J. Ortin. 1996. Identification of two separate domains in the influenza virus PB1 protein involved in the interaction with the PB2 and PA subunits: a model for the viral RNA polymerase structure. Nucleic Acids Res. 244456-4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hara, K., F. I. Schmidt, M. Crow, and G. G. Brownlee. 2006. Amino acid residues in the N-terminal region of the PA subunit of influenza A virus RNA polymerase play a critical role in protein stability, endonuclease activity, cap binding, and virion RNA promoter binding. J. Virol. 807789-7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He, X., J. Zhou, M. Bartlam, R. Zhang, J. Ma, Z. Lou, X. Li, J. Li, A. Joachimiak, Z. Zeng, R. Ge, Z. Rao, and Y. Liu. 2008. Crystal structure of the polymerase PA(C)-PB1(N) complex from an avian influenza H5N1 virus. Nature 4541123-1126. [DOI] [PubMed] [Google Scholar]
- 22.Hutchinson, E. C., M. D. Curran, E. K. Read, J. R. Gog, and P. Digard. 2008. Mutational analysis of cis-acting RNA signals in segment 7 of influenza A virus. J. Virol. 8211869-11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jorba, N., E. Area, and J. Ortin. 2008. Oligomerization of the influenza virus polymerase complex in vivo. J. Gen. Virol. 89520-524. [DOI] [PubMed] [Google Scholar]
- 24.Kozak, M. 1986. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 44283-292. [DOI] [PubMed] [Google Scholar]
- 25.Kreysa, E. M., J. C. von Kirchbach, E. Fodor, and P. Digard. 2008. Intranuclear dynamics of the influenza A virus RNA polymerase as revealed by live cell imaging studies, p. 82-84. In J. M. Katz (ed.), Options for the control of influenza VI. International Medical Press, London, England.
- 26.Kuiken, T., E. C. Holmes, J. McCauley, G. F. Rimmelzwaan, C. S. Williams, and B. T. Grenfell. 2006. Host species barriers to influenza virus infections. Science 312394-397. [DOI] [PubMed] [Google Scholar]
- 27.Landolt, G. A., and C. W. Olsen. 2007. Up to new tricks: a review of cross-species transmission of influenza A viruses. Anim. Health Res. Rev. 81-21. [DOI] [PubMed] [Google Scholar]
- 28.Lee, M. T., K. Bishop, L. Medcalf, D. Elton, P. Digard, and L. Tiley. 2002. Definition of the minimal viral components required for the initiation of unprimed RNA synthesis by influenza virus RNA polymerase. Nucleic Acids Res. 30429-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maeda, Y., H. Goto, T. Horimoto, A. Takada, and Y. Kawaoka. 2004. Biological significance of the U residue at the −3 position of the mRNA sequences of influenza A viral segments PB1 and NA. Virus Res. 100153-157. [DOI] [PubMed] [Google Scholar]
- 30.Mazur, I., D. Anhlan, D. Mitzner, L. Wixler, U. Schubert, and S. Ludwig. 2008. The proapoptotic influenza A virus protein PB1-F2 regulates viral polymerase activity by interaction with the PB1 protein. Cell Microbiol. 101140-1152. [DOI] [PubMed] [Google Scholar]
- 31.McAuley, J. L., F. Hornung, K. L. Boyd, A. M. Smith, R. McKeon, J. Bennink, J. W. Yewdell, and J. A. McCullers. 2007. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2240-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mullin, A. E., R. M. Dalton, M. J. Amorim, D. Elton, and P. Digard. 2004. Increased amounts of the influenza virus nucleoprotein do not promote higher levels of viral genome replication. J. Gen. Virol. 853689-3698. [DOI] [PubMed] [Google Scholar]
- 33.Nieto, A., S. de la Luna, J. Barcena, A. Portela, J. Valcarcel, J. A. Melero, and J. Ortin. 1992. Nuclear transport of influenza virus polymerase PA protein. Virus Res. 2465-75. [DOI] [PubMed] [Google Scholar]
- 34.Obayashi, E., H. Yoshida, F. Kawai, N. Shibayama, A. Kawaguchi, K. Nagata, J. R. Tame, and S. Y. Park. 2008. The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature 4541127-1131. [DOI] [PubMed] [Google Scholar]
- 35.Ohtsu, Y., Y. Honda, Y. Sakata, H. Kato, and T. Toyoda. 2002. Fine mapping of the subunit binding sites of influenza virus RNA polymerase. Microbiol. Immunol. 46167-175. [DOI] [PubMed] [Google Scholar]
- 36.Peiris, J. S., M. D. de Jong, and Y. Guan. 2007. Avian influenza virus (H5N1): a threat to human health. Clin. Microbiol. Rev. 20243-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez, D. R., and R. O. Donis. 1995. A 48-amino-acid region of influenza A virus PB1 protein is sufficient for complex formation with PA. J. Virol. 696932-6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez, D. R., and R. O. Donis. 2001. Functional analysis of PA binding by influenza a virus PB1: effects on polymerase activity and viral infectivity. J. Virol. 758127-8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poole, E. L., L. Medcalf, D. Elton, and P. Digard. 2007. Evidence that the C-terminal PB2-binding region of the influenza A virus PB1 protein is a discrete alpha-helical domain. FEBS Lett. 5815300-5306. [DOI] [PubMed] [Google Scholar]
- 40.Simonsen, L., M. J. Clarke, G. D. Williamson, D. F. Stroup, N. H. Arden, and L. B. Schonberger. 1997. The impact of influenza epidemics on mortality: introducing a severity index. Am. J. Public Health 871944-1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simpson-Holley, M., D. Ellis, D. Fisher, D. Elton, J. McCauley, and P. Digard. 2002. A functional link between the actin cytoskeleton and lipid rafts during budding of filamentous influenza virions. Virology 301212-225. [DOI] [PubMed] [Google Scholar]
- 42.Thompson, C. I., W. S. Barclay, M. C. Zambon, and R. J. Pickles. 2006. Infection of human airway epithelium by human and avian strains of influenza a virus. J. Virol. 808060-8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toyoda, T., D. M. Adyshev, M. Kobayashi, A. Iwata, and A. Ishihama. 1996. Molecular assembly of the influenza virus RNA polymerase: determination of the subunit-subunit contact sites. J. Gen. Virol. 77(Pt. 9)2149-2157. [DOI] [PubMed] [Google Scholar]
- 44.Whiteley, A., D. Major, I. Legastellois, L. Campitelli, I. Donatelli, C. I. Thompson, M. C. Zambon, J. M. Wood, and W. S. Barclay. 2007. Generation of candidate human influenza vaccine strains in cell culture: rehearsing the European response to an H7N1 pandemic threat. Influenza Other Respir. Viruses 1157-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williams, B. J., J. R. Boyne, D. J. Goodwin, L. Roaden, G. M. Hautbergue, S. A. Wilson, and A. Whitehouse. 2005. The prototype gamma-2 herpesvirus nucleocytoplasmic shuttling protein, ORF 57, transports viral RNA through the cellular mRNA export pathway. Biochem. J. 387295-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu, K. M., G. J. Smith, J. Bahl, L. Duan, H. Tai, D. Vijaykrishna, J. Wang, J. X. Zhang, K. S. Li, X. H. Fan, R. G. Webster, H. Chen, J. S. Peiris, and Y. Guan. 2007. The genesis and evolution of H9N2 influenza viruses in poultry from southern China, 2000 to 2005. J. Virol. 8110389-10401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamada, H., R. Chounan, Y. Higashi, N. Kurihara, and H. Kido. 2004. Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 578331-336. [DOI] [PubMed] [Google Scholar]
- 48.Yuan, P., M. Bartlam, Z. Lou, S. Chen, J. Zhou, X. He, Z. Lv, R. Ge, X. Li, T. Deng, E. Fodor, Z. Rao, and Y. Liu. 2009. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 458909-913. [DOI] [PubMed] [Google Scholar]
- 49.Zamarin, D., A. Garcia-Sastre, X. Xiao, R. Wang, and P. Palese. 2005. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 1e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zamarin, D., M. B. Ortigoza, and P. Palese. 2006. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J. Virol. 807976-7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zell, R., A. Krumbholz, A. Eitner, R. Krieg, K. J. Halbhuber, and P. Wutzler. 2007. Prevalence of PB1-F2 of influenza A viruses. J. Gen. Virol. 88536-546. [DOI] [PubMed] [Google Scholar]