

Abstract
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is an inherited cancer syndrome linked to biallelic inactivation of the gene encoding the tricarboxylic acid cycle enzyme fumarate hydratase (FH). Individuals with HLRCC are at risk to develop cutaneous and uterine leiomyomas and an aggressive form of kidney cancer. Pseudohypoxic drive—the aberrant activation of cellular hypoxia response pathways despite normal oxygen tension—is considered to be a likely mechanism underlying the etiology of this tumor. Pseudohypoxia requires the oxygen-independent stabilization of the α subunit of the hypoxia-inducible transcription factor (HIF-1α). Under normoxic conditions, proline hydroxylation of HIF-1α permits VHL recognition and subsequent targeting for proteasomal degradation. Here, we demonstrate that inactivating mutations of FH in an HLRCC-derived cell line result in glucose-mediated generation of cellular reactive oxygen species (ROS) and ROS-dependent HIF-1α stabilization. Additionally, we demonstrate that stable knockdown of FH in immortalized renal epithelial cells results in ROS-dependent HIF-1α stabilization. These data reveal that the obligate glycolytic switch present in HLRCC is critical to HIF stabilization via ROS generation.
Patients with hereditary leiomyomatosis and renal cell cancer (HLRCC) harbor germ line mutations of the FH gene, which encodes the tricarboxylic acid cycle enzyme fumarate hydratase, and affected individuals are at risk for the development of leiomyomas of the skin and uterus (fibroids) as well as kidney cancer (11, 25, 37). Genetic analysis of tumor samples indicates that FH acts as a tumor suppressor gene (37). The renal tumors that develop in HLRCC patients are notable for their aggressiveness, and effective systemic therapies are lacking at this time. Hence, identification of the molecular mechanisms that underlie the pathogenesis of this disease is needed to facilitate the development of targeted therapeutic strategies. Moreover, such studies may provide further insight into the role of mitochondrial metabolism in both normal and aberrant cellular physiology.
FH catalyzes the enzymatic step of the tricarboxylic (TCA) cycle that hydrates fumarate to form malate. Proposed mechanisms for HLRCC tumor formation include apoptotic resistance, oxidative stress, and pseudohypoxic drive (10). Of these, most reports to date support a role for pseudohypoxic drive, based specifically on studies of hypoxia-inducible transcription factor 1α (HIF-1α) expression. Pseudohypoxia is defined as the aberrant activation of hypoxia response pathways under normal oxygen conditions. HIF-1α expression is elevated both in HLRCC tumor specimens and in normoxic cells in which FH expression has been transiently suppressed with small interfering RNA (siRNA) (16). HIF-2α expression is also elevated in HLRCC tumor samples, although to a lesser extent than is HIF-1α. In addition, there is clear evidence of upregulated transcription of HIF target genes in HLRCC tumor samples and in FH siRNA-treated cells (16, 30). Further support for hypoxia-independent HIF activation in HLRCC tumor samples is provided by a recent study of FH knockout mice, in which developing renal cysts were characterized by elevated nuclear HIF-1α expression and increased transcription of the HIF target genes Glut1 and VEGF (32).
Cellular sensing of oxygen levels is tightly regulated by enzymes termed HIF prolyl hydroxylases (HPHs) (3, 5, 17, 18). These enzymes require the cofactors Fe2+ and ascorbate as well as the cosubstrates molecular oxygen (O2) and 2-oxoglutarate (2-OG) (19). The reaction catalyzed by the HPHs results in hydroxylation of conserved HIF proline residues, using molecular oxygen as the hydroxyl group donor and producing succinate as a side reactant. In their hydroxylated form, HIF-α (either HIF-1α or HIF-2α) proteins are recognized by the E3 ubiquitin ligase pVHL, resulting in proteasome targeting and degradation (17). When O2 levels are limiting (e.g., hypoxia) HIF-α remains unhydroxylated and able to heterodimerize with its constitutively expressed partner protein HIF-1β (for a review, see reference 9). This heterocomplex transcriptionally upregulates several genes, including EPO, VEGF, GLUT1, a number of glycolytic enzyme genes, and TGFα, all of which serve to coordinate the cellular and organismal response to hypoxia. Interestingly, VHL mutations are found in the majority of clear cell renal carcinomas, the most common histologic variant of kidney cancer (8). Consistent with the function of VHL, stabilization of HIF-1α is often found in clear cell renal carcinomas with identifiable VHL mutations (41).
Thus far, VHL mutation status has not been characterized in HLRCC tumors. However, the link between FH mutation/loss and normoxic stabilization of HIF may depend instead on the biochemical consequences of reduced or absent FH activity. An FH-deficient tumor is likely to contain elevated levels of fumarate, and, using magnetic resonance spectroscopy, this has been confirmed in HLRCC fibroids (31). We have proposed that fumarate may initiate pseudohypoxia by competitively inhibiting 2-OG-dependent HIF-α hydroxylation (35). Indeed, we recently reported fumarate to be a potent competitive inhibitor of purified HPH and an inhibitor of HIF-α hydroxylation in vitro and in vivo (16).
Oxidative stress has also been proposed as a possible mechanism of tumorigenesis in several model systems, and abundant evidence suggests that reactive oxygen species (ROS) can promote stabilization of HIF-α. As ROS generation is one of the earliest cellular responses to high intracellular glucose (13), the dramatic increase in glucose utilization following experimental abrogation of FH activity (16) suggests that ROS generation may be an inescapable by-product of HLRCC metabolism. Since our previous study examined only the acute impact of diminished FH activity on HIF, we wished to assess the possible contribution of long-term glycolytic addiction to HIF stabilization, as this is more likely to reflect the in vivo situation in HLRCC tumor cells.
In this report, we demonstrate that ROS is constitutively elevated and is an important mediator of HIF-1α stabilization in the first reported HLRCC-derived tumor cell line, as well as in other renal epithelial cells whose FH expression has been stably suppressed. Moreover, we propose that the obligate glycolytic phenotype of HLRCC tumor cells contributes to elevated levels of cellular ROS. These data identify a prominent role for ROS in maintaining the pseudohypoxic gene expression profile characteristic of HLRCC.
MATERIALS AND METHODS
Reagents and cell culture.
All chemical reagents were purchased from Sigma, except dimethyloxalylglycine (DMOG) (Cayman) and PS341 (Millenium). UOK262 cells were obtained from a patient undergoing clinically indicated surgical resection for kidney cancer. Samples were obtained as part of a clinical/research protocol approved by the Institutional Review Board of the National Cancer Institute. Tumor cells were harvested from a metastatic deposit in an HLRCC patient (germ line FH mutation present) with advanced kidney cancer and immediately placed into cell culture without any transformation-based manipulations. UOK262 cells were confirmed to have absent FH enzymatic activity utilizing an assay as previously described (29). The resultant cell line is described in further detail elsewhere (Y. Yang, V. A. Valera Romero, H. M. Padillia-Nash, C. Sourbier, C. D. Vocke, M. A. Vira, M. S. Abu-Asab, G. Bratslavsky, M. Tsokos, M. J. Merino, P. A. Pinto, R. Srinivasan, T. Ried, L. M. Neckers, and W. M. Linehan, submitted for publication). UOK262 cells were stably transfected utilizing retrovirus with a control vector (CV; pBabePuro) or a vector containing wild-type FH with a C-terminal FLAG tag. Puromycin-resistant clones were selected and screened for transgene expression via immunoblotting. All cell lines except HK-2 were cultured in high-glucose (4.5 g/liter) Dulbecco's modified Eagle's medium (DMEM) supplemented with penicillin (100 U/ml), streptomycin (100 mg/ml), 10% heat-inactivated fetal bovine serum, and HEPES (10 mM). HK-2 cells were purchased from ATCC and were cultured in DMEM/Ham's F-12 media with l-glutamine supplemented with penicillin (100 U/ml), streptomycin (100 mg/ml), 10% heat-inactivated fetal bovine serum, and HEPES (10 mM).
Lactate measurements.
A total of 5 × 105 cells were allowed to adhere overnight in standard media. Plates were washed, and cells were incubated in modified Krebs buffer (1.3 mM CaCl2, 1.3 mM MgCl2, 124 mM NaCl, 3.5 mM KCl, 1.25 mM K2HPO4, 26.3 mM NaHCO3 supplemented with 10 mM HEPES) with glucose (5 mM). After 24 h in Krebs buffer, media were harvested and analyzed for lactate levels using a Synchron LX20 analyzer (Beckman Coulter).
Western blotting.
All HIF-1α and CREB immunoblot analyses were performed on nuclear extracts. Nuclear extracts were obtained utilizing differential salt lysis buffers as previously described (16). All other immunoblot analyses were performed on whole-cell lysates prepared with the use of radioimmunoprecipitation assay buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) supplemented with protease inhibitor cocktail (Roche). Thirty micrograms of lysate proteins (either nuclear or whole cell) was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Membranes were blocked with 5% nonfat milk in TBST (10 mM Tris-HCl, 100 mM NaCl, 0.1% Tween 20). Membranes were then incubated with the following primary antibodies: HIF-1α (BD Transduction), CREB (Cell Signaling), hydroxylated HIF-1α (Rockland Immunochemicals), protein kinase C-δ (PKC-δ) (BD Transduction), FH (Genetex), α-tubulin (Abcam), p47phox (Cell Signaling), and glyceraldehyde-3-phosphatedehydrogenase (GAPDH) (Novus Biologicals). Membranes were washed with TBST followed by incubation with the indicated secondary antibody (horseradish peroxidase conjugated). Proteins were visualized with enhanced chemiluminescence.
DCF ROS measurements in cultured cell lines.
Back clear-view 96-well plates (PerkinElmer) were plated with 5,000 cells and treated the following day. The 5-(and -6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) (Invitrogen), becomes fluorescent after oxidation to DCF by ROS in live cells. After the above-indicated treatment, plates were loaded with 10 μM dye in phosphate-buffered saline for 1 hour at 37°C (100 μl/well). The buffer was then removed, and the cells were returned to prewarmed Krebs buffer media with glucose (4.5 mg/ml). After 1 hour of recovery, oxidation of the dye was measured by fluorescence detection using excitation and emission wavelengths of 488 and 520 nm, respectively, in a multiwell fluorescence plate reader (Wallac 1420 Victor; PerkinElmer). To normalize the results, protein was precipitated with 30 μl of acetone and resuspended in 20 μl of H2O. A bicinchoninic acid assay (Pierce Biotechnology, Inc., Rockford, IL) was performed to obtain the protein concentration in each well. The ROS level was then normalized to the protein content.
ROS measurements in Saccharomyces cerevisiae.
Cells were grown in a rotary shaker at 30°C in liquid YPD medium (1% yeast extract, 2% Bacto peptone, 2% glucose supplemented with 40 μg/ml adenine) to a density of 2 × 108 cells/ml in the presence of 5 μg/ml H2-DCFDA. Prior to analysis, cultures were sonicated and then diluted to a density of 1 × 106 cells/ml in 50 mM Tris-HCl (pH 7.5). Fluorescence data from 10,000 cells per sample were then collected using a Becton-Dickinson FACSCalibur flow cytometer and analyzed using Cell Quest Pro software. Fluorescence-activated cell sorter parameters were set at excitation and emission settings of 304 and 551 nm (filter FL-1), respectively. The ROS level is expressed as mean fluorescence channel number (± standard deviation [SD]).
NADPH oxidase activity.
NADPH oxidase activity was measured as previously described for renal epithelial cells utilizing a lucigenin-enhanced chemiluminescence method (2).
RNA interference.
For FH knockdown, an invalidated short hairpin RNA (shRNA) target set (four clones total) was obtained (Sigma). shRNA constructs were screened, and a clone was identified which resulted in significant knockdown of FH at the protein level, referred to as shFH. CV and shFH were transfected into HK-2 renal epithelial cells, using the Nucleofector system (Amaxa) according to the manufacturer's protocol, and stable transfectants (single-cell clones) were obtained following selection in puromycin.
For measurement of NADPH oxidase activity following PKC-δ knockdown, cells were transfected with ON-TARGETplus SMARTpool siRNA reagent (Thermo Fisher) with the Amaxa Nucleofector system according to the manufacturer's protocol. Cells were harvested at 48 h following transfection, and lysates were analyzed for PKC-δ expression to confirm knockdown. For assessment of effects on nuclear HIF-1α levels, either pooled PKC-δ siRNA (Dharmacon) or siRNA duplex (sense, 5′-GGAGUAACAGGAAACAUCAdTdT-3′; antisense, 5′-UGAUGUUUCCUGUUACUCCdTdT-3′) (Sigma-Aldrich) was utilized with similar effects on HIF-1α levels. For p47phox knockdown, pooled siRNA was utilized (Santa Cruz Biotechnology).
Fe measurements.
Fe2+ and Fe3+ ratios were determined using the QuantiChrom iron assay kit (BioAssay Systems) following the manufacturer's instructions. The chromogen in this assay specifically reacts with Fe2+ and can be measured with or without reductant to permit measurement of oxidized iron ions (Fe3+).
Statistics.
Unless otherwise noted, data are presented as the mean ± SD and are representative of at least three independent experiments.
RESULTS
HIF-1α expression in the HLRCC tumor cell line UOK262 is glucose dependent.
Previous reports have established that siRNA-mediated FH knockdown results in HIF-1α stabilization(16, 21). While these studies support a role for pseudohypoxia in HLRCC tumor formation and progression, the recent development of a cell line from an HLRCC tumor specimen, hereafter referred to as UOK262, provides a more biologically relevant model to study HLRCC. These tumor cells were obtained from an abdominal metastatic site in an HLRCC patient with a known germ line mutation of FH. Cells were obtained and immediately placed into culture without the need for any transformation-based manipulations. Genotype and cytogenetic analyses demonstrate that UOK262 cells possess a mutant FH allele present in the germ line (Q132P) and have lost the second wild-type allele. Whereas fibroblasts from HLRCC patients (which contain a single allele alteration) have intermediate FH enzymatic activity, FH enzymatic activity in UOK262 cells is undetectable (Yang et al., submitted for publication). In vitro UOK262 cell growth requires high glucose levels, and the cells express elevated levels of the glucose transporter protein GLUT1 (Yang et al., submitted for publication), as does HLRCC tumor tissue (16). The nonfermentable sugar galactose cannot substitute for glucose in supporting UOK262 cell growth (Fig. 1A), suggesting that these cells lack mitochondrial respiration and are addicted to glycolysis. This phenotype is identical to that of yeast lacking fumarase activity (22). In support of this hypothesis, UOK262 cells produce significantly more extracellular lactate than do HK-2 and HEK293 renal epithelial cells (Fig. 1B). Nuclear HIF-1α levels were readily detectable in normoxic UOK262 cells, in contrast to HEK293 and HK-2 cells, which both express wild-type FH protein (Fig. 1C). Expression of wild-type, FLAG-tagged FH results in a marked reduction in nuclear HIF-1α levels (Fig. 1D). HIF-1α expression in UOK262 cells was glucose dependent, since the protein was rapidly lost from cells cultured in the absence of glucose or in the presence of excess 2-deoxyglucose, a competitive inhibitor of glucose uptake (Fig. 1E, top panel). Moreover, UOK262 cells were not able to sustain nuclear HIF-1α levels if glucose was replaced by galactose for even a brief time period of 1 hour when no cytotoxicity was observed (Fig. 1E, bottom panel). While these data are consistent with the molecular and biochemical profile of HLRCC tumor specimens and cell lines acutely exposed to FH siRNA (16), the glucose dependence of HIF-1α expression and growth in UOK262 cells suggest a more complex relationship between chronic loss of FH activity and maintenance of pseudohypoxia than had been previously considered.
FIG. 1.

HIF-1α expression in UOK262 cells is glucose dependent. (A) UOK262 cells were seeded at a starting density of 104 cells/ml (bold line). Cell counts were determined 12 h after seeding in Krebs buffer with or without glucose or galactose. (B) A total of 5 × 105 cells were allowed to adhere and then were incubated in Krebs buffer containing 10 mM glucose and no lactate in six-well plates. Media were harvested after 24 h of incubation and analyzed for lactate levels using a Synchron LX20 analyzer (Beckman Coulter). Data represent values adjusted for cell count relative to HK-2 cells that were normalized to 1. Statistical significance with a P value of <0.05 is denoted with an asterisk. (C) Adherent UOK262, HEK293, and HK-2 renal cells were incubated in Krebs buffer for 30 min. Nuclear lysates were harvested and immunoblotted for HIF-1α levels. Immunoblotting for CREB is used to confirm equal protein loading. (D) UOK262 cells were stably transfected with CV or vector with FLAG-tagged wild-type FH. Whole-cell and nuclear extracts were immunoblotted for FLAG and HIF-1α, respectively. Immunoblots for GAPDH and CREB confirm equal protein loading in whole-cell and nuclear extracts, respectively. (E) (Top panel) UOK262 cells were treated in plain Krebs buffer (Untreated) or Krebs buffer with glucose (5 mM) ± 2-deoxyglucose (5 mM) (2-DG) for 2 and 4 h. Nuclear lysates were immunoblotted for HIF-1α and CREB (to confirm equal protein loading). (Bottom panel) UOK262 cells were treated in plain Krebs buffer (Untreated) or Krebs buffer with glucose (5 mM) or galactose (5 mM) for 1 h. Nuclear lysates were immunoblotted for HIF-1α and CREB (to confirm equal protein loading).
Oxidative stress mediates HIF-1α stabilization in HLRCC renal cancer cells.
We next examined whether accumulation of glycolytic end products may contribute to the maintenance of these cells’ pseudohypoxic drive. Recently, pyruvate was reported to promote HIF-1α stabilization via inhibition of HPH (27). Given their glucose dependence, we expected that UOK262 cells would contain increased levels of glycolytic metabolites, including pyruvate, which could cooperate with elevated fumarate to further stabilize HIF-1α. Unexpectedly, we found that exogenous administration of pyruvate to UOK262 cells resulted in a rapid loss of nuclear HIF-1α expression (Fig. 2A). In contrast, exogenous lactate did not have this effect. Furthermore, pharmacologic inhibition of endogenous pyruvate conversion to lactate with the lactate dehydrogenase (LDH) inhibitor oxamate also dramatically reduced nuclear HIF-1α levels, suggesting that the high rate of lactic acid production by these cells is critical to the maintenance of HIF-1α expression (Fig. 2B). Indeed, the LDH isoform LDH5 is highly expressed in UOK262 cells (Yang et al., submitted for publication). In colorectal carcinomas, high LDH5 tumor cell content has been directly related to an upregulated HIF pathway, aerobic glycolysis, and an aggressive phenotype (23). To explain the discrepancy between our data and earlier reports, we considered other properties of pyruvate that might be relevant to these cells. Several previously published studies have indicated that pyruvate (but not lactate) is an effective antioxidant (28, 39). We therefore examined whether its ability to deplete HIF-1α could be mimicked by other antioxidants. Indeed, we observed that N-acetylcysteine (NAC) also caused rapid reduction in nuclear HIF-1α levels (Fig. 2C). Both pyruvate and NAC significantly reduced ROS levels in UOK262 cells (Fig. 2D). Furthermore, UOK262 cells had significantly higher basal levels of cellular ROS than did VHL null 786-O renal carcinoma (Fig. 2D). Additionally, expression of wild-type FH in UOK262 cells resulted in a significant reduction in cellular ROS levels (Fig. 2E), indicating that loss of FH activity is associated with increased cellular ROS levels.
FIG. 2.

HIF-1α stabilization in HLRCC tumor cells is ROS dependent. (A) UOK262 cells were incubated in plain Krebs buffer (first lane) or with Krebs buffer containing either lactate (5 mM) or pyruvate (5 mM) for the indicated time period. Nuclear lysates were immunoblotted for HIF-1α and CREB. (B) UOK262 cells were incubated in Krebs buffer with 5 mM glucose with or without 10 mM oxamate (LDH inhibitor) for the indicated time period. Nuclear lysates were immunoblotted for HIF-1α and CREB. (C) UOK262 cells were treated with the indicated amount of NAC in Krebs buffer for 30 min prior to nuclear protein harvest. Nuclear lysates were immunoblotted for HIF-1α and CREB. (D) ROS levels were determined in UOK262 cells and 786-O cells following treatment with pyruvate (5 mM, 1 h) or NAC (10 mM, 1 h), using a spectrophotometric assay measuring fluorescence of the oxidant-sensitive dye DCF. CTL indicates control untreated cells. (E) Relative ROS levels were determined in stable transfectants UOK262/CV and UOK262/FH, using a spectrophotometric assay measuring fluorescence of the oxidant-sensitive dye DCF. (F) UOK262 cells were pretreated with plain Krebs buffer with proteasome inhibitor PS341 (1 μM) or DMOG (500 μM) for 2 h, followed by 30 min of incubation in plain Krebs buffer (B) or Krebs buffer supplemented with 5 mM pyruvate (P). Nuclear lysates were probed for total HIF-1α, hydroxylated HIF-1α, and CREB. (G) The ratio of ferric to ferrous iron in a panel of renal epithelial cell lines. (H) UOK262 cells were not treated (U) or treated with FeCl2 (80 μM) for the indicated time period. Cells treated with 5 mM pyruvate (P) for 30 min are shown for comparison in the last lane. Nuclear lysates were immunoblotted for HIF-1α and CREB. Statistical significance with a P value of <0.05 is denoted with an asterisk.
Pretreatment of UOK262 cells with proteasome inhibitor (PS341) blocked the effect of pyruvate, indicating that loss of HIF-1α was due to pyruvate-induced proteasomal degradation (Fig. 2F). Moreover, in cells pretreated with proteasome inhibitor, pyruvate induced a banding pattern suggestive of posttranslational modification consistent with ubiquitination (Fig. 2F, right panel). Pharmacologic inhibition of the HPH enzymes with DMOG also prevented pyruvate from destabilizing HIF-1α (Fig. 2F, left panel), suggesting that pyruvate may promote HIF-1α hydroxylation. To verify this possibility, we monitored the hydroxylation status of HIF-1α, using a polyclonal antibody specific for the hydroxylated protein. As expected, no hydroxylated HIF-1α was detected in DMOG-treated cells (Fig. 2F, left panel). In contrast, in cells pretreated with proteasome inhibitor, pyruvate increased hydroxylated HIF-1α levels, indicating that pyruvate treatment promoted HPH activity (Fig. 2F, right panel). Thus, pyruvate reduced ROS levels in UOK262 cells while increasing hydroxylation of HIF-1α.
The HPHs require divalent ferrous iron (Fe2+) in order to hydroxylate HIF-1α, and as part of this reaction, Fe2+ is oxidized to trivalent ferric iron (Fe3+). Ascorbate, another required cofactor, restores cellular Fe2+ pools to permit subsequent hydroxylation reactions (33). Hence, elevated cellular ROS levels may skew the balance of reduced and oxidized iron, thereby inhibiting HPH activity and resulting in HIF-1α stabilization. Indeed, excess exogenous ascorbate, like pyruvate and NAC, also reduced the level of HIF-1α expression in UOK262 cells (data not shown). Given the potential link between ROS and the oxidant status of iron, the cellular ratio of iron ions in the UOK262 cell line was compared with that in other renal epithelial cell lines (Fig. 2G). The ratio of oxidized to reduced iron was 0.67 in normal renal epithelial cells (HRCE), 0.9 in 786-O cells, and 0.96 in HEK293 cells. However, in UOK262 cells, the Fe3+/Fe2+ ratio rose to 1.80, consistent with elevated cellular ROS levels in these cells. Moreover, exogenous administration of Fe2+ (in the form of FeCl2) to UOK262 cells resulted in rapid HIF-1α depletion, suggesting that ROS-dependent iron oxidation and the resultant inhibition of HIF hydroxylation is a biologically relevant mechanism underlying chronic HIF-1α stabilization in these cells (Fig. 2H).
Loss of FH activity promotes ROS accumulation in renal epithelial cells and in yeast.
In order to confirm that loss of FH activity is directly associated with increased cellular ROS levels, additional cell models were evaluated. HK-2 immortalized renal epithelial cells were stably transfected with FH-specific shRNA and screened for loss of FH protein expression by immunoblot analysis. Two clones, C7 and D10, were found to contain markedly reduced cellular FH protein levels in comparison to those of HK-2 cells stably transfected with CV (Fig. 3A, left panel). Stable knockdown of FH resulted in an elevated steady-state level of HIF-1α in both clones (Fig. 3A, right panel), and both clones had elevated basal ROS levels compared to those of an empty vector clone (Fig. 3B). In addition, treatment with the antioxidant NAC also resulted in decreased HIF-1α protein levels in FH knockdown HK-2 cells (Fig. 3C).
FIG. 3.

Loss of FH activity promotes ROS in renal epithelial cells and yeast. (A) FH protein levels were determined in parental HK-2 cells in addition to cells stably transfected with a CV and shRNA to FH (C7 and D10). A GAPDH immunoblot is included as a loading control (left panel). Nuclear HIF-1α levels in the various clones were determined by immunoblotting (right panel). (B) ROS levels were determined using DCF fluorescence, as described in Materials and Methods, in parental HK-2 cells as well as in the derived clones described for panel A. (C) FH knockdown clone D10 cells were incubated in DMEM and treated with 10 mM NAC for 1 h or left untreated. Nuclear lysates were immunoblotted for HIF-1α and CREB. (D) Wild-type yeast and derived Δfum1, H153R, and K187R fumarase mutants were assayed for ROS by flow cytometry as described in Materials and Methods. Statistical significance with a P value of <0.05 is denoted with an asterisk.
Yeast cells were also examined for effects of fumarase deletion or mutation on ROS levels (Fig. 3D). fum1 is the sole fumarase gene in yeast and is not essential for viability, but its loss confers an obligate fermentor phenotype (22). The Δfum1 (fumarase knockout mutant) and H153R and K187R (both of which express different human FH mutant proteins as their sole source of fumarase) yeast mutants were previously reported to have depressed fumarase activity in comparison to that of the wild-type fumarase-expressing yeast strains (22). The H153R and K187R mutants represent strains with germ line mutations found in HLRCC kindreds. All three yeast fumarase mutants studied were found to have elevated cellular ROS levels in comparison to those of the wild-type yeast strain. Taken together, these data strongly support the hypothesis that increased cellular ROS levels are a direct consequence of diminished FH enzymatic activity.
Glucose stabilization of HIF-1α is mediated by protein kinase C-δ and NADPH oxidase.
Glucose is critical for growth and for maintenance of HIF-1α expression in UOK262 cells (Fig. 1E), and high intracellular glucose levels have been associated with enhanced ROS production. Indeed, blockade of UOK262 glucose uptake with fasentin, a pharmacologic inhibitor of the glucose transporter GLUT1 (42), resulted in a reduction of ROS to a level equivalent to that seen following wild-type FH replacement (Fig. 4A). Likewise, fasentin treatment of normal renal epithelial HK-2 cells with stably knocked-down FH restores ROS level to that seen in CV-transfected HK-2 cells (Fig. 4A). Recent data have identified cellular NADPH oxidases as a major source of increased ROS in the context of high-glucose states (36). NADPH oxidases catalyze the reduction of molecular oxygen to superoxide anion (O2−) that is subsequently converted to hydrogen peroxide by superoxide dismutase. We observed that UOK262 cells generate NADPH-dependent superoxide at a much higher rate than do HK-2 cells (Fig. 4B), suggesting that increased NADPH oxidase activity may contribute to elevated ROS levels in UOK262 cells. Moreover, expression of wild-type FH in UOK262 cells resulted in a significant reduction of NADPH-dependent superoxide production, suggesting that loss of FH activity is associated with increased NADPH oxidase activity (Fig. 4C). In support of this hypothesis, both cellular ROS (data not shown) and HIF-1α levels (Fig. 4D) in UOK262 were markedly reduced following treatment with the NADPH oxidase inhibitor diphenylene iodonium (DPI). While DPI can inhibit other flavoproteins that can produce cellular ROS, particularly components of the electron transport chain (ETC), no reduction of HIF-1α was noted in UOK262 cells following treatment with the ETC inhibitor rotenone (data not shown). In addition, inhibition of NADPH oxidase via siRNA-mediated knockdown of the p47phox subunit resulted in reduced nuclear HIF-1α levels in UOK262 cells (Fig. 4E).
FIG. 4.

Glucose-dependent ROS generation and stabilization of HIF-1α in UOK262 cells are mediated by NADPH oxidase and PKC-δ. (A) (Left panel) Relative ROS levels based on DCF fluorescence were determined in UOK262/CV (normalized to 1), UOK262/FH, and UOK262/CV cells treated with GLUT1 inhibitor fasentin. (Right panel) Relative ROS levels were determined in HK-2/CV cells (normalized to 1) and HK2/shFH cells with or without fasentin. (B) NADPH-dependent ROS generation in cell homogenates was measured by lucigenin-enhanced chemiluminescence in HK-2 and UOK262 cells. Superoxide anion production was expressed as relative chemiluminescent light units (RLU) normalized to protein content and measured over time. (C) NADPH-dependent superoxide production was determined in UOK262/CV and UOK262/FH cells. Data were obtained 3 min after addition of NADPH. (D) UOK262 cells were treated with the indicated concentrations of NADPH oxidase inhibitor DPI for 30 min in Krebs buffer. Nuclear lysates were immunoblotted for HIF-1α and CREB. Note that the control lane represents the same control lane as that in Fig. 2C to permit direct comparison of antioxidant effect. (E) UOK262 cells were transfected with control siRNA (siCTRL) or siRNA to NADPH oxidase subunit p47phox. Cells were harvested 72 h following transfection, and nuclear lysates were immunoblotted for HIF-1α and CREB. Immunoblotting for p47phox confirms knockdown (GAPDH loading control). (F) UOK262 cells were treated with 10 μM rottlerin for the times shown (left panel). Nuclear lysates were probed for HIF-1α and CREB. Cytoplasmic extracts were also immunoblotted for PKC-δ. UOK262 cells were transiently transfected with siRNA to PKC-δ and scrambled siRNA (si Control) (right panel). Whole-cell lysates were immunoblotted for PKC-δ and α-tubulin (loading control). Nuclear lysates were immunoblotted for HIF-1α and CREB. (G) ROS levels were monitored by DCF fluorescence in untreated UOK262 cells and in cells treated with scrambled control siRNA or siRNA specific for PKC-δ. Knockdown of the PKC-δ protein is demonstrated in panel F. (H) NADPH-dependent ROS generation was measured in cell homogenates by lucigenin-enhanced chemiluminescence in UOK262 cells treated in Krebs buffer with or without rottlerin (10 μM) for 1 h. Superoxide anion was expressed as relative chemiluminescent light units (RLU) normalized to protein content (left panel). NADPH-dependent superoxide production was also measured in UOK262 cells treated with scrambled control siRNA (siCTRL) and in cells treated with siRNA to PKC-δ. Statistical significance with a P value of <0.05 is denoted with an asterisk.
Members of the PKC serine/threonine kinase family, including PKC-δ, have been implicated in glucose-dependent activation of NADPH oxidase (36). We found that pharmacologic inhibition of PKC-δ decreased HIF-1α levels in UOK262 cells (Fig. 4F, left panel). These results were confirmed by siRNA-mediated molecular knockdown of PKC-δ (Fig. 4F, right panel). In addition, PKC-δ knockdown resulted in lower cellular ROS levels (Fig. 4G). Moreover, both pharmacologic inhibition and RNA interference suppression of PKC-δ resulted in reduced NADPH oxidase activity, confirming a role for PKC-δ in mediating ROS generation and HIF-1α stabilization in UOK262 cells (Fig. 4H).
DISCUSSION
The data presented in this report describe a more detailed model for metabolic regulation of HIF-1α stabilization in HLRCC tumors. Of key importance to ROS-mediated HIF-1α stabilization is the glycolytic switch that occurs rapidly in cells following loss of FH activity. While the mechanism underlying this phenomenon remains to be fully elucidated, initial fumarate accumulation that occurs in cells following loss of FH enzymatic activity may be a contributing factor. It is now well established that acute accumulation of intracellular fumarate following either pharmacologic inhibition of FH or its siRNA-mediated knockdown leads to HIF-1α stabilization mediated by competitive inhibition (fumarate competing with 2-OG) of HIF-1α proline hydroxylation. In turn, HIF-1α accumulation is associated with the rapid appearance of a glycolytic phenotype (16). Our current findings suggest the possibility that an initial HIF-dependent increase in glucose uptake and metabolism occurring as an immediate and obligate response to reduced FH activity leads directly to heightened cellular ROS production, which further promotes HIF-1α stabilization by depleting cellular Fe2+ stores, thereby depriving HPH of an additional necessary cofactor. Elevated HIF-1α expression may drive glycolysis to establish a feed-forward signaling loop, and glucose-dependent ROS accumulation is, at least in part, mediated by NADPH oxidase and PKC-δ (Fig. 5). Although there may be alternate mechanisms that permit the glycolytic switch independent of HIF, we believe it is likely that HIF-1α is integrally involved in this metabolic shift, given its established role in promoting glycolysis as opposed to oxidative phosphorylation.
FIG. 5.
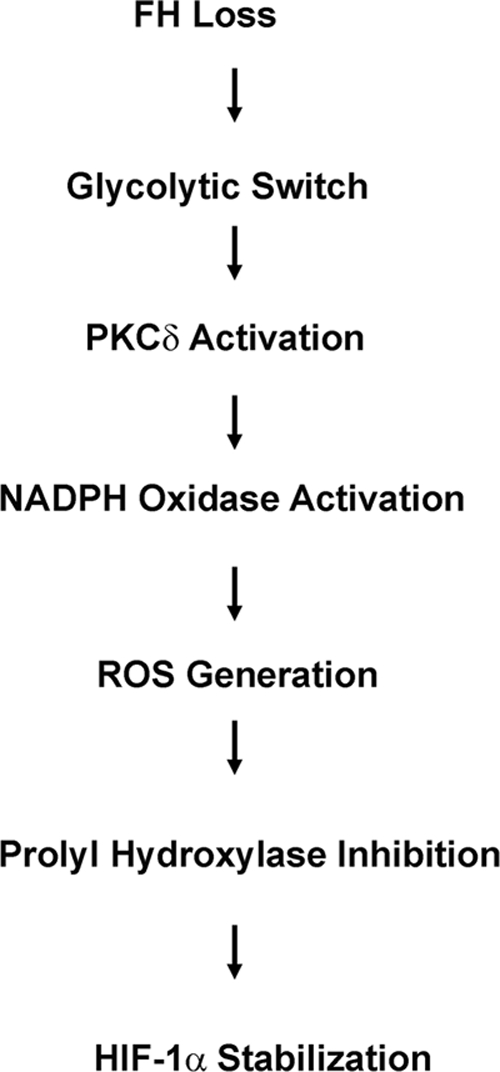
Schema for ROS-mediated HIF-1α stabilization in HLRCC.
The biologic significance of these findings is severalfold. Since they are usually diagnosed with advanced disease, HLRCC patients have limited treatment options and suffer a poor prognosis. Thus, elucidation of the molecular pathogenesis of tumor formation will better inform the development of effective therapeutic strategies. Our current data highlight an important role for ROS in maintaining HIF-1α stabilization in these tumors and suggest various metabolic approaches to treatment. A rationale for the use of antioxidants in HIF-1α-driven tumors was recently provided by Gao et al., who examined the antitumorigenic effect of the antioxidant NAC (7). They reported that NAC treatment resulted in reduced HIF-1α expression and in inhibition of in vivo tumor formation in a HIF-driven model of tumorigenesis. Their data support a role for ROS-mediated inhibition of HPH activity, as NAC was ineffective in a tumor model expressing a mutant HIF-1α allele that was resistant to HPH-dependent degradation.
In addition, our data add to the growing body of evidence linking derangements in mitochondrial metabolism to carcinogenesis. First described many years ago by Otto Warburg, the “Warburg effect” refers to the preference of cancer cells to obtain energy via glycolysis and not oxidative phosphorylation, even in normoxia (40). HIF-1α has been suggested as a key regulator of this phenomenon, since it transcriptionally drives many components of glycolysis. In addition, HIF-1α shunts metabolic intermediates away from the TCA cycle by upregulating pyruvate dehydrogenase kinase 1 (PDK1) (20). PDK1 phosphorylates and inhibits pyruvate dehydrogenase (PDH), thus limiting the ability of PDH to supply acetyl coenzyme A to the TCA cycle (by conversion from pyruvate). When the TCA cycle is genetically compromised, as is the case in HLRCC, glycolytic addiction of the tumor cells is ensured. This may prove to be an Achilles’ heel of HLRCC, even more so than in the case of other solid tumors. Because HLRCC tumors have become genetically obligated to glycolysis (as evidenced by their inability to substitute galactose for glucose), they must rapidly convert the pyruvate accumulating as a result of this metabolic process to lactic acid. Previous reports have suggested that failure to do so may slow glycolysis by-product feedback inhibition (4). Others have suggested that rapid conversion of pyruvate to lactic acid is necessary to regenerate cellular NAD+, which is required to support additional cycles of glycolysis (6). Our data suggest a third, not mutually exclusive, possibility—that accumulation of pyruvate causes a reduction in cellular ROS level sufficient to negatively impact HIF-1α stabilization. Since lactate dehydrogenase is itself transcriptionally regulated by HIF-1α, as are glucose uptake, glycolysis, and downregulation of oxidative phosphorylation, interference in this process (e.g., LDH inhibition, ROS scavenging) provides the potential to convert a feed-forward stimulatory signaling loop into a reverse inhibitory loop and offers several potential treatment strategies. An initial report describing the impact of LDH knockdown on the viability of cells experimentally knocked down for FH supports this hypothesis (43).
The initial link between loss of FH activity and ROS generation could be HIF-1α itself, but this hypothesis does not explain the elevated ROS levels that we observed in fumarase-deficient yeast (which do not express HIF proteins). However, like UOK262 cells, yeast lacking fumarase activity cannot use mitochondrial respiration to grow on nonfermentable carbon sources (22), suggesting that addiction to glycolysis in the absence of FH activity is a highly conserved property of eukaryotic cells. Indeed, the enforced glycolysis and resultant rapid generation and secretion of lactic acid characteristic of FH-deficient cells may underlie the high metastatic potential of HLRCC, as lactic acid is reported to stimulate endothelial cell migration, which is a crucial component of a tumor's metastatic phenotype (1, 24). In patients with cervical cancer, high lactate levels have been directly correlated with likelihood of tumor metastasis and reduced patient survival (38).
Exposure to high glucose levels stimulates NADPH-mediated ROS production in vascular smooth muscle and endothelial cells (15, 26), and this has been associated with increased activity of PKC isozymes, including PKC-δ (14). Furthermore, ROS production is elevated in adipocytes obtained from high-fat-diet-induced obese and insulin-deficient mice, and both increased PKC-δ activity and NADPH oxidase have been proposed to mediate high glucose-dependent ROS production in these settings (36). Our data identify both NADPH oxidase and PKC-δ as contributors to the elevated HIF-1α level found in UOK262 cells, as inhibition of both proteins led to decreased HIF-1α expression. A similar effect on HIF-1α was noted following RNA interference knockdown of the NADPH oxidase subunit p47phox and following molecular knockdown of PKC-δ.
Inhibition of HPH activity by either excess fumarate or elevated ROS causes the phenotype we have described in this study. While ROS levels are clearly elevated following experimental FH knockdown, it is unclear whether fumarate levels are elevated sufficiently in HLRCC renal tumor cells to inhibit HPH. These results parallel findings with SDH mutations (SDHB, SDHC, SDHD) where both succinate and ROS have been proposed to lead to HIF-1α stabilization (12, 34). However, more recent evidence suggests that elevation of the succinate level alone, in the absence of ROS, is not sufficient to support HIF-1α stabilization (12).
Additional mechanisms also may account for the pseudohypoxic expression profile of HLRCC. As FH is a mitochondrial enzyme, another potential source for ROS may be the electron transport chain, which is the source of ROS generation in the context of SDH mutations. Alternatively, the mTOR pathway has been implicated in HIF-1α synthesis. Although neither the inhibition of the electron transport chain nor the inhibition of mTOR impacts HIF-1α expression over the short time frame of our experiments (data not shown), our results do not exclude the possibility that HIF-1α in UOK262 cells may be impacted by these or other inputs over a longer time scale.
In addition, the role of pseudohypoxia in HLRCC tumorigenesis, while implied, has not been proven. The HPH enzymes belong to a family of dioxygenases that uniformly require 2-OG and Fe2+ as cosubstrate and cofactor, respectively. These enzymes include collagen hydroxylases and certain histone demethylases. Thus, elevated levels of fumarate and/or ROS may impact additional cellular processes unrelated to HIF-1α. It is certainly possible that inhibition of these or other dioxygenases, whether in concert with or independent of HIF-1α stabilization, may contribute to HLRCC tumor formation.
In summary, our findings link loss of FH activity to glycolytic addiction, chronic ROS production and HIF-1α stabilization, and we show that these phenomena are interdependent. The UOK262 cell line represents a unique model system by which to further evaluate the impact on HLRCC tumorigenesis of various strategies aimed at interdicting this signaling axis, with the potential to identify novel therapeutic approaches to target this disease.
Acknowledgments
This work was supported by the Intramural Research Program of the NIH (National Cancer Institute, Center for Cancer Research). S.S. was supported by a matching gift from Charles Butt and employees of the H-E-B and the University of Texas System Board of Regents, and also by the Cancer Therapy and Research Center at the University of Texas Health Science Center at San Antonio. K.B. was supported by NIH grant R01 NCI CA131272 and a Veterans Administration Career Development Award.
We gratefully acknowledge Nadja Rehak (NIH) for assistance with biochemical measurements. We also acknowledge Paul Hoover and Hilal Ahmad (University of Texas Health Science Center) for technical assistance.
Footnotes
Published ahead of print on 26 May 2009.
REFERENCES
- 1.Beckert, S., F. Farrahi, R. S. Aslam, H. Scheuenstuhl, A. Konigsrainer, M. Z. Hussain, and T. K. Hunt. 2006. Lactate stimulates endothelial cell migration. Wound Repair Regen. 14321-324. [DOI] [PubMed] [Google Scholar]
- 2.Block, K., Y. Gorin, P. Hoover, P. Williams, T. Chelmicki, R. A. Clark, T. Yoneda, and H. E. Abboud. 2007. NAD(P)H oxidases regulate HIF-2alpha protein expression. J. Biol. Chem. 2828019-8026. [DOI] [PubMed] [Google Scholar]
- 3.Bruick, R. K., and S. L. McKnight. 2001. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2941337-1340. [DOI] [PubMed] [Google Scholar]
- 4.Denko, N. C. 2008. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8705-713. [DOI] [PubMed] [Google Scholar]
- 5.Epstein, A. C., J. M. Gleadle, L. A. McNeill, K. S. Hewitson, J. O'Rourke, D. R. Mole, M. Mukherji, E. Metzen, M. I. Wilson, A. Dhanda, Y. M. Tian, N. Masson, D. L. Hamilton, P. Jaakkola, R. Barstead, J. Hodgkin, P. H. Maxwell, C. W. Pugh, C. J. Schofield, and P. J. Ratcliffe. 2001. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 10743-54. [DOI] [PubMed] [Google Scholar]
- 6.Fantin, V. R., J. St-Pierre, and P. Leder. 2006. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9425-434. [DOI] [PubMed] [Google Scholar]
- 7.Gao, P., H. Zhang, R. Dinavahi, F. Li, Y. Xiang, V. Raman, Z. M. Bhujwalla, D. W. Felsher, L. Cheng, J. Pevsner, L. A. Lee, G. L. Semenza, and C. V. Dang. 2007. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 12230-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gnarra, J. R., K. Tory, Y. Weng, L. Schmidt, M. H. Wei, H. Li, F. Latif, S. Liu, F. Chen, F. M. Duh, et al. 1994. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 785-90. [DOI] [PubMed] [Google Scholar]
- 9.Gordan, J. D., and M. C. Simon. 2007. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr. Opin. Genet. Dev. 1771-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gottlieb, E., and I. P. Tomlinson. 2005. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat. Rev. Cancer 5857-866. [DOI] [PubMed] [Google Scholar]
- 11.Grubb, R. L., III, M. E. Franks, J. Toro, L. Middelton, L. Choyke, S. Fowler, C. Torres-Cabala, G. M. Glenn, P. Choyke, M. J. Merino, B. Zbar, P. A. Pinto, R. Srinivasan, J. A. Coleman, and W. M. Linehan. 2007. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J. Urol. 1772074-2079. (Discussion, 177:2079-2080.) [DOI] [PubMed] [Google Scholar]
- 12.Guzy, R. D., B. Sharma, E. Bell, N. S. Chandel, and P. T. Schumacker. 2008. Loss of the SdhB, but not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell. Biol. 28718-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ha, H., and H. B. Lee. 2000. Reactive oxygen species as glucose signaling molecules in mesangial cells cultured under high glucose. Kidney Int. Suppl. 77S19-S25. [DOI] [PubMed] [Google Scholar]
- 14.Haller, H., E. Baur, P. Quass, M. Behrend, C. Lindschau, A. Distler, and F. C. Luft. 1995. High glucose concentrations and protein kinase C isoforms in vascular smooth muscle cells. Kidney Int. 471057-1067. [DOI] [PubMed] [Google Scholar]
- 15.Inoguchi, T., T. Sonta, H. Tsubouchi, T. Etoh, M. Kakimoto, N. Sonoda, N. Sato, N. Sekiguchi, K. Kobayashi, H. Sumimoto, H. Utsumi, and H. Nawata. 2003. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. J. Am. Soc. Nephrol. 14S227-S232. [DOI] [PubMed] [Google Scholar]
- 16.Isaacs, J. S., Y. J. Jung, D. R. Mole, S. Lee, C. Torres-Cabala, Y. L. Chung, M. Merino, J. Trepel, B. Zbar, J. Toro, P. J. Ratcliffe, W. M. Linehan, and L. Neckers. 2005. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8143-153. [DOI] [PubMed] [Google Scholar]
- 17.Ivan, M., K. Kondo, H. Yang, W. Kim, J. Valiando, M. Ohh, A. Salic, J. M. Asara, W. S. Lane, and W. G. Kaelin, Jr. 2001. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292464-468. [DOI] [PubMed] [Google Scholar]
- 18.Jaakkola, P., D. R. Mole, Y. M. Tian, M. I. Wilson, J. Gielbert, S. J. Gaskell, A. Kriegsheim, H. F. Hebestreit, M. Mukherji, C. J. Schofield, P. H. Maxwell, C. W. Pugh, and P. J. Ratcliffe. 2001. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292468-472. [DOI] [PubMed] [Google Scholar]
- 19.Kaelin, W. G., Jr. 2005. The von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem. Biophys. Res. Commun. 338627-638. [DOI] [PubMed] [Google Scholar]
- 20.Kim, J. W., I. Tchernyshyov, G. L. Semenza, and C. V. Dang. 2006. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3177-185. [DOI] [PubMed] [Google Scholar]
- 21.Koivunen, P., M. Hirsila, A. M. Remes, I. E. Hassinen, K. I. Kivirikko, and J. Myllyharju. 2007. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 2824524-4532. [DOI] [PubMed] [Google Scholar]
- 22.Kokko, A., S. S. Ylisaukko-Oja, M. Kiuru, M. S. Takatalo, P. Salmikangas, J. Tuimala, D. Arango, A. Karhu, L. A. Aaltonen, and J. Jantti. 2006. Modeling tumor predisposing FH mutations in yeast: effects on fumarase activity, growth phenotype and gene expression profile. Int. J. Cancer 1181340-1345. [DOI] [PubMed] [Google Scholar]
- 23.Koukourakis, M. I., A. Giatromanolaki, C. Simopoulos, A. Polychronidis, and E. Sivridis. 2005. Lactate dehydrogenase 5 (LDH5) relates to up-regulated hypoxia inducible factor pathway and metastasis in colorectal cancer. Clin. Exp. Metastasis 2225-30. [DOI] [PubMed] [Google Scholar]
- 24.Kumar, V. B., R. I. Viji, M. S. Kiran, and P. R. Sudhakaran. 2007. Endothelial cell response to lactate: implication of PAR modification of VEGF. J. Cell Physiol. 211477-485. [DOI] [PubMed] [Google Scholar]
- 25.Launonen, V., O. Vierimaa, M. Kiuru, J. Isola, S. Roth, E. Pukkala, P. Sistonen, R. Herva, and L. A. Aaltonen. 2001. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc. Natl. Acad. Sci. USA 983387-3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee, H. S., S. M. Son, Y. K. Kim, K. W. Hong, and C. D. Kim. 2003. NAD(P)H oxidase participates in the signaling events in high glucose-induced proliferation of vascular smooth muscle cells. Life Sci. 722719-2730. [DOI] [PubMed] [Google Scholar]
- 27.Lu, H., C. L. Dalgard, A. Mohyeldin, T. McFate, A. S. Tait, and A. Verma. 2005. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J. Biol. Chem. 28041928-41939. [DOI] [PubMed] [Google Scholar]
- 28.Moriguchi, N., E. Hinoi, Y. Tsuchihashi, S. Fujimori, M. Iemata, T. Takarada, and Y. Yoneda. 2006. Cytoprotection by pyruvate through an anti-oxidative mechanism in cultured rat calvarial osteoblasts. Histol. Histopathol. 21969-977. [DOI] [PubMed] [Google Scholar]
- 29.Pithukpakorn, M., M. H. Wei, O. Toure, P. J. Steinbach, G. M. Glenn, B. Zbar, W. M. Linehan, and J. R. Toro. 2006. Fumarate hydratase enzyme activity in lymphoblastoid cells and fibroblasts of individuals in families with hereditary leiomyomatosis and renal cell cancer. J. Med. Genet. 43755-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pollard, P., N. Wortham, E. Barclay, A. Alam, G. Elia, S. Manek, R. Poulsom, and I. Tomlinson. 2005. Evidence of increased microvessel density and activation of the hypoxia pathway in tumours from the hereditary leiomyomatosis and renal cell cancer syndrome. J. Pathol. 20541-49. [DOI] [PubMed] [Google Scholar]
- 31.Pollard, P. J., J. J. Briere, N. A. Alam, J. Barwell, E. Barclay, N. C. Wortham, T. Hunt, M. Mitchell, S. Olpin, S. J. Moat, I. P. Hargreaves, S. J. Heales, Y. L. Chung, J. R. Griffiths, A. Dalgleish, J. A. McGrath, M. J. Gleeson, S. V. Hodgson, R. Poulsom, P. Rustin, and I. P. Tomlinson. 2005. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 142231-2239. [DOI] [PubMed] [Google Scholar]
- 32.Pollard, P. J., B. Spencer-Dene, D. Shukla, K. Howarth, E. Nye, M. El-Bahrawy, M. Deheragoda, M. Joannou, S. McDonald, A. Martin, P. Igarashi, S. Varsani-Brown, I. Rosewell, R. Poulsom, P. Maxwell, G. W. Stamp, and I. P. Tomlinson. 2007. Targeted inactivation of fh1 causes proliferative renal cyst development and activation of the hypoxia pathway. Cancer Cell 11311-319. [DOI] [PubMed] [Google Scholar]
- 33.Qutub, A. A., and A. S. Popel. 2006. A computational model of intracellular oxygen sensing by hypoxia-inducible factor HIF1 alpha. J. Cell Sci. 1193467-3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selak, M. A., S. M. Armour, E. D. MacKenzie, H. Boulahbel, D. G. Watson, K. D. Mansfield, Y. Pan, M. C. Simon, C. B. Thompson, and E. Gottlieb. 2005. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 777-85. [DOI] [PubMed] [Google Scholar]
- 35.Sudarshan, S., W. M. Linehan, and L. Neckers. 2007. HIF and fumarate hydratase in renal cancer. Br. J. Cancer 96403-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Talior, I., T. Tennenbaum, T. Kuroki, and H. Eldar-Finkelman. 2005. PKC-delta-dependent activation of oxidative stress in adipocytes of obese and insulin-resistant mice: role for NADPH oxidase. Am. J. Physiol. Endocrinol. Metab. 288E405-E411. [DOI] [PubMed] [Google Scholar]
- 37.Tomlinson, I. P., N. A. Alam, A. J. Rowan, E. Barclay, E. E. Jaeger, D. Kelsell, I. Leigh, P. Gorman, H. Lamlum, S. Rahman, R. R. Roylance, S. Olpin, S. Bevan, K. Barker, N. Hearle, R. S. Houlston, M. Kiuru, R. Lehtonen, A. Karhu, S. Vilkki, P. Laiho, C. Eklund, O. Vierimaa, K. Aittomaki, M. Hietala, P. Sistonen, A. Paetau, R. Salovaara, R. Herva, V. Launonen, and L. A. Aaltonen. 2002. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 30406-410. [DOI] [PubMed] [Google Scholar]
- 38.Walenta, S., M. Wetterling, M. Lehrke, G. Schwickert, K. Sundfor, E. K. Rofstad, and W. Mueller-Klieser. 2000. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 60916-921. [PubMed] [Google Scholar]
- 39.Wang, X., E. Perez, R. Liu, L. J. Yan, R. T. Mallet, and S. H. Yang. 2007. Pyruvate protects mitochondria from oxidative stress in human neuroblastoma SK-N-SH cells. Brain Res. 11321-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warburg, O. 1956. On the origin of cancer cells. Science 123309-314. [DOI] [PubMed] [Google Scholar]
- 41.Wiesener, M. S., P. M. Munchenhagen, I. Berger, N. V. Morgan, J. Roigas, A. Schwiertz, J. S. Jurgensen, G. Gruber, P. H. Maxwell, S. A. Loning, U. Frei, E. R. Maher, H. J. Grone, and K. U. Eckardt. 2001. Constitutive activation of hypoxia-inducible genes related to overexpression of hypoxia-inducible factor-1alpha in clear cell renal carcinomas. Cancer Res. 615215-5222. [PubMed] [Google Scholar]
- 42.Wood, T. E., S. Dalili, C. D. Simpson, R. Hurren, X. Mao, F. S. Saiz, M. Gronda, Y. Eberhard, M. D. Minden, P. J. Bilan, A. Klip, R. A. Batey, and A. D. Schimmer. 2008. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol. Cancer Ther. 73546-3555. [DOI] [PubMed] [Google Scholar]
- 43.Xie, H., V. A. Valera, M. J. Merino, A. M. Amato, S. Signoretti, W. M. Linehan, V. P. Sukhatme, and P. Seth. 2009. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol. Cancer Ther. 8626-635. [DOI] [PMC free article] [PubMed] [Google Scholar]