

Abstract
Rac1 is a member of the Rho family of small GTPases that not only regulates signaling pathways involved in cell adhesion and migration but also regulates gene transcription. Here we show that the transcriptional repressor BCL-6 is regulated by Rac1 signaling. Transfection of active Rac1 mutants into colorectal DLD-1 cells led to increased expression of a BCL-6-controlled luciferase reporter construct. Conversely, inhibition of endogenous Rac1 activation by the Rac1 inhibitor NSC23766 decreased reporter activity. Moreover, BCL-6 lost its typical localization to nuclear dots upon activation of Rac1 and became predominantly soluble in a non-chromatin-bound cell fraction. Rac1 signaling also regulated the expression of endogenous BCL-6-regulated genes, including the p50 precursor NF-κB1/p105 and the cell adhesion molecule CD44. Interestingly, these effects were not stimulated by the alternative splice variant Rac1b. The mechanism of BCL-6 inhibition does not involve formation of a stable Rac1/BCL-6 complex and is independent of Rac-induced reactive oxygen species production or Jun NH2-terminal kinase activation. We show that PAK1 mediates inhibition downstream of Rac and can directly phosphorylate BCL-6. Together, these data provide substantial evidence that Rac1 signaling inhibits the transcriptional repressor BCL-6 in colorectal cells and reveal a novel pathway that links Rac1 signaling to the regulation of gene transcription.
The Rho family of small GTPases contains 20 different members (7, 69); RhoA, Rac1, and Cdc42 have been the best characterized members of the family (20, 27, 53). These GTPases typically cycle between an inactive, GDP-bound state and an active, GTP-bound state. The transition between these two states is controlled by three distinct types of proteins in vivo, the guanine nucleotide exchange factors, which activate, and the GTPase-activating proteins or the guanine nucleotide dissociation inhibitors, which both inactivate GTPases (reviewed in references 6 and 14). Once in the GTP-loaded conformation, Rho GTPases become able to interact with downstream effector proteins that initiate further signaling events in the cell. The corresponding cellular responses range from changes in cell morphology to changes in gene expression.
Rac1, in particular, has been documented to stimulate the polymerization of actin filaments leading to the formation of lamellipodia and affecting the stability of adherens junctions (23). Rac signaling further activates the protein kinase PAK (p21-activated kinase), the Jun NH2-terminal kinase (JNK), and the production of reactive oxygen species (ROS) (57). Moreover, recent data have revealed that Rac1 also has distinct roles in the regulation of gene transcription (3). For instance, stimulation of JNK by Rac signaling can lead to activation of its target transcription factors, c-Jun, ATF, ELK, and AP1. Also, Rac signaling can activate proteins of the Stat family (49, 57), and the formation of protein complexes between Rac1 and Stat3 or Stat5 has been reported (36, 61, 67). Likewise, an active Rac1 mutant amplifies the transcriptional activation mediated by β-catenin and TCF/LEF (18).
A further important transcription factor stimulated by Rac1 is NF-κB. The NF-κB family is composed of five transcription factors that form homodimers or heterodimers with each other, namely, RelA, RelB, c-Rel, p50, and p52. Unlike the three Rel proteins, p50 and p52 are produced through proteolytic processing from two inhibitory precursor proteins, NF-κB1/p105 and NF-κB2/p100, respectively. The NF-κB dimers remain transcriptionally inactive as long as they are associated with an NF-κB inhibitor protein, such as IκBα or the NF-κB2/p100 precursor protein. Signaling from GTP-bound Rac1 activates the IKK protein kinase complex, resulting in the phosphorylation of both IκBα and NF-κB2/p100 proteins. In addition, the inhibitory complexes are recruited to sites at the plasma membrane where Rac is activated and brings them into proximity with the SCF ubiquitin ligase complex (8, 44). This leads to proteolytic degradation of IκBα and subsequent nuclear translocation of the transcriptionally competent RelA/p50 dimer (the canonical NF-κB pathway) but also promotes proteolytic processing of NF-κB2/p100 to p52, with subsequent transcriptional activation of RelB/p52 dimers (44). The canonical, IκBα-regulated NF-κB pathway is also stimulated by Rac1b, an alternative splice variant that exists predominantly in the active GTP-bound state in cell lines (21, 33, 44, 62). Whereas Rac1b does not activate several classical Rac signaling pathways, including lamellipodium formation or the activation of PAK1 or JNK activities, it retains the ability to induce IκBα phosphorylation, nuclear translocation of RelA, and transcriptional stimulation of luciferase reporter constructs containing either a consensus NF-κB binding motif or the native cyclin D1 promoter (19, 43, 44).
BCL-6 is a transcriptional repressor (59) and was identified as one of the most frequently translocated genes in B-cell non-Hodgkin's lymphomas (12, 64). BCL-6 contains carboxy-terminal zinc finger modules that bind DNA in a sequence-specific manner, especially the high-affinity site TTCCT(A/C)GAA (10, 31). The genes repressed by BCL-6 in germinal center B cells are involved in lymphocyte activation and differentiation, immunoglobulin (Ig) isotype switching, and regulation of inflammation or cell cycle progression (13, 47, 60). The repressor activity of BCL-6 can be regulated by posttranslational modifications. Both acetylation and phosphorylation events were shown to downregulate the ability of BCL-6 to repress transcription, the former impairing its recruitment of histone deacetylases (4) and the latter leading to its proteasomal degradation (46, 50).
Here we describe a novel link of Rac1 signaling to the regulation of gene transcription. We found that the transcriptional repressor BCL-6 is inhibited in colorectal tumor cells following Rac1 activation. This leads to increased expression of endogenous BCL-6-regulated genes, including NF-κB1/p105, the p50 precursor, and the cell adhesion molecule CD44. The mechanism of BCL-6 inactivation requires PAK1-mediated phosphorylation of BCL-6 downstream of Rac1 and is not triggered by splice variant Rac1b.
MATERIALS AND METHODS
Cell culture and transfection.
DLD-1 colorectal cells were maintained in Dulbecco's minimal essential medium supplemented with 10% (vol/vol) fetal calf serum (Invitrogen) and regularly checked for mycoplasma infection. Cells were transfected at 60 to 80% confluence using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions, and analyzed 16 to 20 h later. The total amounts of transfected plasmid DNA were 4 μg per 60-mm dish for immunoprecipitation and 2 μg of DNA per 35-mm dish for immunofluorescence, cell fractionation, and reporter assays. When required, the amount of DNA was adjusted with empty vector. Transfection efficiency in DLD-1 cells was 50 to 70% as judged microscopically by expression of 2 μg of green fluorescent protein (GFP) expression vector. For RNA interference experiments, DLD-1 cells at 30 to 40% confluence were transfected in 35-mm dishes with 200 pmol of the indicated small interfering RNAs (siRNAs) using Lipofectamine 2000, transfected again after 24 h with expression vectors or reporter constructs, and analyzed 24 h later. The predesigned siRNA oligonucleotides were from Santa Cruz Biotechnology (Santa Cruz, CA) with the following references: αPAK siRNA (sc29700), γPAK (sc36183), BCL-6 (sc29791), and scramble control oligonucleotide (5′-AGG UAG UGU AAU CGC CUU GTT). For drug treatments, cells were incubated for 16 to 20 h with 200 μM Rac inhibitor NSC23766 (Calbiochem) or with 25 μM NADPH oxidase inhibitor diphenyleneiodonium chloride (DPI) (Sigma), or 10 μM PAK inhibitor IPA-3 (Calbiochem).
DNA plasmids and constructs.
The following published constructs were received as gifts: pcDNA3-HA-IκBα (A32A36) from M. Karin (University of California, San Diego); pcDNA3-HA-RelB from C. V. Paya (Mayo Clinic, Rochester, MN), SAPKβ-MKK7 from U. Rapp (Würzburg, Germany); PAK1-wt, PAK1-K299R, and PAK1-T423E from J. Chernoff (Fox Chase Cancer Center, Philadelphia, PA); the 3x-κB-luc vector (three copies of the Igκ-κB motif immediately upstream of the β-globin TATA box) (39) from B. Baumann (University of Ulm); and the 5xBCL-6 vector (32) from V. J. Bardwell (University of Minnesota). Rac1 and Rac1b cDNAs as well as their Q61L mutants in pcDNA3-Myc or pEGFP vectors were as described previously (43, 44). For their subcloning into pDsRed, the respective pEGFP vectors were cut using the EcoRI/BamHI restriction sites. pEGFP-BCL-6 was generated by PCR amplification of the BCL-6 cDNA from pmT2T-HA-BCL6, provided by R. Dalla-Favera (Columbia University), using a forward primer (5′-GGT ACC ATG GCC TCG CCG GCT GAC A) and reverse primer (5′-TCA GCA GGC TTT GGG GAG CT), followed by subcloning into pEGFP-C3 using KpnI and SmaI. All PAK1 constructs were subcloned into pEGFP-C3 vector using HindIII/EcoRI restriction sites. All constructs were confirmed by automated DNA sequencing.
Analysis of transcript expression and semiquantitative reverse transcription-PCR (RT-PCR).
Total RNA was extracted from cell lysates with the RNeasy kit (Qiagen), and 1-μg samples were reverse transcribed using random primers (Invitrogen) and Ready-to-Go You-Prime First-Strand beads (GE Healthcare, Buckinghamshire, United Kingdom). Primers for the specific amplification of Rac1/Rac1b were described previously (43). The primers (F, forward; R, reverse) for the specific amplification of other proteins, genes, or transcription factors were as follows: for BCL-6, Bcl6-F (5′-AGA GCC CAT AAA ACG GTC CT) and Bcl6-R (5′-AGT GTC CAC AAC ATG CTC CA); for NFKB1, p105-F (5′-CCT GGA TGA CTC TTG GGA AA) and p105-R (5′-TCA GCC AGC TGT TTC ATG TC); for CD44, CD44-F (5′-TCT GTG CAG CAA ACA ACA CA) and CD44-R (5′-TAG GGT TGC TGG GGT AGA TG); for PAK1, PAK1-444F (5′-GTC AGC TGA GGA TTA CAA TTC) and PAK1-661R (5′-GAG ATG TAG CCA CGT CCC GAG); for PAK2, PAK2-431F (5′-CTC CTG AGA AAG ATG GCT TTC) and PAK2-632R (5′-ACA TGT GAA TCA CCA ACT GGT); for PAK3, PAK3-437F (5′-GTG CAC ATG GAT ACA TAG CAG) and PAK3-663R (5′-TGT GAC CTC TTT ATT TGG TAC); for BAZF, BAZF-e1F (5′-AGA GCA CAC AAG GCA GTT CTC) and BAZF-e2R (5′-GTG CAG TGG CTG GAG AGA GG); and for RNA polymerase II (Pol II), Pol II-F (5′-GAG CGG GAA TTT GAG CGG ATG C) and Pol II-R (5′-GAA GGC GTG GGT TGA TGT GGA AGA). Amplification reactions were performed using AmpliTaq polymerase (Perkin-Elmer, Wellesley, MA) using the following basic program: 30 s at 94°C, 30 s at the annealing temperature, and 30 s at 72°C. The annealing temperature and number of cycles for each PCR were as follows: 62°C and 30 cycles for Rac1/Rac1b, 58°C and 30 cycles for BCL-6, 64°C and 30 cycles for BAZF, 56°C and 29 cycles for PAK1 and PAK2, 58°C and 35 cycles for PAK3, 58°C and 28 cycles for NFKB1, 60°C and 30 cycles for CD44, and 64°C and 28 cycles for Pol II. All reactions included an initial denaturation step of 5 min at 94°C and a final extension step of 10 min at 72°C. To allow a semiquantitative analysis of transcript levels, all amplification conditions were experimentally optimized to correspond to the linear amplification phase, using serial dilutions of control cDNAs. The products were separated on 2% agarose gels containing ethidium bromide, and band intensities were quantified on digitalized images using ImageJ software (NIH) followed by normalization to Pol II expression levels. No amplification was obtained when RNA was mock reverse transcribed without adding reverse transcriptase.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting.
Samples were prepared and detected as described previously (44). The antibodies used for Western blots were as follows: rabbit polyclonal anti-c-Myc A14, anti-histone H2b (sc-10808), and rabbit anti-BCL-6 from Santa Cruz Biotechnology; rabbit antihemagglutinin (anti-HA) and anti-β-tubulin clone Tub2.1 (as a loading control) from Sigma; rabbit anti-GFP ab290 and anti-PAK1 ab40795 from Abcam; anti-Rac1 from Upstate Biotechnologies, anti-PAK1/2/3 (catalog no. 2604) from Cell Signaling Technology; and anti-p50 and anti-RelA from Hypromatrix. For densitometric analysis, films from at least three independent experiments were digitalized and analyzed using ImageJ software (NIH).
Cell fractionation.
Nuclear proteins were separated into a soluble pool not retained in the nucleus and into a chromatin-bound insoluble pool according to previously described procedures (63). Briefly, cells were washed in cold phosphate-buffered saline (PBS), scraped off, and lysed on ice for 10 min in 200 μl of fractionation buffer (50 mM Tris-HCl [pH 7.9], 0.1% [vol/vol] NP-40, 1.5 mM MgCl2, 10 mM KCl, and a protease inhibitor cocktail [Sigma]). The soluble fraction was collected by centrifuging the lysate at 3,500 × g for 5 min and adding the supernatant to 50 μl of 5× Laemmli SDS sample buffer. The pellet containing the insoluble nuclear fraction was washed once in fractionation buffer and then resuspended in 250 μl of 1× Laemmli sample buffer supplemented with 5 mM MgCl2 and 50 U endonuclease (Benzonase; Sigma) to digest nucleic acids. Equal volumes of both fractions were analyzed side by side on Western blots. Results were confirmed in at least three independent experiments.
Immunoprecipitation.
Approximately 2 × 106 DLD-1 cells were seeded in 60-mm dishes, transfected as indicated, and assayed 16 to 20 h later. For coprecipitation experiments, cells were washed in cold PBS and lysed on ice in 250 μl of lysis buffer (50 mM Tris-HCl [pH 7.5], 1% [vol/vol] NP-40, 100 mM NaCl, 10% [vol/vol] glycerol, 5 mM MgCl2, and a protease inhibitor cocktail [Sigma]). Total lysates were then sonicated on ice (10 pulses of 20 s at 40% power on a Sonics Vibra Cell sonicator) and cleared by centrifugation at 2,500 × g for 5 min. An aliquot of 0.1 volume was added to 2× Laemmli sample buffer. The remaining lysate was incubated for 1 h at 4°C with mouse monoclonal anti-c-Myc (9E10; Sigma) or anti-GFP (ab1218; Abcam) antibodies at 2 μg ml−1, precoupled to protein G-agarose beads (Roche). Beads were then washed five times with an excess of lysis buffer containing 300 mM of NaCl, and the precipitated protein complexes were analyzed on Western blots as described above. Immunoprecipitation of protein substrates for in vitro kinase assays followed the same methodology, except that cell lysis was performed using radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.5], 1% [vol/vol] NP-40, 150 mM NaCl, 0.5% [wt/vol] sodium deoxycholate, 0.1% [wt/vol] SDS, and a protease inhibitor cocktail [Sigma]). All results were confirmed in at least three independent experiments.
Confocal immunofluorescence microscopy.
Cells were grown on glass coverslips (10 by 10 mm), transfected, and incubated as indicated above, then washed twice in PBS, immediately fixed with 4% (vol/vol) formaldehyde in PBS for 30 min at room temperature, and subsequently permeabilized with 0.2% (vol/vol) Triton X-100 in PBS for 10 min at room temperature. Myc-tagged Pak (Myc-Pak) was detected with mouse anti-c-Myc (9E10; Sigma), followed by goat anti-mouse Texas Red (Jackson ImmunoResearch Laboratories). Cells were then briefly stained with 0.5 ng/ml 4′,6′-diamidino-2-phenylindole (DAPI) (Sigma) and washed in PBS, and the coverslips were mounted in VectaShield (Vector Laboratories) and sealed with nail polish. Images were recorded with the 405-nm, 488-nm, and 532-nm laser lines of a Leica TCS-SPE confocal microscope and processed with Leica and Adobe Photoshop software.
Luciferase reporter assay.
Approximately 5 × 105 DLD-1 cells were seeded in 35-mm dishes, transfected with 50 ng of the pRL-TK luciferase reporter (for constitutive expression of Renilla luciferase as internal control; Promega) and 1 μg of either standard NF-κB or pGL3-5xBCL-6 or pGL3 control reporter. For experiments titrating individual proteins, 500 to 1,000 ng of the indicated construct was cotransfected, whereas for the coexpression of two proteins, the amount of construct was previously adjusted to yield comparable expression levels. At 16 to 20 h posttransfection in the absence or presence of the NADPH oxidase inhibitor DPI or the Rac inhibitor NSC23766, cells were lysed, assayed with the Dual-Luciferase reporter assay (Promega) following the manufacturer's instructions, and measured in an Anthos Lucy-2 luminometer. Lysates were assayed in duplicate samples, and additional aliquots were analyzed by Western blotting to document protein expression levels. All firefly luciferase values were first normalized to the internal control values obtained for Renilla luciferase and then plotted as the increase over the value of untreated or vector control. The values displayed were from at least three independent transfection assays.
In vitro protein kinase assays.
For in vitro protein kinase assays, either 1 μg of a recombinant fragment of human BCL-6 (amino acids 3 to 484 lacking the C-terminal zink finger domains) (sc-858; Santa Cruz) or the beads containing immunoprecipitated GFP-tagged BCL-6 (GFP-BCL-6) protein were resuspended in 20 μl of kinase reaction buffer (30 mM Tris-HCl [pH 7.5], 10% [vol/vol] glycerol, 1 mM dithiothreitol, 1 mM Na3VO4, 37.5 mM MgCl2, and 250 μM ATP) and incubated in the presence of 5 μCi [γ-32P]ATP at 30°C for 60 min with 25, 50, 100, or 200 ng recombinant PAK1 (Proquinase, Freiburg, Germany). Then, 5× Laemmli SDS sample buffer was added to the reaction mixtures, and proteins were separated by SDS-PAGE and then transferred to a polyvinylidene difluoride membrane. The membrane was first analyzed by autoradiography, followed by Western blotting using the indicated antibodies.
RESULTS
Rac1 activation leads to an increase in NFKB1/p50 protein levels.
Previously we reported that Rac1 signaling stimulates NF-κB transcriptional activity through both the canonical RelA/p50 pathway and the RelB/p52-dependent pathway in colorectal cells (44). In the course of these studies, we noticed that the expression of active Rac1 not only increased NF-κB reporter vector activity but also the protein level of p50, whereas expression of its dimerization partner RelA remained unaffected (Fig. 1). Moreover, we observed that p50 levels decreased when activation of endogenous Rac1 was repressed by the inhibitor NSC23766 (24).
FIG. 1.

Rac1 activation modulates NFKB1/p50 protein levels. DLD-1 colorectal cells were transfected or treated with drug as indicated in the figure (+, transfected or treated with drug; −, not transfected or treated with drug) and lysed 24 h later. In one lysate aliquot, the luciferase activity of the cotransfected NF-κB reporter plasmid was measured, whereas in another aliquot, the indicated protein levels were determined by Western blotting. Note that the presence of an active Rac1 mutant was particularly efficient in stimulating reporter gene activity and in increasing p50 protein levels, while the amount of RelA remained unchanged (tubulin levels served as a loading control). Whereas reporter gene transcription was strongly inhibited by DPI (inhibitor of NADPH oxidase and ROS formation) or by the superrepressor IκBα (A32A36), both treatments had no effect on the Rac1-L61 stimulated p50 increase. (NSC23766 is an inhibitor of endogenous Rac1 activation.)
The p50 subunit is produced through constitutive proteolytic processing of the precursor protein p105, which is transcribed from the NFKB1 gene. A previous report has shown evidence that the NFKB1 gene promoter could be stimulated by RelA/p50 itself in hematopoietic cells (11). In order to test whether the RelA complex is involved in regulating NFKB1/p50 expression in colorectal cells, we first cotransfected DLD-1 cells with expression vectors encoding a constitutively active Rac1-L61 mutant and the nondegradable superrepressor IκBα (A32A36) (16), which inhibits RelA/p50 activation. We found that the presence of the superrepressor significantly inhibited the Rac1-mediated activation of the NF-κB transcriptional reporter, but the increase in p50 levels was still detected (Fig. 1). This increase in p50 was also observed when the Rac1-mediated production of ROS, an upstream event of RelA/p50 activation, was inhibited by the NADPH oxidase inhibitor DPI.
Rac1 regulates NFKB1 expression by releasing BCL-6-mediated transcriptional repression.
A previous report has demonstrated that the NFKB1 promoter contains binding sites for the transcription factor BCL-6 (40), a repressor identified in B-cell lymphoma (12, 59, 64). In addition, a highly related repressor protein, BAZF/BLC6b, has been identified (56), which binds the same promoter sites as BCL-6 does. To analyze the contribution of both factors to NFKB1 expression, we first determined by RT-PCR whether endogenous BCL-6 and BAZF transcripts were expressed in three colorectal cell lines as well as in the B-cell precursor leukemia 697 cell line and erythroleukemia HEL cells as positive controls. We found endogenous BCL-6 expression in the three colorectal cell lines and the erythroleukemia cell line, whereas BAZF was expressed only in HEL cells (Fig. 2A). These data identified BCL-6 as a candidate regulator of NFKB1/p50 expression in colorectal cells, prompting us to transfect cells with increasing amounts of an expression vector encoding the BCL-6 protein. Intriguingly, the endogenous NFKB1 transcript expression (Fig. 2B), as well as the corresponding NFKB1/p50 protein levels (Fig. 2C), were clearly inhibited by the expression of BCL-6. In contrast, expression of active Rac1 led to increased expression of both the NFKB1 transcript (Fig. 2B) and NFKB1/p50 protein (Fig. 2C). Conversely, when the endogenous Rac1 activation in DLD-1 cells was impaired by treating cells with the Rac inhibitor NSC23766, expression of NFKB1 transcript and NFKB1/p50 protein was inhibited (Fig. 2B and C). These data indicated that Rac1 signaling could modulate NFKB1 gene expression via the transcriptional repressor BCL-6.
FIG. 2.

Rac1 modulates NFKB1 expression via BCL-6. (A) The expression of endogenous BCL-6 and the highly related repressor protein BAZF/BLC6b was tested by RT-PCR in three colorectal cell lines and two hematopoietic cell lines, as indicated. The amplification of RNA polymerase II (Pol II) served as internal control. (B and C) Role of BCL-6 overexpression or modulation of Rac1 signaling in NFKB1/p50 expression. DLD-1 cells were transfected with either GFP control vector versus increasing amounts (indicated by the height of the black triangle) of GFP-BCL-6 or with Myc control vector versus increasing amounts of Myc-Rac1-L61 or mock transfected and treated with the Rac1 inhibitor NSC23766. Cells were lysed following 24 h to isolate either total RNA or whole protein. Symbols: +, transfected or treated with drug; −, not transfected or treated with drug. (B, left) NFKB1 or control Pol II transcripts were amplified by semiquantitative RT-PCR, and (right) band intensities were quantified from digital images by densitometry. (C, left) Western blot showing NFKB1/p50 protein levels as well as levels of transfected GFP-BCL-6 or Myc-Rac1-L61. α-GFP, anti-GFP antibody. (Right) Detection of endogenous Rac1 served as a loading control, and band intensities were quantified by densitometry.
In order to test whether Rac1 activation can regulate the transcriptional activity of BCL-6, we utilized a previously described BCL-6-controlled reporter gene (32). When this reporter was coexpressed with BCL-6, a clear repression of transcriptional activity was observed (Fig. 3A). Repression was also evident when activation of endogenous Rac1 was inhibited with NSC23766. In contrast, transcriptional activity of the reporter was clearly promoted by siRNA-mediated depletion of endogenous BCL-6 or upon cotransfection with Rac1-L61, in a dose-dependent manner (Fig. 3A). These data provide substantial evidence that Rac1 signaling regulates gene expression via BCL-6. Interestingly, we observed that the splice variant Rac1b could not significantly affect the BCL-6-controlled reporter gene (Fig. 3A).
FIG. 3.

Rac1 releases transcriptional repression by BCL-6. (A) DLD-1 cells were transfected with a transcriptional luciferase reporter vector under the control of five consensus BCL-6 binding motifs (32) or the respective empty pGL3 control vector. Cells were cotransfected with the indicated expression vectors and siRNAs or mock transfected and treated with the Rac1 inhibitor NSC23766. The increasing or decreasing amount of vector is indicated by the height of the black triangle. Symbols: +, transfected or treated with drug; −, not transfected or treated with drug. siBCL-6, BCL-6-specific siRNAs. (B) Effect of BCL-6 or Rac1 signaling on the endogenous BCL-6 target gene CD44. (Left) Cells were treated as described in the legend to Fig. 2, CD44 or control Pol II transcripts were amplified by semiquantitative RT-PCR, and band intensities were quantified from digital images by densitometry (right).
We next asked whether another endogenous BCL-6 target gene, the cell adhesion molecule CD44 (60), was modulated by Rac1 activation in DLD-1 cells. We observed that ectopic expression of BCL-6 or inhibition of endogenous Rac1 activity led to decreased CD44 transcript expression, whereas transfection of Rac1-L61 promoted an increase (Fig. 3B). Together, our results strongly indicate that Rac1 activation releases BCL-6 repression from target genes including NFKB1, CD44, and a BCL-6-specific luciferase reporter.
Active Rac1 induces nuclear redistribution and chromatin release of BCL-6.
In order to obtain mechanistic insights into how Rac1 activation would influence BCL-6 activity, we first studied its effect on the subcellular localization of BCL-6 using immunofluorescence microscopy and cell fractionation. As shown in Fig. 4A, expression of BCL-6 alone revealed a strictly nuclear localization in DLD-1 cells with the typical concentration of BCL-6 in numerous nuclear dots that has previously been described (9, 32). In the presence of Rac1-L61, BCL-6 lost accumulation in nuclear dots and appeared diffuse in the nucleoplasm. In contrast, splice variant Rac1b-L61 had little effect on the accumulation of BCL-6 in dots in the nucleus (Fig. 4A). This is in agreement with the poor stimulation of the BCL-6-controlled reporter gene that we observed for Rac1b-L61 (Fig. 3A).
FIG. 4.
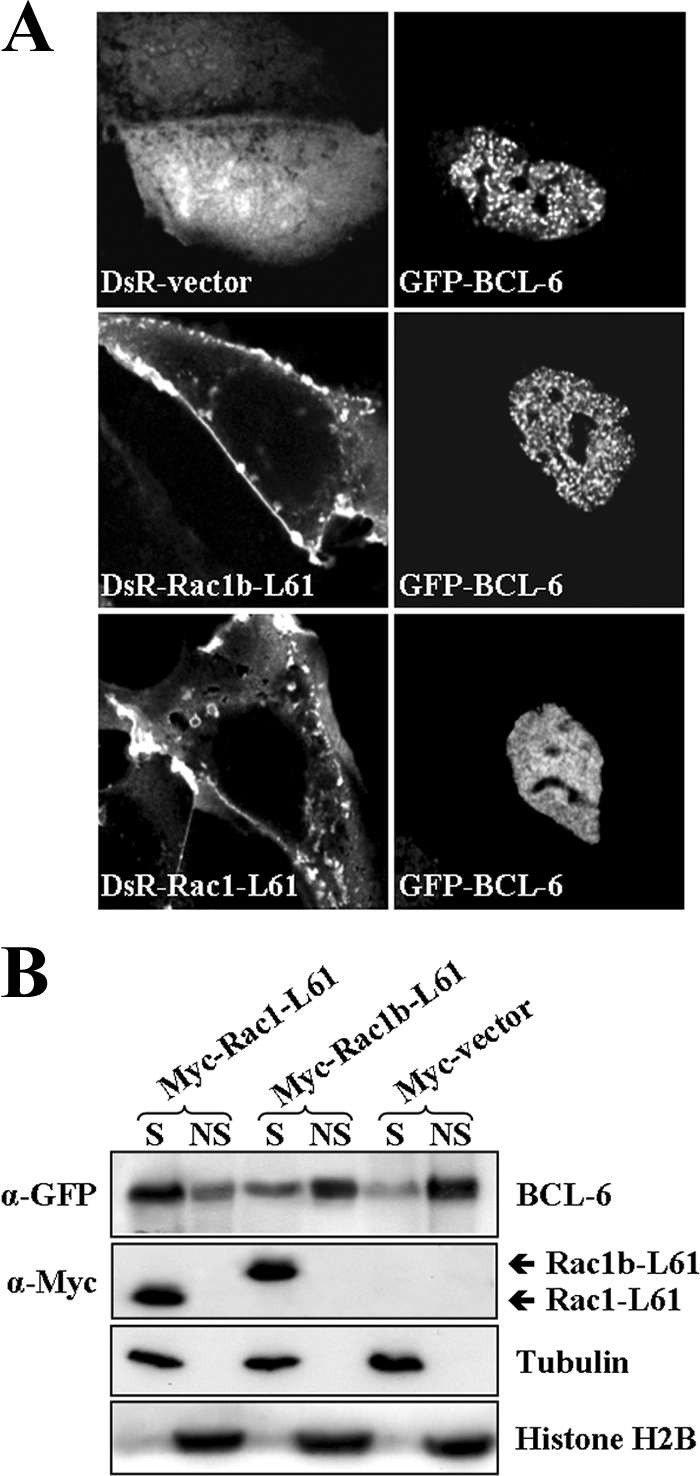
Active Rac1 affects subnuclear location and chromatin binding of BCL-6. (A) DLD-1 cells were cotransfected with GFP-BCL-6 and either DsRed empty vector, DsRed-Rac1-L61, or DsRed-Rac1b-L61 as indicated. Cells were fixed after 24 h, nuclei were counterstained with DAPI, and fluorescent signals were recorded by confocal microscopy. (B) Presence of BCL-6 in a soluble or chromatin-bound form. DLD-1 cells were cotransfected with GFP-BCL-6 and the indicated Myc-tagged vectors and lysed after 24 h so that a soluble (S) and a nonsoluble (NS) chromatin-bound fraction was obtained. Western blot analysis of these fractions is shown. Histone 2B was detected as a marker for insoluble chromatin-bound proteins, and β-tubulin was detected as a marker for soluble factor. α-GFP, anti-GFP antibody.
In order to test whether these differences in localization would represent altered chromatin binding, we applied a previously described cell fractionation protocol (15, 37, 63), which separates transcription factors into a soluble pool that is extracted from the nucleus and into a chromatin-bound pool that remains insoluble. Under these experimental conditions, the expression of BCL-6 alone revealed the majority of the protein in the insoluble chromatin-bound fraction (Fig. 4B). This is compatible with its role as a transcriptional repressor and corroborates the inhibition of NFKB1 and CD44 gene expression observed in Fig. 2B and 3B. When we determined the fractionation of BCL-6 in cells coexpressing an active mutant of the splice variant Rac1b, only a very small increase in the soluble fraction was observed. In contrast, coexpression of activated Rac1 led to a remarkable transition of BCL-6 from the chromatin-bound insoluble fraction into the soluble pool (Fig. 4B). In these experiments, the total amount of BCL-6 protein apparently remained unaffected.
Altogether, these data demonstrate that upon activation of Rac1, the transcription factor BCL-6 becomes relocalized within the nucleus, is no longer retained in the chromatin-bound fraction, and loses its activity to repress target genes.
Modulation of BCL-6 by Rac1 signaling requires PAK1.
For further insights into the effect of Rac1 on BCL-6 activity, we tested the interaction of Rac1 and BCL-6 by coimmunoprecipitation. Whereas Rac1-L61 coprecipitated with RelB, in agreement with previously described data (44), we found no evidence for the formation of a stable complex between BCL-6 and active Rac1 under the same experimental conditions (Fig. 5). The lack of interaction between Rac1 and BCL-6 suggested that Rac1 affects BCL-6 activity indirectly through a downstream signaling pathway.
FIG. 5.
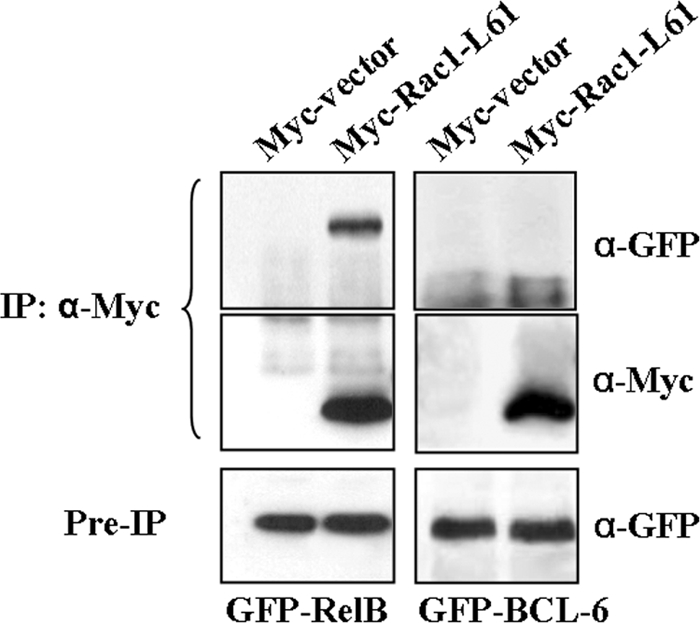
Active Rac1 and BCL-6 are not found in a protein complex. DLD-1 cells expressing Myc control vector or Myc-Rac1-L61 were cotransfected with either GFP-BCL-6 or GFP-RelB. Whereas RelB coimmunoprecipitated with Myc-Rac1-L61, confirming previously described data (44), no such complex was detected between Rac1-L61 and BCL-6. IP: α-Myc, immunoprecipitation with anti-Myc antibody.
The generation of ROS through the stimulation of NADPH oxidase activity is a Rac function conserved in immune and epithelial cells. Moreover, ROS are known to modulate the activity of several transcription factors (72), including NF-κB downstream of Rac1 and Rac1b (44). We therefore treated Rac1-L61-expressing cells with DPI, a cell-permeable inhibitor of the NADPH oxidase widely used to block the generation of ROS (5, 44, 65, 66). We found that although this treatment clearly inhibited activation of an NF-κB-driven luciferase reporter (Fig. 1), it had no effect on the increase in the BCL-6-driven reporter activity (Fig. 6A).
FIG. 6.
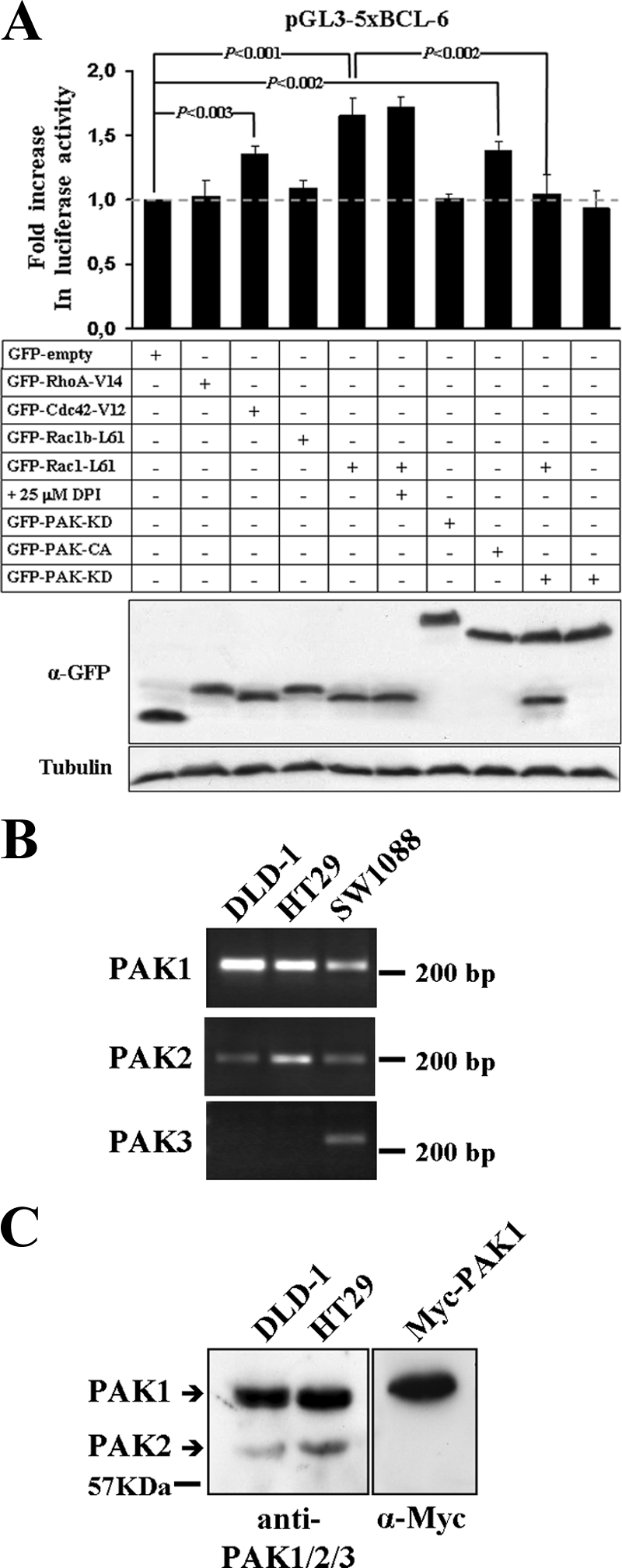
PAK1 acts downstream of Rac1 in the release of transcriptional repression by BCL-6. (A) DLD-1 cells were transfected with the transcriptional BCL-6 luciferase reporter vector and one of the indicated GFP-tagged expression vectors encoding either activated small GTPase mutants or protein kinase mutants. Luciferase activity was determined in cell lysates, and expression of transfected proteins was documented by Western blotting. A graph with the observed changes in luciferase activity relative to GFP empty vector-transfected control cells (top panel) and immunoblots with the expression levels of the GFP-tagged proteins (middle panel) and β-tubulin as a loading control (bottom panel) are shown. The migration of molecular size markers is indicated. Note that transcriptional repression by BCL-6 was released in the presence of Rac1-L61, Cdc42-V12, and a constitutively active (CA) PAK1 mutant, whereas a kinase-dead (KD) PAK1 prevented the Rac1-L61 mediated increase in luciferase activity. α-GFP, anti-GFP antibody. Symbols: +, transfected or treated with drug; −, not transfected or treated with drug. (B) RT-PCR analysis to determine the expression of PAK1, PAK2, and PAK3 in DLD-1 and HT29 colorectal cells compared to SW1088 glioblastoma cells. (C) Western blot analysis to directly compare the expression levels of PAK1 and PAK2 in DLD-1 or HT29 cells using an anti-PAK1/2/3 antibody. Note that PAK1 is the most prominent isoform expressed.
In order to determine whether the observed modulation of BCL-6 repression was Rac1 specific, we compared the effects of activated RhoA, Rac1, and Cdc42. We found that active Cdc42 also produced a moderate but significant stimulation of the BCL-6 reporter (Fig. 6A).
Stimulation of the protein kinases PAK and JNK are two classical downstream pathways that are stimulated by Rac1 and Cdc42, but not by RhoA or by the Rac1b splice variant (43, 62), both of which failed to inhibit BCL-6 activity (Fig. 6A). Thus, the activity of the BCL-6 luciferase reporter was analyzed in cells transfected with previously described constitutively active mutants of JNK (SAPKβ-MKK7) (52) and PAK1 (PAK1-T423E) (58). As shown in Fig. 6A, active JNK had no effect, whereas the expression of constitutively active PAK1 significantly stimulated transcription from the BCL-6 reporter. We further determined whether the catalytic activity of PAK1 was involved in the observed BCL-6 reporter stimulation. The reporter vector was cotransfected with active Rac1-L61 in the presence of a dominant-negative, kinase-dead PAK1 mutant (PAK1-K299R) (58). These experiments revealed a clear reduction in Rac1-mediated transcriptional stimulation (Fig. 6A), suggesting that the Rac1-L61-stimulated transcription from the BCL-6 reporter required Rac1-induced PAK activation.
GTP-bound Rac1 can activate PAK-1 (α-PAK), PAK-2 (γ-PAK), and PAK-3 (β-PAK). Thus, we determined which endogenous PAK isoform could be mediating the observed effects downstream of active Rac1 in DLD-1 cells. Using RT-PCR, we found that only PAK-1 and PAK-2 were expressed in colorectal cells, whereas PAK-3 was detected in a glioblastoma cell line (Fig. 6B). In order to directly compare the expression levels of PAK1 and PAK2, colorectal cell lysates were analyzed by Western blotting using an anti-PAK1/2/3 antibody. We found that PAK1 was by far the most prominent isoform expressed (Fig. 6C).
We then determined whether the Rac1-L61-stimulated transcription from the BCL-6 reporter required endogenous PAK1. Expression of endogenous PAK1 or expression of PAK2 was depleted by transfection of cells with specific siRNAs. As shown in Fig. 7A, these oligonucleotides specifically depleted either PAK1 or PAK2; however, only the depletion of PAK1 affected both the endogenous and Rac1-L61-stimulated 5xBCL-6 activation (Fig. 7B). In addition, prior incubation of DLD-1 cells with IPA-3, a specific inhibitor that prevents activation of group I PAKs by allosteric targeting of their autoregulatory domain, blocked the effect of Rac1-L61 on the 5xBCL-6 reporter (Fig. 7B). In these experiments, no detectable changes in the total amount of BCL-6 protein were observed (not shown).
FIG. 7.
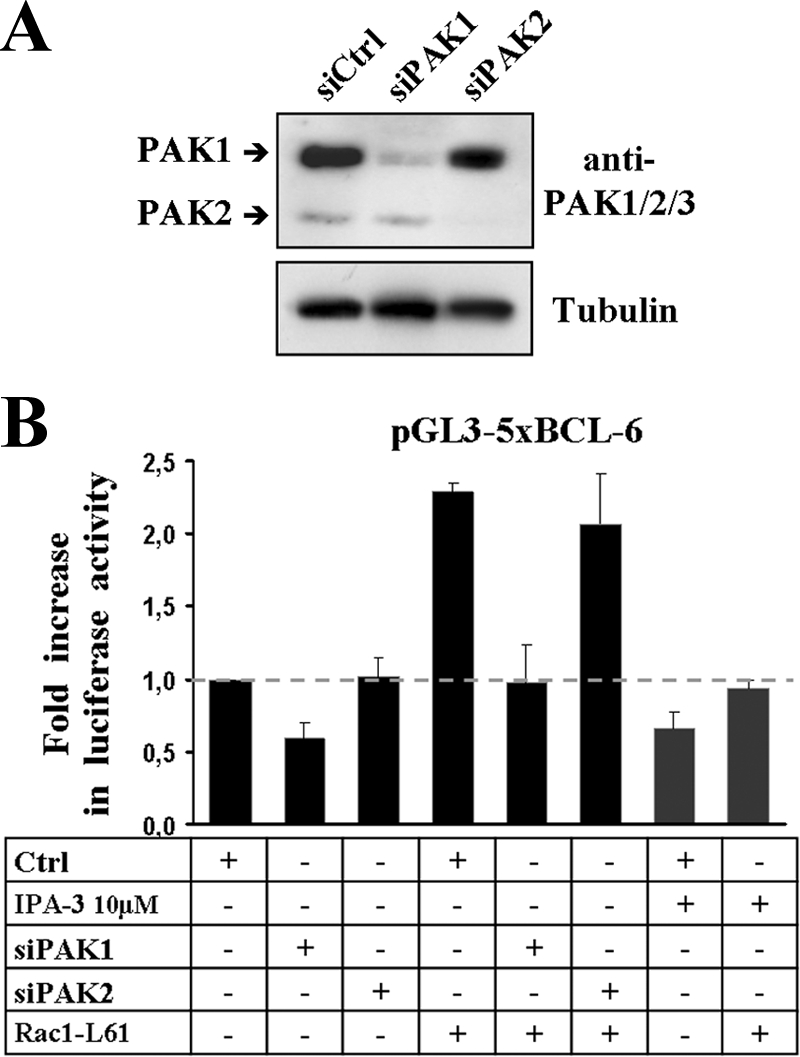
Interference with PAK1 by depletion or inhibitor treatment blocks Rac1-mediated activation of BCL-6. (A) Western blot showing the efficiency and specificity of PAK1- or PAK2-specific siRNAs transfected into DLD-1 cells. Detection of β-tubulin served as a loading control. (B) DLD-1 cells were transfected with the indicated siRNAs and 24 h later transfected again with the 5xBCL-6 transcriptional luciferase reporter vector in the presence (+) or absence (−) of Rac1-L61. When indicated, cells were incubated with 10 μM PAK inhibitor IPA-3. Symbols: +, transfected or treated with drug; −, not transfected or treated with drug. Ctrl, control.
Altogether, these data indicate that PAK1 is a critical link between Rac1 activation and transcriptional repression by BCL-6.
PAK1 binds to and phosphorylates BCL-6.
Since PAK1 overexpression stimulated the BCL-6 reporter, we used immunofluorescence microscopy to test whether PAK1 could also affect the nuclear redistribution of BCL-6 observed in the presence of active Rac1. As shown in Fig. 8A, overexpression of the kinase-dead Myc-PAK1-K299R mutant apparently enhanced the dot-like localization pattern of BCL-6 in the nucleus, whereas overexpression of a kinase-competent PAK1 redistributed BCL-6 to a more diffuse nucleoplasmic pattern (Fig. 8B). In addition, the expression of PAK1 decreased the amount of BCL-6 remaining in the insoluble chromatin-bound cell fraction, whereas kinase-dead PAK1 did not affect chromatin binding (Fig. 8C). PAK1 also clearly localized to the nucleus. In fact, we observed a correlation between the expression level of BCL-6 and the recruitment of PAK1 from the cytoplasm into the nucleus. In particular, a pixel intensity analysis in confocal images revealed that cells with an equivalent overall level of ectopic PAK1 expression (Fig. 8D, top graph) differed in their nuclear PAK1 signal (Fig. 8D, bottom graph), depending on the expression level of BCL-6 (Fig. 8D, middle graph).
FIG. 8.

Effect of PAK1 on nuclear BCL-6. (A and B) DLD-1 cells were cotransfected with GFP-BCL-6 and the kinase-dead Myc-PAK1-K299R mutant (A) or with wild-type Myc-PAK1 (B). The cells were fixed after 20 h and analyzed by confocal microscopy. Note in panel A, the increase in nuclear dot localization of BCL-6 in the presence of dominant-negative PAK1 but the diffuse nucleoplasmic pattern with wild-type PAK1 in panel B. ROI 1, region of interest 1. (C) Presence of BCL-6 in the chromatin-bound fraction. DLD-1 cells were cotransfected with GFP-BCL-6 and the indicated Myc-tagged vectors and separated into a soluble (S) and a nonsoluble (NS) chromatin-bound fraction, as described in the legend to Fig. 4B. α-GFP, anti-GFP antibody. (D) The intensity of the overall fluorescent PAK1 signal in the two representative cells shown in panel B, with different amounts of BCL-6 expression, was determined (top panel, compare regions of interest ROI 1 and ROI 2) and found to be equivalent. Then the intensities of the nuclear versus cytoplasmic PAK1 and BCL-6 signals were compared along the axes indicated as ROI 3 and ROI 4. Note that the distribution of PAK1 signal between the nucleus and cytoplasm (ROI 3 and ROI 4 in the red channel [bottom panel]) correlates with the signal intensity of BCL-6 in the nucleus (ROI 3 and ROI 4 in the green channel [middle panel]). (E) BCL-6 and PAK1 coimmunoprecipitate. DLD-1 cells were cotransfected with Myc-PAK1 and either GFP control vector or GFP-BCL-6 or GFP-Rac1-L61 as a positive control. Cells were lysed after 24 h, extracts were incubated with anti-GFP antibodies (α-GFP), and the presence of coprecipitated Myc-PAK1 was analyzed (top panel). Successful precipitation of GFP-tagged proteins (middle panel) as well as equal expression of Myc-PAK1 in total cell extracts (preimmunoprecipitation [Pre-IP]; bottom panel) is also shown.
The generation of pixel overlap maps from the confocal images allowed the calculation of Pearson's correlation values, which suggested colocalization between nuclear PAK1 and BCL-6 (data not shown). We thus analyzed whether a PAK1/BCL-6 complex could be isolated by coimmunoprecipitation from DLD-1 colorectal cells. Using the previously described coprecipitation of Rac1-L61 with PAK1 (43) as a positive control, we demonstrate that PAK1 can form a stable complex with BCL-6 (Fig. 8E).
Since BCL-6 can be phosphorylated by mitogen-activated protein kinases (46), we asked whether it could be a direct substrate for PAK1. Using an in vitro phosphorylation assay, we added increasing amounts of full-length, recombinant PAK1 to a recombinant 484-amino-acid fragment of the BCL-6 protein. Under these conditions, a concomitant increase of BCL-6 phosphorylation was observed (Fig. 9A, top panel), which generated electrophoretic band shifts that were confirmed by Western blot analysis with an anti-BCL-6 serum (Fig. 9A, middle panel). These data suggest the presence of multiple phosphorylation sites for PAK1 in the BCL-6 fragment.
FIG. 9.
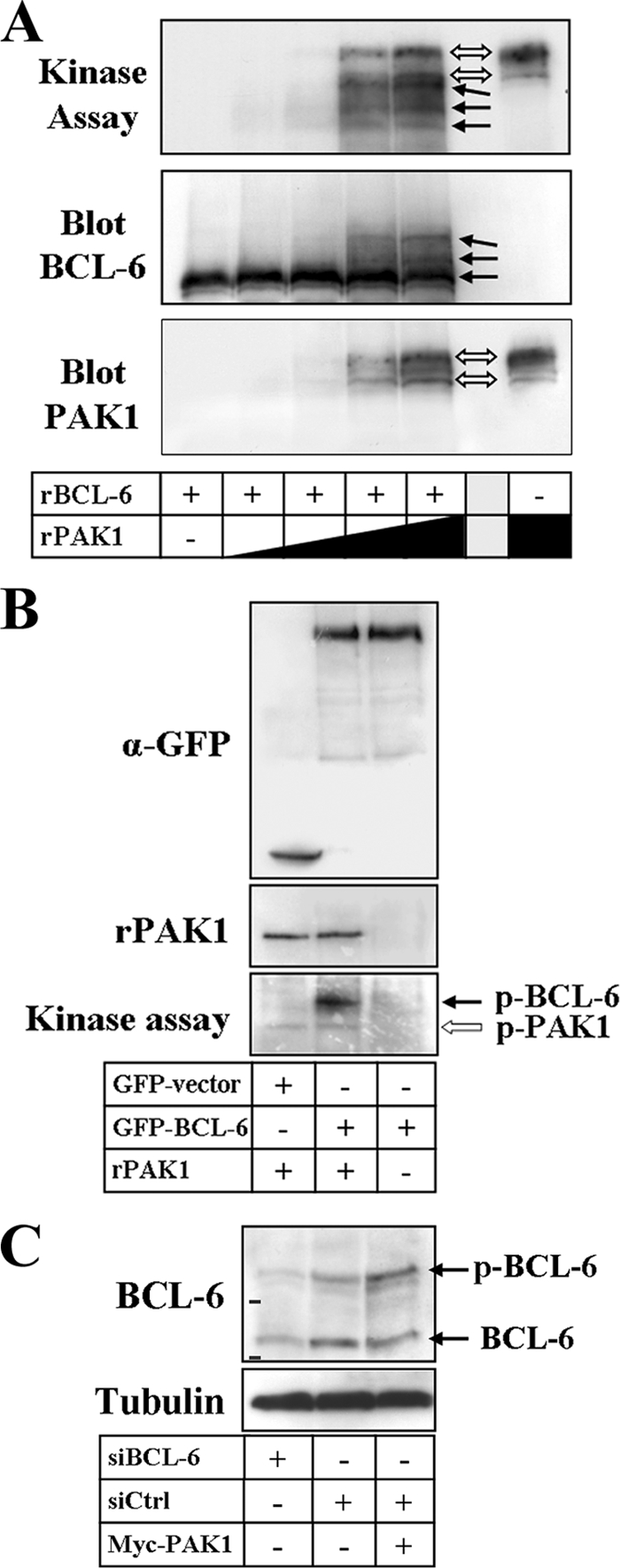
PAK1 phosphorylates BCL-6. (A) A recombinant N-terminal BCL-6 fragment (rBCL-6) and increasing amounts of recombinant PAK1 (rPAK1) were incubated in an in vitro protein kinase assay before proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Membranes were exposed to X-ray films (top panel), followed by sequential immunoblot detection of BCL-6 (middle panel) and PAK1 (bottom panel). Note the shifts in electrophoretic migration of the phosphorylated recombinant BCL-6 as well as of autophosphorylated PAK1 bands. The presence (+) or absence (−) of rBCL-6 and the increasing amount of rPAK1 is indicated by the height of the black triangle or black rectangle. (B) GFP control vector or full-length GFP-BCL-6 were transfected into DLD-1 cells, immunoprecipitated with anti-GFP antibodies (α-GFP) using RIPA buffer and then incubated in vitro in the presence (+) or absence (−) of 200 ng recombinant PAK1. Western blots to document successful protein precipitation (top panel) and the presence of recombinant PAK1 (middle panel) are shown. The corresponding autoradiograph shows phosphorylation of GFP-BCL-6 by PAK1 as well as PAK1 autophosphorylation (bottom panel). (C) Detection of endogenous BCL-6 by Western blotting in DLD-1 cells transfected with either BCL-6-specific siRNAs (siBCL-6) or control siRNA (siCtrl) in the presence or absence of Myc-PAK1. Two specific bands were detected, and the shift in electrophoretic migration upon transfection of PAK1 indicates that the top BCL-6 band is endogenous phospho-BCL-6 (p-BCL-6) (bars show migration of 75- and 100-kDa markers).
To confirm that PAK1 also phosphorylates the full-length protein, GFP-BCL-6 was immunoprecipitated from DLD-1 cells using stringent RIPA buffer conditions to avoid background phosphorylation events and then incubated in vitro with recombinant PAK1. As shown in Fig. 9B, full-length GFP-BCL-6 became clearly phosphorylated by PAK1, although the higher molecular weight of the GFP-tagged protein did not allow the detection of possible band shifts. We then analyzed whole-cell lysates and detected two BCL-6 protein bands of about 80 and 120 kDa by Western blotting, both of which became specifically depleted upon transfection of cells with BCL-6 siRNAs (Fig. 9C). The higher-molecular-mass band was clearly increased upon transfection of cells with PAK1, indicating a phosphorylation-dependent band shift of the endogenous BCL-6 protein.
DISCUSSION
The main finding in this work is that Rac1 regulates the transcription factor BCL-6 via PAK1 and counteracts repression of its target genes.
The BCL-6 transcriptional repressor is one of the most frequently translocated genes in B-cell non-Hodgkin's lymphomas (12, 64). BCL-6 translocations do not alter the BCL-6 coding sequence but associate the gene with other promoter region, such as the IgH enhancer, and this deregulates BCL-6 expression. Moreover, the activity of BCL-6 can be modulated through posttranslational modifications. For instance, acetylation of lysine 379 downregulates its ability to repress transcription, probably due to impaired recruitment of histone deacetylases (4), while phosphorylation by mitogen-activated protein kinases in B cells was shown to target BCL-6 for rapid degradation by the ubiquitin-proteasome pathway (46, 50).
More recently, evidence has accumulated that BCL-6 is also expressed in nonhematopoietic cells. Its expression was detected in olfactory sensory neurons (48), in healthy skin and epidermal neoplasms (34), in uroepithelial cells (30, 41), and in epithelial cells of the mammary gland (42). BCL-6 expression has also been detected in HeLa cells (1), and in this study, we document for the first time its expression in colorectal cells.
Here we found that Rac1 signaling affects the subnuclear localization and transcriptional repressor activity of BCL-6 and demonstrated these effects using four different approaches. First, a reporter construct expressing the luciferase gene under the control of five BCL-6 binding sites immediately upstream of the simian virus 40 promoter (32) was used and shown to become repressed in cells cotransfected with BCL-6, as expected. Repression of reporter activity was further observed when endogenous Rac1 activation was diminished by treating cells with the Rac1-specific inhibitor NSC23766. In contrast, expression of an active Rac1 mutant led to increased luciferase activity. These data clearly indicate a modulation of BCL-6 in response to Rac1 signaling. Second, the expression of two previously described endogenous BCL-6 target genes, NFKB1 (40) and CD44 (60), was analyzed. As was observed with the BCL-6 reporter, overexpression of BCL-6 and inhibition of endogenous Rac1 activation by NSC23766 decreased expression of these genes. In contrast, transfection of activated Rac1 increased their expression up to 2- and 2.6-fold, respectively. Third, we determined the subnuclear distribution of BCL-6, which has previously been reported to accumulate in characteristic nuclear foci (9, 32). In the presence of active Rac1, a clear redistribution of BCL-6 from these nuclear foci to a more diffuse, homogenous nucleoplasmic localization was observed (Fig. 4A). Fourth, our cell fractionation studies corroborate these results by showing the transition of BCL-6 from an insoluble, chromatin-bound form into a soluble form in the presence of active Rac1 (Fig. 4B). Together, these data provide substantial evidence that BCL-6-mediated gene repression is negatively regulated by Rac1 signaling.
The mechanism of how Rac1 affects BCL-6 activity apparently does not involve formation of a stable complex between BCL-6 and active Rac1, because both proteins did not coimmunoprecipitate (Fig. 5). Although one cannot disregard the possibility of a transient interaction occurring between the two proteins, these data suggest that BCL-6 rather responds to a signaling pathway downstream of Rac1.
Rac signaling activates the production of ROS via NADPH oxidases (57), and ROS are known to modulate the activity of several transcription factors, such as AP1, Ets, Smad, Snail, and NF-κB (72). However, we demonstrate that Rac1-induced production of NADPH oxidases had no effect on BCL-6 activity. Overexpression of activated Cdc42 also induced a weak but significant stimulation of the BCL-6 reporter, and Cdc42 shares the downstream effectors PAK and JNK with Rac1 (28). Because BCL-6 can be downregulated by phosphorylation in B cells (46, 50), we tested the effect of constitutively active kinase mutants on BCL-6 activity.
We found that the activation of PAK was required for the observed changes in BCL-6 activity. PAK1 was found to be the predominantly expressed PAK isoform in the colorectal cells studied, and its overexpression mimics the effect of active Rac1 on nuclear distribution, chromatin binding, or transcriptional activity of BCL-6. Moreover, interfering with PAK1 function by siRNA-mediated depletion, cell treatment with inhibitor IPA-3, or expression of a dominant-negative PAK1 mutant strongly inhibited the effect of Rac1-L61 on the BCL-6 reporter. In all these experiments, no detectable changes in the total amount of BCL-6 protein were observed. PAK1 could be isolated in a protein complex with BCL-6 and was able to phosphorylate BCL-6 in vitro (Fig. 9). PAK1 was further recruited to the cell nucleus in BCL-6-overexpressing cells and colocalized with BCL-6 in the nucleoplasm. Together, these data identify PAK1 as the critical mediator between Rac1 activation and BCL-6 downregulation.
PAK1 phosphorylation has been found to modulate various transcriptional regulators with respect to their transcriptional activity, subnuclear location, and nuclear import or export (38). Our results therefore reveal that the regulation of BCL-6 repressor activity by PAK1 constitutes yet another pathway through which this kinase exerts its control over specific transcriptional events. In addition, the identification of PAK1 as the link between Rac1 and BCL-6 is in agreement with our observation that splicing variant Rac1b, which was previously shown unable to stimulate PAK1 activation (44, 62), had no significant effect on the BCL-6 transcriptional reporter activity, on the subnuclear localization of BCL-6, and on its transition into a soluble nuclear fraction.
One of the physiological target genes for BCL-6 repression is NFKB1 encoding the p105 precursor protein for the NF-κB member p50. In response to Rac1 signaling, we found increased levels of p105 transcripts and of p50 protein, which is generated from p105 via a constitutive proteolytic pathway (35, 45). p50 can dimerize with various Rel partner proteins, and the resulting protein complexes can either activate or repress transcription (2, 17, 22, 26, 68, 73). Therefore, the physiological effect of increasing p50 levels in response to Rac1 activation is not readily apparent. We speculate that a short-term effect will be an increased availability of transcriptionally competent Rel protein/p50 dimers that make any NF-κB stimulation more robust. On a longer term, however, increasing p50 levels may favor the formation of p50/p50 homodimers that can act as repressors and downregulate the NF-κB response, similar to what has been described during the inflammatory response (2, 17).
Another endogenous BCL-6 target gene that is expressed in the colon is CD44 (60). The CD44 family is a family of cell-surface glycoproteins involved in cell-matrix adhesion and growth factor presentation and was shown to influence cell growth, survival, and differentiation. Members of the CD44 family have been implicated in the progression and metastasis of tumors (51), including colorectal tumors (25, 29, 54, 70, 71). Our data therefore suggest that a deregulation of Rac1 signaling may contribute to the altered CD44 expression described in colorectal tumors.
Acknowledgments
We thank M. Karin (University of California, San Diego), C. V. Paya (Mayo Clinic, Rochester, MN), U. Rapp (Würzburg, Germany), J. Chernoff (Fox Chase Cancer Center, Philadelphia, PA), B. Baumann (University of Ulm), R. Dalla-Favera (Columbia University), and V. J. Bardwell (University of Minnesota) for generously providing plasmids.
This work was supported by the Fundação para a Ciência e Tecnologia, Portugal (Programas POCI 2010 SAU-OBS/57660/2004; Financiamento Plurianual do CIGMH; fellowships BD 29789/2006 to P.B. and BPD 20531/2004 to P.M.).
Footnotes
Published ahead of print on 1 June 2009.
REFERENCES
- 1.Allman, D., A. Jain, A. Dent, R. R. Maile, T. Selvaggi, M. R. Kehry, and L. M. Staudt. 1996. BCL-6 expression during B-cell activation. Blood 875257-5268. [PubMed] [Google Scholar]
- 2.Baer, M., A. Dillner, R. C. Schwartz, C. Sedon, S. Nedospasov, and P. F. Johnson. 1998. Tumor necrosis factor alpha transcription in macrophages is attenuated by an autocrine factor that preferentially induces NF-κB p50. Mol. Cell. Biol. 185678-5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benitah, S. A., P. F. Valeron, L. van Aelst, C. J. Marshall, and J. C. Lacal. 2004. Rho GTPases in human cancer: an unresolved link to upstream and downstream transcriptional regulation. Biochim. Biophys. Acta 1705121-132. [DOI] [PubMed] [Google Scholar]
- 4.Bereshchenko, O. R., W. Gu, and R. Dalla-Favera. 2002. Acetylation inactivates the transcriptional repressor BCL6. Nat. Genet. 32606-613. [DOI] [PubMed] [Google Scholar]
- 5.Bonizzi, G., J. Piette, S. Schoonbroodt, R. Greimers, L. Havard, M. P. Merville, and V. Bours. 1999. Reactive oxygen intermediate-dependent NF-κB activation by interleukin-1β requires 5-lipoxygenase or NADPH oxidase activity. Mol. Cell. Biol. 191950-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bos, J. L., H. Rehmann, and A. Wittinghofer. 2007. GEFs and GAPs: critical elements in the control of small G proteins. Cell 129865-877. [DOI] [PubMed] [Google Scholar]
- 7.Boureux, A., E. Vignal, S. Faure, and P. Fort. 2007. Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol. Biol. Evol. 24203-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyer, L., S. Travaglione, L. Falzano, N. C. Gauthier, M. R. Popoff, E. Lemichez, C. Fiorentini, and A. Fabbri. 2004. Rac GTPase instructs nuclear factor-κB activation by conveying the SCF complex and IkBα to the ruffling membranes. Mol. Biol. Cell 151124-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cattoretti, G., C. C. Chang, K. Cechova, J. Zhang, B. H. Ye, B. Falini, D. C. Louie, K. Offit, R. S. Chaganti, and R. Dalla-Favera. 1995. BCL-6 protein is expressed in germinal-center B cells. Blood 8645-53. [PubMed] [Google Scholar]
- 10.Chang, C. C., B. H. Ye, R. S. Chaganti, and R. Dalla-Favera. 1996. BCL-6, a POZ/zinc-finger protein, is a sequence-specific transcriptional repressor. Proc. Natl. Acad. Sci. USA 936947-6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cogswell, P. C., R. I. Scheinman, and A. S. Baldwin, Jr. 1993. Promoter of the human NF-kappa B p50/p105 gene. Regulation by NF-kappa B subunits and by c-REL. J. Immunol. 1502794-2804. [PubMed] [Google Scholar]
- 12.Dalla-Favera, R., A. Migliazza, C. C. Chang, H. Niu, L. Pasqualucci, M. Butler, Q. Shen, and G. Cattoretti. 1999. Molecular pathogenesis of B cell malignancy: the role of BCL-6. Curr. Top. Microbiol. Immunol. 246257-263. [DOI] [PubMed] [Google Scholar]
- 13.Dent, A. L., F. H. Vasanwala, and L. M. Toney. 2002. Regulation of gene expression by the proto-oncogene BCL-6. Crit. Rev. Oncol. Hematol. 411-9. [DOI] [PubMed] [Google Scholar]
- 14.DerMardirossian, C., and G. M. Bokoch. 2005. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 15356-363. [DOI] [PubMed] [Google Scholar]
- 15.Dhordain, P., O. Albagli, N. Honore, F. Guidez, D. Lantoine, M. Schmid, H. D. The, A. Zelent, and M. H. Koken. 2000. Colocalization and heteromerization between the two human oncogene POZ/zinc finger proteins, LAZ3 (BCL6) and PLZF. Oncogene 196240-6250. [DOI] [PubMed] [Google Scholar]
- 16.DiDonato, J., F. Mercurio, C. Rosette, J. Wu-Li, H. Suyang, S. Ghosh, and M. Karin. 1996. Mapping of the inducible IκB phosphorylation sites that signal its ubiquitination and degradation. Mol. Cell. Biol. 161295-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Driessler, F., K. Venstrom, R. Sabat, K. Asadullah, and A. J. Schottelius. 2004. Molecular mechanisms of interleukin-10-mediated inhibition of NF-kappaB activity: a role for p50. Clin. Exp. Immunol. 13564-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esufali, S., and B. Bapat. 2004. Cross-talk between Rac1 and dysregulated Wnt signaling pathway leads to cellular redistribution of β-catenin and TCF/LEF-mediated transcriptional activation. Oncogene 238260-8271. [DOI] [PubMed] [Google Scholar]
- 19.Esufali, S., G. S. Charames, V. V. Pethe, P. Buongiorno, and B. Bapat. 2007. Activation of tumor-specific splice variant Rac1b by Dishevelled promotes canonical Wnt signaling and decreased adhesion of colorectal cancer cells. Cancer Res. 672469-2479. [DOI] [PubMed] [Google Scholar]
- 20.Etienne-Manneville, S., and A. Hall. 2002. Rho GTPases in cell biology. Nature 420629-635. [DOI] [PubMed] [Google Scholar]
- 21.Fiegen, D., L. C. Haeusler, L. Blumenstein, U. Herbrand, R. Dvorsky, I. R. Vetter, and M. R. Ahmadian. 2004. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J. Biol. Chem. 2794743-4749. [DOI] [PubMed] [Google Scholar]
- 22.Fujita, T., G. P. Nolan, H. C. Liou, M. L. Scott, and D. Baltimore. 1993. The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through NF-kappa B p50 homodimers. Genes Dev. 71354-1363. [DOI] [PubMed] [Google Scholar]
- 23.Fukata, M., and K. Kaibuchi. 2001. Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat. Rev. Mol. Cell Biol. 2887-897. [DOI] [PubMed] [Google Scholar]
- 24.Gao, Y., J. B. Dickerson, F. Guo, J. Zheng, and Y. Zheng. 2004. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 1017618-7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gotley, D. C., J. Fawcett, M. D. Walsh, J. A. Reeder, D. L. Simmons, and T. M. Antalis. 1996. Alternatively spliced variants of the cell adhesion molecule CD44 and tumour progression in colorectal cancer. Br. J. Cancer 74342-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guan, H., S. Hou, and R. P. Ricciardi. 2005. DNA binding of repressor nuclear factor-kappaB p50/p50 depends on phosphorylation of Ser337 by the protein kinase A catalytic subunit. J. Biol. Chem. 2809957-9962. [DOI] [PubMed] [Google Scholar]
- 27.Hall, A. 1998. Rho GTPases and the actin cytoskeleton. Science 279509-513. [DOI] [PubMed] [Google Scholar]
- 28.Hall, A. 2005. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 33891-895. [DOI] [PubMed] [Google Scholar]
- 29.Herrlich, P., S. Pals, and H. Ponta. 1995. CD44 in colon cancer. Eur. J. Cancer 311110-1112. [DOI] [PubMed] [Google Scholar]
- 30.Huang, Y. C., W. C. Hung, W. Y. Kang, W. T. Chen, and C. Y. Chai. 2007. Expression of STAT3 and Bcl-6 oncoprotein in sodium arsenite-treated SV-40 immortalized human uroepithelial cells. Toxicol. Lett. 17357-65. [DOI] [PubMed] [Google Scholar]
- 31.Huynh, K. D., and V. J. Bardwell. 1998. The BCL-6 POZ domain and other POZ domains interact with the co-repressors N-CoR and SMRT. Oncogene 172473-2484. [DOI] [PubMed] [Google Scholar]
- 32.Huynh, K. D., W. Fischle, E. Verdin, and V. J. Bardwell. 2000. BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 141810-1823. [PMC free article] [PubMed] [Google Scholar]
- 33.Jordan, P., R. Brazão, M. G. Boavida, C. Gespach, and E. Chastre. 1999. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 186835-6839. [DOI] [PubMed] [Google Scholar]
- 34.Kanazawa, N., M. Moriyama, T. Onizuka, K. Sugawara, and S. Mori. 1997. Expression of bcl-6 protein in normal skin and epidermal neoplasms. Pathol. Int. 47600-607. [DOI] [PubMed] [Google Scholar]
- 35.Karin, M., Y. Cao, F. R. Greten, and Z. W. Li. 2002. NF-κB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer 2301-310. [DOI] [PubMed] [Google Scholar]
- 36.Kawashima, T., Y. C. Bao, Y. Nomura, Y. Moon, Y. Tonozuka, Y. Minoshima, T. Hatori, A. Tsuchiya, M. Kiyono, T. Nosaka, H. Nakajima, D. A. Williams, and T. Kitamura. 2006. Rac1 and a GTPase-activating protein, MgcRacGAP, are required for nuclear translocation of STAT transcription factors. J. Cell Biol. 175937-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kazansky, A. V., E. B. Kabotyanski, S. L. Wyszomierski, M. A. Mancini, and J. M. Rosen. 1999. Differential effects of prolactin and src/abl kinases on the nuclear translocation of STAT5B and STAT5A. J. Biol. Chem. 27422484-22492. [DOI] [PubMed] [Google Scholar]
- 38.Kumar, R., A. E. Gururaj, and C. J. Barnes. 2006. p21-activated kinases in cancer. Nat. Rev. Cancer 6459-471. [DOI] [PubMed] [Google Scholar]
- 39.Lernbecher, T., U. Müller, and T. Wirth. 1993. Distinct NF-kappa B/Rel transcription factors are responsible for tissue-specific and inducible gene activation. Nature 365767-770. [DOI] [PubMed] [Google Scholar]
- 40.Li, Z., X. Wang, R. Y. Yu, B. B. Ding, J. J. Yu, X. M. Dai, A. Naganuma, E. R. Stanley, and B. H. Ye. 2005. BCL-6 negatively regulates expression of the NF-kappaB1 p105/p50 subunit. J. Immunol. 174205-214. [DOI] [PubMed] [Google Scholar]
- 41.Lin, Z., H. Kim, H. Park, Y. Kim, J. Cheon, and I. Kim. 2003. The expression of bcl-2 and bcl-6 protein in normal and malignant transitional epithelium. Urol. Res. 31272-275. [DOI] [PubMed] [Google Scholar]
- 42.Logarajah, S., P. Hunter, M. Kraman, D. Steele, S. Lakhani, L. Bobrow, A. Venkitaraman, and S. Wagner. 2003. BCL-6 is expressed in breast cancer and prevents mammary epithelial differentiation. Oncogene 225572-5578. [DOI] [PubMed] [Google Scholar]
- 43.Matos, P., J. Collard, and P. Jordan. 2003. Tumour-related alternative-spliced Rac1b is not regulated by Rho-GDI and exhibits selective downstream signalling. J. Biol. Chem. 27850442-50448. [DOI] [PubMed] [Google Scholar]
- 44.Matos, P., and P. Jordan. 2006. RAC1, but not RAC1B, stimulates RELB-mediated gene transcription in colorectal cancer cells. J. Biol. Chem. 28113724-13732. [DOI] [PubMed] [Google Scholar]
- 45.Moorthy, A. K., O. V. Savinova, J. Q. Ho, V. Y. Wang, D. Vu, and G. Ghosh. 2006. The 20S proteasome processes NFkappaB1 p105 into p50 in a translation-independent manner. EMBO J. 251945-1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niu, H., B. H. Ye, and R. Dalla-Favera. 1998. Antigen receptor signaling induces MAP kinase mediated phosphorylation and degradation of the BCL-6 transcription factor. Genes Dev. 121953-1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niu, H. 2002. The proto-oncogene BCL-6 in normal and malignant B cell development. Hematol. Oncol. 20155-166. [DOI] [PubMed] [Google Scholar]
- 48.Otaki, J. M., D. T. Fearon, and H. Yamamoto. 2005. The proto-oncogene BCL-6 is expressed in olfactory sensory neurons. Neurosci. Res. 53189-200. [DOI] [PubMed] [Google Scholar]
- 49.Park, E. J., K. A. Ji, S. B. Jeon, W. H. Choi, I. O. Han, H. J. You, J. H. Kim, I. Jou, and E. H. Joe. 2004. Rac1 contributes to maximal activation of STAT1 and STAT3 in IFN-gamma-stimulated rat astrocytes. J. Immunol. 1735697-5703. [DOI] [PubMed] [Google Scholar]
- 50.Phan, R. T., M. Saito, Y. Kitagawa, A. R. Means, and R. Dalla-Favera. 2007. Genotoxic stress regulates expression of the proto-oncogene Bcl6 in germinal center B cells. Nat. Immunol. 81132-1139. [DOI] [PubMed] [Google Scholar]
- 51.Ponta, H., L. Sherman, and P. A. Herrlich. 2003. CD44: from adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 433-45. [DOI] [PubMed] [Google Scholar]
- 52.Rennefahrt, U. E., B. Illert, E. Kerkhoff, J. Troppmair, and U. R. Rapp. 2002. Constitutive JNK activation in NIH 3T3 fibroblasts induces a partially transformed phenotype. J. Biol. Chem. 27729510-29518. [DOI] [PubMed] [Google Scholar]
- 53.Ridley, A. J. 2001. Rho family proteins: coordinating cell responses. Trends Cell Biol. 11471-477. [DOI] [PubMed] [Google Scholar]
- 54.Ropponen, K. M., M. J. Eskelinen, P. K. Lipponen, E. Alhava, and V. M. Kosma. 1998. Expression of CD44 and variant proteins in human colorectal cancer and its relevance for prognosis. Scand. J. Gastroenterol. 33301-309. [DOI] [PubMed] [Google Scholar]
- 55.Reference deleted.
- 56.Sakashita, C., T. Fukuda, S. Okabe, H. Kobayashi, S. Hirosawa, T. Tokuhisa, N. Miyasaka, O. Miura, and T. Miki. 2002. Cloning and characterization of the human BAZF gene, a homologue of the BCL6 oncogene. Biochem. Biophys. Res. Commun. 291567-573. [DOI] [PubMed] [Google Scholar]
- 57.Schuringa, J. J., L. V. Dekker, E. Vellenga, and W. Kruijer. 2001. Sequential activation of Rac-1, SEK-1/MKK-4, and protein kinase C delta is required for interleukin-6-induced STAT3 Ser-727 phosphorylation and transactivation. J. Biol. Chem. 27627709-27715. [DOI] [PubMed] [Google Scholar]
- 58.Sells, M. A., U. G. Knaus, S. Bagrodia, D. M. Ambrose, G. M. Bokoch, and J. Chernoff. 1997. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 7202-210. [DOI] [PubMed] [Google Scholar]
- 59.Seyfert, V. L., D. Allman, Y. He, and L. M. Staudt. 1996. Transcriptional repression by the proto-oncogene BCL-6. Oncogene 122331-2342. [PubMed] [Google Scholar]
- 60.Shaffer, A. L., X. Yu, Y. He, J. Boldrick, E. P. Chan, and L. M. Staudt. 2000. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity 13199-212. [DOI] [PubMed] [Google Scholar]
- 61.Simon, A. R., H. G. Vikis, S. Stewart, B. L. Fanburg, B. H. Cochran, and K. Guan. 2000. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science 290144-147. [DOI] [PubMed] [Google Scholar]
- 62.Singh, A., A. E. Karnoub, T. R. Palmby, E. Lengyel, J. Sondek, and J. C. Der. 2004. Rac1b, a tumour associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 239369-9380. [DOI] [PubMed] [Google Scholar]
- 63.Solan, N. J., H. Miyoshi, E. M. Carmona, G. D. Bren, and C. V. Paya. 2002. RelB cellular regulation and transcriptional activity are regulated by p100. J. Biol. Chem. 2771405-1418. [DOI] [PubMed] [Google Scholar]
- 64.Staudt, L. M., A. L. Dent, A. L. Shaffer, and X. Yu. 1999. Regulation of lymphocyte cell fate decisions and lymphomagenesis by BCL-6. Int. Rev. Immunol. 18381-403. [DOI] [PubMed] [Google Scholar]
- 65.Sulciner, D. J., K. Irani, Z. X. Yu, V. J. Ferrans, P. Goldschmidt-Clermont, and T. Finkel. 1996. Rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-κB activation. Mol. Cell. Biol. 167115-7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sundaresan, M., Z. X. Yu, V. J. Ferrans, D. J. Sulciner, J. S. Gutkind, K. Irani, P. J. Goldschmidt-Clermont, and T. Finkel. 1996. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem. J. 318379-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tonozuka, Y., Y. Minoshima, Y. C. Bao, Y. Moon, Y. Tsubono, T. Hatori, H. Nakajima, T. Nosaka, T. Kawashima, and T. A. Kitamura. 2004. GTPase-activating protein binds STAT3 and is required for IL-6-induced STAT3 activation and for differentiation of a leukemic cell line. Blood 1043550-3557. [DOI] [PubMed] [Google Scholar]
- 68.Wessells, J., M. Baer, H. A. Young, E. Claudio, K. Brown, U. Siebenlist, and P. F. Johnson. 2004. BCL-3 and NF-kappaB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J. Biol. Chem. 27949995-50003. [DOI] [PubMed] [Google Scholar]
- 69.Wherlock, M., and M. Mellor. 2002. The Rho GTPase family: a Racs to Wrchs story. J. Cell Sci. 115239-240. [DOI] [PubMed] [Google Scholar]
- 70.Wielenga, V. J., K. H. Heider, G. J. Offerhaus, G. R. Adolf, F. M. van den Berg, H. Ponta, P. Herrlich, and S. T. Pals. 1993. Expression of CD44 variant proteins in human colorectal cancer is related to tumor progression. Cancer Res. 534754-4756. [PubMed] [Google Scholar]
- 71.Wielenga, V. J., R. Smits, V. Korinek, L. Smit, M. Kielman, R. Fodde, H. Clevers, and S. T. Pals. 1999. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am. J. Pathol. 154515-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu, W. S. 2006. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 25695-705. [DOI] [PubMed] [Google Scholar]
- 73.Zhong, H., M. J. May, E. Jimi, and S. Ghosh. 2002. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 9625-636. [DOI] [PubMed] [Google Scholar]