

Abstract
The yeast Candida glabrata is an opportunistic pathogen of humans. C. glabrata is a NAD+ auxotroph, and its growth depends on the availability of niacin (environmental vitamin precursors of NAD+). We have previously shown that a virulence-associated adhesin, encoded by EPA6, is transcriptionally induced in response to niacin limitation. Here we used transcript profiling to characterize the transcriptional response to niacin limitation and the roles of the sirtuins Hst1, Hst2, and Sir2 in mediating this response. The majority of genes transcriptionally induced by niacin limitation are regulated by Hst1, suggesting that it is the primary sensor of niacin limitation in C. glabrata. We show that three highly induced genes, TNA1, TNR1, and TNR2, encode transporters which are necessary and sufficient for high-affinity uptake of NAD+ precursors. Strikingly, if a tna1 tnr1 tnr2 mutant is starved for niacin, it exhibits an extended lag phase, suggesting a central role for the transporters in restoring NAD+ homeostasis after niacin limitation. Lastly, we had previously shown that the adhesin encoded by EPA6 is induced during experimental urinary tract infection (UTI); we show here that EPA6 transcriptional induction during UTI is strongly enhanced in the tna1 tnr1 tnr2 mutant strain, implicating the transporters in the growth of C. glabrata during infection.
Candida glabrata is an opportunistic yeast pathogen of humans that causes both mucosal surface and disseminated invasive infections. In the United States, it is a major cause of hospital-acquired fungal bloodstream infections, second only to Candida albicans. Candida species also account for about 25% of all urinary tract infections (UTIs) related to indwelling catheters, with C. glabrata accounting for approximately 15% of all Candida isolates.
Adherence to host cells is likely an important trait for C. glabrata in establishing infection. In this yeast, adherence to epithelial cells in vitro is mediated by a cell wall-associated adhesin encoded by the EPA1 gene (9). EPA1 is part of a large gene family with at least 23 members, most of which are located at subtelomeric loci, where they are subject to SIR-dependent transcriptional silencing (6, 10). In Saccharomyces cerevisiae, gene silencing at the telomere is initiated by recruitment of the Sir complex (Sir2, Sir3, and Sir4) to the telomeric repeats, followed by spreading into the adjacent subtelomeric region. The spread of silencing is dependent on the function of the sirtuin Sir2, a NAD+-dependent histone deacetylase which deacetylates the N termini of histones H3 and H4 to provide high-affinity binding sites for Sir3 and Sir4. The binding of Sir3 and Sir4, which is coextensive with the region of silencing, is thought to facilitate the remodeling of chromatin structure to a repressive state (22). The molecular mechanism for telomeric gene silencing described above is likely conserved in C. glabrata. Disruption of the silencing machinery by deletion of the C. glabrata SIR2, SIR3, or SIR4 gene results in the two normally silent subtelomeric EPA genes (EPA6 and EPA7) being transcribed, and this contributes strongly to the overall adherence of the mutant strain (10, 11, 13).
Are there environmental cues that induce the expression of usually silent EPA genes? We found previously that EPA6 is transcribed during infection in a murine UTI model in which C. glabrata is delivered through a catheter into the bladder. We further demonstrated that EPA1, EPA6, and EPA7 transcription can be induced by growth of C. glabrata in synthetic urine or human urine samples (11). In these experiments, the important environmental signal was limitation for vitamin precursors of NAD+. In S. cerevisiae, NAD+ is synthesized by two routes; one is the de novo kynurenine pathway that originates with tryptophan (21), and the other is salvaging pathways from vitamin precursors, including nicotinic acid (NA), nicotinamide (NAM), and NAM riboside (NR) (reference 5 and references therein). The commonly used term niacin refers to a mixture of NA and NAM and will be used in this paper simply to refer nonspecifically to all available NAD+ precursors. Like S. cerevisiae, C. glabrata can salvage NAD+ from NA, NAM, and NR (19). Unlike S. cerevisiae, however, C. glabrata does not possess genes for the de novo pathway and therefore requires supplementation with niacin (11). Consequently, the environmental niacin supply affects the intracellular NAD+ concentration in C. glabrata. In our working model, because Sir2 requires NAD+ as a cosubstrate for the deacetylation reaction that it catalyzes, a low intracellular NAD+ concentration resulting from limitation of environmental niacin causes the derepression of EPA genes in C. glabrata. Under conditions of NAD+ limitation, we believe that Sir2 activity is abrogated and transcription of the silent EPA genes is derepressed (11).
As in S. cerevisiae, C. glabrata has, in addition to SIR2, four SIR2 homologues, HST1 to HST4. In S. cerevisiae, these sirtuins have various roles: Sir2 mediates gene silencing at the silent mating loci, rDNA genes, and telomeric regions (22); Hst1 and Hst2 mediate gene-specific silencing (12, 25); and Hst3 and Hst4 catalyze the deacetylation of Lys56 of histone H3, which is involved in DNA replication and cell cycle control (7, 20). The sirtuins all use NAD+ as a cosubstrate for the deacetylation reactions they catalyze, with each deacetylation being accompanied by the consumption of one NAD+ molecule. Thus, in C. glabrata, low environmental niacin levels could, in principle, abrogate the activity of any or all of the five sirtuins.
Since niacin limitation acts as a signal for regulating the transcription of virulence-associated adhesin genes in C. glabrata, we have used transcriptional profiling to carry out a genome-wide analysis of genes whose transcription is altered under conditions of niacin limitation. We further characterized the functions of three genes, TNA1, TNR1, and TNR2, which are among the most highly induced by niacin limitation. We show that these three genes encode high-affinity transporters of NAD+ precursors and that their functions are required for establishing and maintaining NAD+ homeostasis in C. glabrata, as well as for allowing this yeast to grow in human urine. Since Sir2, Hst1, and Hst2 are all involved in transcriptional control, we have carried out transcriptional profiling to identify the C. glabrata genes whose expression is regulated by Sir2, Sir4, Hst1, and Hst2. We show that the expression of TNA1, TNR1, and TNR2 is primarily regulated by Hst1. Furthermore, our data suggest that Hst1, not Sir2, is the primary sirtuin that mediates the transcriptional response of C. glabrata to NAD+ limitation.
MATERIALS AND METHODS
Strains.
Escherichia coli strain DH10 was used for plasmid transformation and preparation. C. glabrata strain BG2 was used as the wild type. All C. glabrata deletion strains were derived from a ura3− derivative of BG2, BG14 (8). The relevant genotypes of the yeast strains used in this study are presented in Table 1.
TABLE 1.
C. glabrata strains used in this study
| Strain | Parent | Relevant genotype | Source |
|---|---|---|---|
| BG2 | Clinical isolate | ||
| BG14 | BG2 | ura3Δ::Tn903 G418r | |
| BG1326 | BG14 | ura3 tnr1Δ::hph Hygr (made with pBM11) | This study |
| BG1327 | BG14 | ura3 tnr2Δ::hph Hygr (made with pBM12) | This study |
| BG1333 | BG1327 | ura3 tnr1Δ::hph Hygrtnr2Δ::hph Hygr (made with pBM11) | This study |
| BG1335 | BG1327 | URA3 tnr2Δ::hph Hygr (made with pBC34.1) | This study |
| BG1337 | BG1333 | URA3 tnr1Δ::hph Hygrtnr2Δ::hph Hygr (made with pBC34.1) | This study |
| BG1338 | BG1326 | URA3 tnr1Δ::hph Hygr (made with pBC34.1) | This study |
| BG1348 | BG1333 | ura3 tnr1Δ tnr2Δ (made with pRD16) | This study |
| BG1349 | BG1348 | ura3 tnr1Δ tnr2Δ CAGL0G08448gΔ::hph Hygr (made with pBM13) | This study |
| BG1366 | BG1349 | ura3 tnr1Δ tnr2Δ tna1Δ CAGL0G08448gΔ::hph Hygr (made with pRD35) | This study |
| BG1386 | BG1366 | URA3 tnr1Δ tnr2Δ tna1Δ CAGL0G08448gΔ::hph Hygr (made with pBC34.1) | This study |
| BG1391 | BG1366 | ura3 tnr1Δ tnr2Δ tna1Δ CAGL0G08448gΔ::hph Hygr CAGL0F00209gΔ (made with pRK5) | This study |
| BG1404 | BG1391 | URA3 tnr1Δ tnr2Δ tna1Δ CAGL0G08448gΔ::hph Hygr CAGL0F00209gΔ (made with pBC34.1) | This study |
| BG1438 | BG14 | ura3 tna1Δ (made with pRD35) | This study |
| BG1441 | BG1438 | URA3 tna1Δ (made with pBC34.1) | This study |
| BG1442 | BG1333 | ura3Δ::Tn903 G418rtnr1Δ::hph Hygrtnr2Δ::hph Hygrtna1Δ (made with pRD35) | This study |
| BG1447 | BG1442 | URA3 tnr1Δ::hph Hygrtnr2Δ::hph Hygrtna1Δ (made with pBC34.1) | This study |
| BG1554 | BG1442 | ura3Δ::Tn903 G418rtnr1Δ::hph Hygrtnr2Δ::hph Hygrtna1Δ/[URA3PPGK1-CgTNA1(pBM28)] | This study |
| BG1555 | BG1442 | ura3Δ::Tn903 G418rtnr1Δ::hph Hygr/[URA3PPGK1-TNR1(pBM29)] tnr2Δ::hph Hygrtna1Δ | This study |
| BG1556 | BG1442 | ura3Δ::Tn903 G418rtnr1Δ::hph Hygrtnr2Δ::hph Hygr/[URA3PPGK1-TNR2(pBM30)] tna1Δ | This study |
| BG1557 | BG1442 | ura3Δ::Tn903 G418rtnr1Δ::hph Hygrtnr2Δ::hph Hygrtna1Δ/[URA3PPGK1-ScTNA1(pBM31)] | This study |
| BG1558 | BG1442 | ura3Δ::Tn903 G418rtnr1Δ::hph Hygrtnr2Δ::hph Hygrtna1Δ/[URA3PPGK1-ScTHI7(pBM32)] | This study |
| BG1559 | BG1442 | ura3Δ::Tn903 G418rtnr1Δ::hph Hygrtnr2Δ::hph Hygrtna1Δ/[URA3PPGK1-YOR071c(pBM33)] | This study |
| BG1560 | BG1442 | ura3Δ::Tn903 G418rtnr1Δ::hph Hygrtnr2Δ::hph Hygrtna1Δ/[URA3PPGK1-YOR192c(pBM34)] | This study |
| BG1695 | BG14 | ura3Δ::Tn903 G418repa6Δ::FLP1 (made with pRD11) | This study |
| BG1696 | BG1695 | URA3 epa6Δ::FLP1 (made with pBC34.1) | This study |
| BG1697 | BG1696 | URA3 epa6Δ::FLP aqy1Δ::FRT-hph Hygr (made with pBM1) | This study |
| BG1698 | BG1442 | ura3Δ::Tn903 G418rtna1Δ tnr1Δ tnr2Δ epa6Δ::FLP1 (made with pRD11) | This study |
| BG1699 | BG1698 | URA3 tna1Δ tnr1Δ tnr2Δ epa6Δ::FLP1 (made with pBC34.1) | This study |
| BG1700 | BG1699 | URA3 tna1Δ tnr1Δ tnr2Δ epa6Δ::FLP1 aqy1Δ::FRT-hph Hygr (made with pBM1) | This study |
| BG1216 | BG14 | URA3 sir2Δ::hph Hygr (made with pAP596) | This study |
| BG1217 | BG14 | URA3 sir4Δ::hph Hygr (made with pAP598) | This study |
| BG1218 | BG14 | URA3 hst1Δ (made with pAP628) | This study |
| BG1219 | BG14 | URA3 hst2Δ::hph Hygr (made with pAP633) | This study |
| BG1445 | BG1218 | URA3 hst1Δ/[pClonNATr-HST1(pBM17)] | This study |
| BG1462 | BG2 | URA3 sum1Δ::hph Hygr (made with pRD61) | This study |
Medium and chemicals.
All of the bacteria used in this study were routinely grown in LB medium. For culturing C. glabrata, synthetic complete (SC) medium without niacin (SC-NA) was prepared by following the recipe from the Qbiogene catalog (Irvine, CA) where niacin was left out. Various NAD+ precursors were added to this medium as specified in Results and the figure legends. When needed, uracil was left out to make SC-Ura medium. NA (N-4126), [carboxy-14C]NA (N-2267; 47 mCi/mmol), NAM (N-5535), and NR (N-4035) were purchased from Sigma-Aldrich and dissolved in H2O. [Carbonyl-14C]NAM (MC-1427; 55 mCi/mmol) and 4-3H(N)-labeled β-NAM mononucleotide (NMN) (MT-516; 1.8 Ci/mmol) were purchased from Moravek (Brea, CA). 4-3H(N)-labeled NR was generated by incubating 0.2 mCi 4-3H(N)-labeled NMN with 1,000 U calf intestinal alkaline phosphatase (New England BioLabs) in 1 ml 100 mM NaCl-20 mM Tris (pH 7.9) at 37°C for 2 h. Subsequently, the reaction mixture was heat inactivated by incubation at 75°C for 10 min and the phosphatase was removed through an Ultrafree-CL filter (5-kDa cutoff; Millipore).
Gene deletion and restoration in C. glabrata.
Gene deletion in C. glabrata was achieved by a previously described two-step method (10, 11, 13). The sequence information for generating gene deletion constructs was downloaded from the Génolevures website (http://cbi.labri.fr/Genolevures/blast.php). For the primers used to make gene deletion constructs, see Table S1 in the supplemental material. For descriptions of the plasmids containing gene deletion constructs, see Table S2 in the supplemental material. In the deletion strains, the open reading frame (ORF) of each target gene was either replaced with a hygromycin resistance (Hygr) cassette or completely deleted. To restore a gene of interest on self-replicating plasmids in C. glabrata, the ORF of the gene was amplified by PCR (for the primers used, see Table S1 in the supplemental material). The PCR product was subsequently restriction digested and subcloned into pGRB2.2, a URA3 CEN/ARS plasmid, between the S. cerevisiae PGK1 promoter and the C. glabrata HIS3 3′ untranslated region. For the plasmids used for gene restoration, see Table S2 in the supplemental material.
Growth assay and NAD+ precursor uptake assay.
All yeast cells were grown at 30°C. The wild type and all deletion strains were routinely kept on yeast extract-peptone-dextrose (YPD) plates or SC plates containing 10 μM NA, while all strains that carry self-replicating plasmids were kept on SC-Ura plates containing 10 μM NA. For growth assays on plates, 3-μl samples of overnight cultures of various strains grown in liquid SC or SC-Ura medium containing 8 μM NA were spotted onto SC or SC-Ura plates with different NAD+ precursors as specified in Results. When needed, yeast cells were starved for NAD+ by transferring cells from overnight cultures in SC medium containing 8 μM NA to the same medium but without any NAD+ source until growth arrested. For the growth assay in human urine, fresh individual urine samples were supplemented with 2% dextrose and filter sterilized. Yeast cells from overnight cultures in SC medium containing 8 μM NA were collected, washed with phosphate-buffered saline (PBS), and inoculated into 15 ml sterile urine in test tubes to an initial optical density at 600 nm (OD600) of 0.1. The cultures were grown at 30°C on a rolling drum, and OD600 was measured at 8 h.
To measure the uptake of various NAD+ precursors, the tna1Δ tnr1Δ tnr2Δ mutant strain carrying pGRB2.2 alone (control) or carrying self-replicating plasmids that express TNA1, TNR1, or TNR2 were grown in SC-Ura medium containing 3 μM NA for 16 h. The cells were reinoculated into SC-Ura medium without any NAD+ precursor and grown for an additional 5 h from an initial OD600 of 0.1. Afterward, these cells were washed twice in cold PBS and resuspended in SC-Ura medium without any NAD+ precursor at an OD600 of 1.0. Various amounts of [14C]NA, [14C]NAM, or [3H]NR were added to 500-μl aliquots of cells in microcentrifuge tubes to final concentrations ranging from 0.01 to 100 μM. For each NAD+ precursor concentration, the cell suspensions were incubated at room temperature on a rolling drum for various lengths of time (from 15 s to 4 h) and the cells were immediately spun down at 4°C for 5 s at 15,000 rpm. Subsequently, the supernatant was removed by aspiration and the cell pellets were washed twice with cold PBS and resuspended in scintillant fluid for counting on a Beckman LS600SE scintillation counter. The uptake resulting from TNA1, TNR1, or TNR2 was calculated by subtracting the base level of counts associated with control cells from the counts of those cells expressing TNA1, TNR1, or TNR2 on plasmids. Uptake curves (uptake versus time) at each NAD+ precursor concentration were drawn, and uptake rates were determined from the linear range of the curves. The Km value was calculated from fitted curves of uptake rates versus NAD+ precursor concentrations by using Prism 4 software (GraphPad Software).
Transcriptional profiling by microarray.
The C. glabrata microarray comprises 5,908 69- to 70-mer oligonucleotides that correspond to nearly all of the putative C. glabrata ORFs and some noncoding sequences. The sequence information of the oligonucleotides and the microarray setup can be found at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL3922. The details of the yeast growth conditions, microarray hybridization method, and experiment setup for this study can be found at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE6582 and http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE6626. For each experimental condition, dye swap experiments were performed on two or three sets of independent biological samples. The hybridized microarrays were scanned with an Axon 4000B scanner, and data were extracted with Axon GenePix software. For each hybridized microarray, the log2(experimental/control) value of each spot on the microarray was calculated. Subsequently, for each dye swap experiment, the log2(experimental/control) values of each spot were averaged. Finally, four or six averaged log2(experimental/control) values corresponding to each oligonucleotide on the microarray (from two or three biological repeats × duplicate spots of each oligonucleotide on the microarray) were imported into SAM (significance analysis of microarrays) software (downloaded from http://www-stat.stanford.edu/∼tibs/SAM/) for statistical analysis. For genes that are considered significantly upregulated or downregulated, the median false-discovery rate equals 0 while the 90% false-discovery rate is less than 0.19.
S1 nuclease protection assay.
The S1 nuclease protection assay was carried out as described previously (6, 10). The sequences of the probes used are as follows: for ACT1, 5′-CTCAAAATAGCGTGTGGCAAAGAGAAACCGGCGTAAATTGGAACAACGTGGGTAACACCGTCACCAGAGTCCTTTTG-3′; for EPA6, 5′-TG TACGATAGTTATAAGCTTGTGGTAATGTATCAAACAGCGAAGTACAC CCCATTGGGCGTAC-3′; for TNA1, 5′-AGCCCGCCACTTCGGCGTTACCAATATTCGACTTATCCAAGTTAGACAAGAAGTACATCACTGGG-3′; for both TNR1 and TNR2, 5′-CAACTGTCAATGCTGCTGTAGCGCCCATCCATGTACCTACTGTAAAGGCAATTGTACCCCTCATC-3′.
Purification of recombinant Sir2 (rSir2) and Hst1 (rHst1) and histone deacetylation (HDAC) enzymatic assays.
Maltose-binding-protein-tagged rHst1 or rSir2 was expressed in E. coli by using pMALDEST, which is derived from pMAL-c2G (New England BioLabs) and adapted for use in the Gateway system (Invitrogen) (gift of C. Wolberger, Johns Hopkins University School of Medicine). DNA fragments which encode full-length C. glabrata Sir2 (ORF CAGL0K01463g) and Hst1 (ORF CAGL0C05357g) were amplified by PCR with genomic DNA as the template. Additional sequences including sequences which encode an N-terminal tobacco etch virus protease cleavage site and a C-terminal six-His tag, as well as attB recombination sites, were included in the PCR primers and incorporated in the final construct. Gateway recombination reactions were performed to insert the PCR products into pMALDEST according to the manufacturer's manual (Invitrogen). The resulting plasmid, pMALSir2 or pMALHst1, encodes the full-length protein fused to a tobacco etch virus-cleavable N-terminal maltose-binding protein tag and a C-terminal six-His tag. Either plasmid was transformed into E. coli BL21 cells for induction of recombinant protein expression by addition of isopropyl-β-d-thiogalactopyranoside (IPTG; 1 mM) and incubation for 14 h at 15°C. The proteins were purified by Ni-Sepharose and amylose chromatography, and protein concentrations were measured with a Bradford assay kit (Pierce). The HDAC assay was carried out with an HDAC kit (catalog no. 17-320; Millipore). Briefly, biotin-conjugated histone H4 peptide (amino acids 2 to 24) was acetylated with 3H-labeled acetyl coenzyme A (NET290; 1 Ci/mmol; Perkin-Elmer) by using recombinant histone acetyltransferase PCAF and following the kit's instruction. Subsequently, the labeled peptide was bound to streptavidin-agarose beads and free labels were removed by extensive washes with Tris-buffered saline. The [3H]acetate incorporation rate in the labeling reaction was 60 to 80%. HDAC activity of purified rHst1 or rSir2 was assessed by following the release of 3H label from the 3H-labeled acetyl histone H4 peptide. HDAC reactions were carried out in a final volume of 100 μl comprising 10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% glycerol, 1 mM dithiothreitol, 8 μg purified rHst1 or rSir2, 100,000 cpm agarose beads carrying [3H]acetyl histone H4 peptide, and various concentrations of NAD+ (N1511; Sigma), ranging from 0 to 3,200 μM. The reactions were stopped at various time points, and the supernatant was extracted with 400 μl ethyl acetate, from which 200 μl was transferred to a scintillation vial containing scintillation fluid, mixed thoroughly, and counted in a Beckman LS600SE scintillation counter. At each tested [NAD+], reaction curves of rHst1 or rSir2 HDAC activities versus time were drawn. We verified that for rHst1, the increase in HDAC activity was in the linear range within 15 min following the initiation of the reaction, while for rSir2, it was in the linear range within 4 h. The initial reaction rates were calculated from the fitted reaction curves, and the apparent Km values were calculated with Prism software.
Animal studies.
For disseminated-infection studies, yeast cells grown for 16 h in SC medium supplemented with 50 μM NA and 50 μM NR were collected, washed twice with cold PBS, and then resuspended in PBS at a concentration of approximately 2.5 × 108/ml. Groups of seven 6- to 8-week-old BALB/c mice (Taconic) were individually injected through the tail vein with 100 μl of a cell suspension (2.5 × 107 cells). For the UTI model, a similar culturing protocol was followed and 2.5 × 107 yeast cells in 50 μl of PBS supplemented with 10% glycerol were delivered transurethrally via catheter as described previously (11). Mice were maintained on a complete diet but without added niacin, which is a usual dietary supplement. For both models, mice were sacrificed via CO2 asphyxiation on day 7 postinfection. Kidneys, livers, spleens (for the disseminated-infection model), bladders, and kidneys (for the UTI model) were harvested and homogenized in 1 ml of PBS. Appropriate dilutions were plated on YPD plates supplemented with 100 U/ml penicillin and 0.1 mg/ml streptomycin. Colonies were counted after growth for 40 h at 30°C.
For the FLP1 reporter experiments, we used a strain in which the ORF of EPA6 was replaced with S. cerevisiae FLP1. The induction of FLP1 was monitored via recombination occurring between two FRT sites flanking a hygromycin resistance (HYGr) cassette that is integrated into the C. glabrata AQY1 (ORF CAGL0A01221g) locus. We have shown previously that the disruption of AQY1, which is one of two duplicated copies in the C. glabrata genome, behaves similarly to the wild-type C. glabrata BG2 strain both in vitro and in vivo (data not shown). The induction of FLP1 results in loss of the Hygr cassette and the yeast becomes hygromycin sensitive (Hygs). The FLP1 reporter strains were grown in SC medium supplemented with 50 μM NA and 50 μM NR for 15 h, and the subsequent UTI was performed as described above. The percentage of Hygs colonies in the yeast colonies recovered from infected kidneys and bladders was measured by replica plating onto YPD plates supplemented with 400 μg/ml hygromycin (Calbiochem).
Intracellular NAD+ and NADH concentrations.
The acid and alkaline extraction for NAD+ and NADH was modified from the method described by Lin et al. (17). Briefly, 1 ml of ice-cold 0.05 N NaOH-1 mM EDTA was added to 1 × 108 to 2 × 108 frozen C. glabrata cells. For acid extraction, 300 μl of 0.125 N HCl was added to 300 μl of the above alkali extract. The remaining alkali extract and the acid extract were frozen, and cells were broken up with a bead beater. Following incubation at 60°C for 30 min, 100 μl of 0.4 M Tris base was added to 400 μl of acid extract and 100 μl of 100 mM Tris-HCl (pH 8.1)-0.1 N HCl was added to 200 μl of alkali extract for neutralization. The mixtures were centrifuged, and the supernatants were stored at −80°C prior to use.
The enzyme cycling method for measuring intracellular NAD+/NADH was adapted from Shah et al. (24). The reaction mixture containing 0.17 M bicine (pH 7.8), 0.85 M ethanol, 7.1 mM EDTA, 1.4 mg/ml bovine serum albumin, 0.71 mM 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide, and 2.8 mM phenazine ethosulfate was made fresh each time. A 100-μl volume of this reaction mixture was added to 50-μl samples or 0 to 80 pmol standard NAD+ or NADH in a 96-well plate. The reaction was started by the addition of 20 μl of 1 U of alcohol dehydrogenase (0.168 mg/ml in 0.1 M bicine, pH 7.8). The color was developed in the dark at 30°C, and the absorbance was measured at 570 nm 15 and 30 min after the addition of the enzyme.
RESULTS
CAGL0F08371g, CAGL0L13354g, and CAGL0M14113g are highly induced in C. glabrata under niacin limitation.
To understand the transcriptional response of C. glabrata to niacin limitation, we compared the transcriptional profiles of log-phase wild-type BG2 cells grown in SC medium containing 0.016 μM NA versus 3.25 μM NA with whole-genome microarrays. The latter concentration is equivalent to the niacin level in commercially available SC medium for yeast. Under conditions of niacin limitation, 315 and 197 C. glabrata ORFs showed significant induction and repression (≥2.5-fold), respectively. Our previous study showed that growth of C. glabrata in human urine results in the induction of EPA6 in response to low niacin levels. We therefore also compared the transcriptional profiles of log-phase BG2 cells grown in three individual human urine samples versus in SC medium containing 3.25 μM NA. In this experiment, 87 and 125 C. glabrata ORFs were significantly induced or repressed (twofold or more), respectively. There are 60 ORFs that exhibit significant upregulation under the two different niacin-limiting conditions described above, while 16 ORFs show significant downregulation under both conditions. The overlap between the lists of significantly upregulated ORFs derived from two different niacin limitation treatments suggests that there is a specific gene induction profile in response to niacin limitation. The significant ORF lists are deposited at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE6582.
Three ORFs, CAGL0F08371g, CAGL0L13354g, and CAGL0M14113g, are among those exhibiting the highest induction by niacin limitation. This induction was confirmed by the S1 nuclease protection assay detecting steady-state transcript levels. As shown in Fig. 1A, elevated transcript levels of the three ORFs were observed in cells grown either in SC medium containing limiting levels of NA or in human urine. Since the coding sequences of CAGL0L13354g and CAGL0M14113g share 98% homology, the probe used in the S1 nuclease protection assay does not differentiate the two ORFs. However, when probing for the CAGL0L13354g transcript in a C. glabrata strain in which the entire ORF of CAGL0M14113g has been deleted, or vice versa, transcription profiles similar to those seen in the wild-type cells were observed, suggesting that both ORFs are induced under niacin limitation conditions (Fig. 1B).
FIG. 1.

TNA1, TNR1, and TNR2 are transcriptionally induced by niacin limitation. (A) S1 nuclease analysis of TNA1, TNR1/TNR2, and ACT1 transcript levels. RNA prepared from strain BG2 growing in SC medium containing 3.25 μM or 16 nM NA or in three different urine samples. The OD600 of all cultures was 0.5. (B) S1 nuclease analysis of TNA1 and TNR1/TNR2 transcript levels of the BG2, tnr1 mutant (BG1338), tnr2 mutant (BG1335), and tnr1 tnr2 mutant (BG1337) strains. Strains were grown in SC medium containing16 nM NA. The OD600 of all cultures was 0.5. (C) S1 nuclease analysis of TNA1 and TNR1/TNR2 transcript levels of strain BG2 growing in SC medium with limiting amounts of niacin (SC-NA, 16 nM), thiamine (SC-Thi, 6 nM), or pyridoxine (SC-Pyr, 0.19 nM). At these vitamin levels, the terminal OD600 of all of the cultures was 1.5.
CAGL0F08371g (TNA1), CAGL0L13354g (TNR1), and CAGL0M14113g (TNR2) encode high-affinity transporters of NAD+ precursors.
Sequence comparison shows that CAGL0F08371g is an orthologue of S. cerevisiae TNA1. The two genes share 71% amino acid identity. S. cerevisiae TNA1 encodes a high-affinity transporter of NA and belongs to the anion:cation symporter family. From the genomic sequence, CAGL0L13354g and CAGL0M14113g are located at the ends of chromosomes L and M, respectively, and are not syntenic with any S. cerevisiae genes. However, both ORFs share 60 to 62% amino acid identity with three S. cerevisiae genes, THI7, YOR071c, and YOR192c. S. cerevisiae THI7 encodes a high-affinity thiamine transporter and belongs to the nucleobase:cation symporter 1 family. We then measured the transcript levels of CAGL0F08371g, CAGL0L13354g, and CAGL0M14113g in C. glabrata wild-type cells grown in SC medium containing only limiting amounts of thiamine (6 nM) or pyridoxine (0.19 nM). As shown in Fig. 1C, CAGL0F08371g was strongly induced under niacin-limiting conditions and slightly under pyridoxine-limiting conditions. Significant induction of CAGL0L13354g and CAGL0M14113 was observed specifically under niacin limitation conditions but not under limitation of thiamine or pyridoxine. Furthermore, to test rigorously if CAGL0L13354g and CAGL0M14113g might function in thiamine or pyridoxine uptake, we generated strains with a deletion(s) of either or both ORFs. Since C. glabrata is an auxotroph for both thiamine and pyridoxine, slow growth can be observed if the supply of either vitamin in the medium is limiting (≤3 nM for thiamine and ≤0.09 nM for pyridoxine). However, no additional phenotype was observed for the deletion strains compared to wild-type cells (data not shown). In fact, a strain (BG1404) with all five related C. glabrata ORFs (CAGL0F08371g, CAGL0L13354g, CAGL0M14113g, and two additional orthologues of S. cerevisiae THI7, CAGL0G08448g and CAGL0F00209g) deleted showed no defect in growth under thiamine or pyridoxine limitation conditions in comparison to wild-type cells (data not shown). These results suggest that CAGL0L13354g and CAGL0M14113g may not play roles in thiamine or pyridoxine uptake.
Since all three genes, CAGL0F08371g, CAGL0L13354g, and CAGL0M14113g, are induced by niacin limitation and niacin is the precursor for the synthesis of NAD+, we reasoned that the three ORFs may encode transporters of NAD+ precursors. Previously, we showed that C. glabrata can use three compounds, NA, NAM, and NR, as NAD+ precursors; by contrast, C. glabrata is unable to access NAD+ or NMN (19). Thus, we examined the growth of mutant strains with deletion of each of the three ORFs (BG1338, BG1335, BG1441), both CAGL0L13354g and CAGL0M14113g (BG1337), or all three ORFs (BG1447) on various concentrations of NA, NAM, and NR. As shown in Fig. 2A, a strain with a deletion of CAGL0F08371g (BG1441) was unable to grow on plates supplemented with 0.5 μM NA but showed robust growth on plates supplemented with 8 μM NA. This is consistent with the observation that the S. cerevisiae orthologue of CAGL0F08371g, TNA1, functions as a high-affinity transporter of NA (18). Based on these data, we have tentatively assigned the gene name TNA1 to CAGL0F08371g. While strains with a deletion of either CAGL0L13354g (BG1338) or CAGL0M14113g (BG1335) showed no growth phenotype on plates with 0.5 μM of each of the three NAD+ precursors, the strain with deletions of both CAGL0L13354g and CAGL0M14113g (BG1337) showed defective growth on plates with 0.5 μM NR but not on 0.5 μM NA or 0.5 μM NAM. Supplementation of the medium with 32 μM NR permitted robust growth of the double-deletion strain. This suggests that both ORFs may function in the high-affinity uptake of NR;accordingly, we have assigned the gene names TNR1 (trans-porter of NR) and TNR2 to CAGL0L13354g and CAGL0M14113g, respectively. The strain with TNA1, TNR1, and TNR2 deleted showed no growth on plates with 0.5 μM NA, NAM, or NR, strongly suggesting that the TNA1, TNR1, and TNR2 genes encode the major high-affinity transporters of NAD+ precursors. Since the tna1Δ tnr1Δ tnr2Δ mutant strain can still grow robustly on 8 μM NA, 4 μM NAM, or 32 μM NR, the cell can apparently acquire these NAD+ precursors by a separate low-affinity uptake mechanism or by passive diffusion.
FIG. 2.

Growth of tna1, tnr1, and tnr2 mutant strains is compromised at low concentrations of NAD+ vitamin precursors. (A) Growth of six strains on medium with 0.5 μM NA, NAM, or NR as the sole source of NAD+. The relevant genotypes of the strains are wild type (WT; strain BG2), tna1 (BG1441), tnr1 (BG1338), tnr2 (BG1335), tnr1 tnr2 (BG1337), and tna1 tnr1 tnr2 (BG1447). The cells are able to grow equally well if supplemented with excess NAD+ precursors (8 μM NA, 4 μM NAM, and 32 μM NR; at concentrations of one-half of these levels, growth of the relevant transporter mutants is compromised relative to that of the wild type). (B) Complementation of the growth defect of tna1 tnr1 tnr2 mutants. Strain1442 was transformed with an empty expression vector or with vectors expressing TNA1, TNR1, or TNR2, and growth was assessed on SC plates containing 0.5 μM NA, NAM, or NR. (C) The tna1 tnr1 tnr2 mutant C. glabrata strain was transformed with plasmids expressing S. cerevisiae ORFs YGR260w (TNA1), YOR192c (THI72), YOR071c (NRT1), and YLR237w (THI7). These strains were plated on SC-URA-NA plates supplemented with 0.5 μM NA, NAM, or NR.
Our data demonstrate some specificity in the uptake activities of TNA1, TNR1, and TNR2 for various NAD+ precursors. In particular, uptake of NA may depend on TNA1, uptake of NAM may depend on all three genes, and uptake of NR may depend primarily on TNR1 and TNR2 and secondarily on TNA1. These specificities in function were further confirmed by restoring each of the three genes on self-replicating plasmids in the tna1Δ tnr1Δ tnr2Δ mutant strain. To do this, we cloned the ORF of each gene between a constitutive PGK1 promoter and a HIS3 3′ untranslated region in a C. glabrata CEN/ARS plasmid, pGRB2.2. As shown in Fig. 2B, ectopic expression of TNA1 in the tna1Δ tnr1Δ tnr2Δ mutant background restored growth on NA, on NAM, and to a much lesser extent on NR, while ectopic expression of either TNR1 or TNR2 restored growth on both NAM and NR but not at all on NA. To further characterize these specificities, we measured the uptake of radiolabeled NA, NAM, and NR by Tna1, Tnr1, or Tnr2 expressed in the tna1Δ tnr1Δ tnr2Δ mutant background as described above. As shown in Table 2, the apparent Km values of Tna1 for NA and NAM and of both Tnr1 and Tnr2 for NR and NAM are in the low micromolar range. The apparent Km value of Tna1 for NR was much higher than for NA and NAM, again suggesting that TNA1 encodes only weak NR uptake activity. No NA uptake activity encoded by TNR1 or TNR2 could be observed in these experiments.
TABLE 2.
Apparent Km values of Tna1, Tnr1, and Tnr2 for NA, NAM, and NRa
| Protein | Mean Km (μM) ± SEM
|
||
|---|---|---|---|
| NA | NAM | NR | |
| Tna1 | 0.59 ± 0.09 | 6.3 ± 1.7 | 110 ± 50 |
| Tnr1 | No uptake observed | 7.7 ± 1.2 | 1.6 ± 0.4 |
| Tnr2 | No uptake observed | 5.5 ± 1.1 | 1.5 ± 0.4 |
Uptake of radiolabeled precursors into a tna1 tnr1 tnr2 mutant strain (BG1442) expressing the gene for the indicated protein was measured.
We also heterologously expressed the ORFs of S. cerevisiae TNA1 and the S. cerevisiae homologues of the TNR genes THI7 (YLR237W), NRT1 (YOR071c), and THI72 (YOR192c) in the C. glabrata tna1Δ tnr1Δ tnr2Δ mutant strain. Similar to C. glabrata TNA1, S. cerevisiae TNA1 (BG1557) could restore growth on NA, on NAM, and to a lesser extent on NR (Fig. 2C), demonstrating functional conservation between the two species. Among the S. cerevisiae homologues of TNR1 and TNR2, YOR071c (strain BG1559) restored growth on NR and NAM but not NA. YLR237w and YOR192c (BG1558 and BG1560) did not complement for growth on plates with NA, NAM, or NR. Our data are consistent with previous data showing that YOR071c encodes a high-affinity NR transporter in S. cerevisiae (4).
TNA1, TNR1, and TNR2 are required for establishing NAD+ homeostasis in C. glabrata.
C. glabrata lacks the ability to synthesize NAD+ de novo, and its growth requires an environmental supply of niacin. When C. glabrata cells are grown in the absence of NAD+ precursors, they cease growing but show no loss of viability over 72 h (data not shown). Providing these NAD+-starved cells with NA, NAM, and NR restores growth.
Since the tna1Δ tnr1Δ tnr2Δ mutant strain (BG1447) lacks the high-affinity uptake systems for NAD+ precursors, higher concentrations of NAD+ precursors (8 μM NA, 4 μM NAM, or 32 μM NR) are required to support its robust growth than are required for the wild-type strain (Fig. 2A). We noticed, however, that if tna1Δ tnr1Δ tnr2Δ mutant cells were starved for niacin before plating, the appearance of colonies on plates, even at the high concentrations of NAD+ precursors, was delayed by approximately 24 h relative to that of starved wild-type cells (BG2). This suggested that niacin-starved tna1Δ tnr1Δ tnr2Δ mutant cells display a lag in recovering from niacin starvation. As shown in Fig. 3A, we compared the growth of niacin-starved cells (grown in SC-NA medium for 48 h) to that of stationary-phase cells (grown in SC medium for 48 h) once they were inoculated into fresh SC medium with 8 μM NA. While the doubling time (1.3 h) of the wild-type and tna1Δ tnr1Δ tnr2Δ mutant strains was the same regardless of whether they were grown under niacin starvation or grown in stationary phase, there is a substantial lag phase of 9 h specifically for niacin-starved tna1Δ tnr1Δ tnr2Δ mutant cells. During this period, these cells showed no growth. Moreover, in SC medium containing 3 μM NA, while the doubling time of the niacin-starved wild-type strain is 1.3 h and that of the starved tna1Δ tnr1Δ tnr2Δ mutant strain is 1.8 h, the lag phase of the latter is 13 h (data not shown). Wild-type and tna1Δ tnr1Δ tnr2Δ mutant cells starved for 48 h for other nutrients such as glucose or nitrogen both exhibit only a short lag phase (1 h) when transferred to fresh medium (SC medium plus 8 μM NA) (data not shown). This suggests that the phenotype of a long lag period is specific for niacin starvation. One interpretation of these data is that in the absence of the high-affinity uptake systems, even when the environmental supply of niacin is sufficient to support growth, C. glabrata cells are highly compromised in the ability to acquire NAD+ precursors for restoring growth, resulting in a prolonged lag phase. From these data, we conclude that TNA1, TNR1, and TNR2 are required not only for maintaining NAD+ homeostasis in C. glabrata during growth with a limited niacin supply but also for the rapid reestablishment of NAD+ homeostasis after niacin starvation.
FIG. 3.

Growth of the tna1 tnr1 tnr2 mutant strain is compromised after starvation for NAD+ precursors. (A) Wild-type (WT; BG2) and tna1 tnr1 tnr2 mutant (BG1447) strains in SC liquid medium containing 8 μM NA. The strains were inoculated from overnight cultures grown in SC or SC-NA medium for 48 h. Note that there is no difference in growth rate once the cells are growing; however, the lag phase for starved cells is approximately 2 h for BG2 and 9 h for BG1447. (B) Growth of transporter mutant strains BG1441 (tna1Δ), BG1337 (tnr1Δ tnr2Δ), and BG1447 (tna1Δ tnr1Δ tnr2Δ) in human urine cultures. Cultures were inoculated at an OD600 of 0.1 and grown overnight at 30°C.
Because we have previously shown that EPA6 is transcriptionally induced in response to niacin limitation during UTI, we also tested the requirement of TNA1, TNR1, and TNR2 for the growth of C. glabrata in human urine. Wild-type or mutant cells were grown at 30°C in four individual urine samples. As shown in Fig. 3B, for samples 2 to 4, significant growth was observed for wild-type cells and for tna1Δ or tnr1Δ tnr2Δ mutant strains while no growth was observed in tna1Δ tnr1Δ tnr2Δ mutant cells under the same conditions. Addition of excess (30 μM) niacin (a mixture of NA and NR) to these cultures permitted growth saturation for all strains (OD600, >20), showing that in all samples the limiting nutrient was niacin (data not shown). Moreover, ectopic expression of TNA1 or TNR1 in the tna1Δ tnr1Δ tnr2Δ mutant background restored growth in samples 2 to 4 (data not shown). In the fourth urine sample (sample 1), all four strains could grow, showing that the transporters were not required for growth in that sample, suggesting that niacin levels in this sample might be higher. We used a bioassay (10) to determine levels of NAD+ precursors in the four samples. Samples 2 to 4 contained 90 ± 16, 74 ± 3, and 168 ± 21 nM available niacin, respectively; by contrast, sample 1 contained 30 ± 7 μM available niacin (data not shown). The donor of sample 1, it was determined, was taking high-dose niacin supplements, which likely explains the dramatically higher excreted niacin levels. There is a good correlation between the requirement for the transporters for growth in urine and the levels of available excreted niacin. These data and our transcriptional profiling showing the induction of TNA1, TNR1, and TNR2 when grown in human urine suggest that these three genes are generally required for this yeast to grow in human urine.
To test whether the NAD+ precursor transporters were required during disseminated infection, we infected groups of 10 mice with the wild-type (BG2) or tna1Δ tnr1Δ tnr2Δ (BG1447) strain via tail vein injection. After 7 days, we assessed fungal burdens in three target organs—the liver, kidney, and spleen. However, we found no difference in the colonization of these organs by the wild-type and mutant strains (data not shown).
We tested if these three genes play any role during UTI. Previously, we showed that the expression of EPA6 was significantly induced during UTI and hypothesized that this was due in part to the limitation of niacin. One prediction from this model is that the induction of EPA6 should be exacerbated in mutants lacking the functions of TNA1, TNR1, and TNR2, since these cells are strongly compromised in the ability to access limiting levels of niacin. To test this hypothesis in the wild-type and tna1Δ tnr1Δ tnr2Δ mutant backgrounds, we replaced the entire EPA6 ORF with the ORF of S. cerevisiae FLP1, which encodes Flp1, a site-specific recombinase (reviewed in reference 14). In addition, we inserted a HYGr cassette flanked by two FRT sites (the recognition sites for Flp1) into the AQY1 locus in both wild-type and mutant EPA6::FLP1 reporter strains. C. glabrata AQY1 has a duplicate copy (ORF CAGL0D00154g) in the genome. The aqy1Δ mutant strains behave similarly to the wild-type C. glabrata BG2 strain both in vitro and in vivo (data not shown). In the EPA6::FLP1 reporter strains, the induction of EPA6 can be monitored by following the Flp1-mediated recombination at the FRT sites, which results in loss of the HYGr cassette and renders the cells Hygs, in contrast to the Hygr parental strains. For both the wild-type and tna1Δ tnr1Δ tnr2Δ EPA6::FLP1 reporter strains, after overnight growth in SC medium containing excess levels of NA (25 μM), approximately 0.5 to 1% of the cells were Hygs (data not shown). However, after overnight growth in SC medium containing 3 μM NA, 91% of the cells from the tna1Δ tnr1Δ tnr2Δ mutant reporter strain became Hygs, whereas only 0.5% of the wild-type reporter strain cells became Hygs. Thus, the induction of EPA6 in vitro, as measured by the FLP1 reporter, is much greater at modest environmental NA concentrations in the tna1Δ tnr1Δ tnr2Δ mutant background than in the wild-type background, a finding that conforms to our hypothesis. We next tested the wild-type and tna1Δ tnr1Δ tnr2Δ EPA6::FLP1 mutant reporter strains in UTI. Prior to infection, both strains were grown in SC medium containing excess levels of niacin (50 μM NA plus 50 μM NR). Among the 2.5 × 107 yeast cells that were delivered to mice transurethrally via catheters, less than 0.5% were Hygs. On day 7 postinfection, yeast cells were cultured from the bladder and kidneys. In the infected kidneys, an average of 58% of the yeast cells from the tna1Δ tnr1Δ tnr2Δ EPA6::FLP1 mutant reporter strain were Hygs, compared to an average of 14% Hygs cells from the wild-type reporter strain (P = 0.0007) (Fig. 4). Our data suggest that, in the context of murine UTI, the TNA1-, TNR1-, and TNR2-encoded transporters of NAD+ precursors play important roles in maintaining cellular levels of NAD+ sufficient to repress the EPA6 locus. In addition, these data provide evidence supporting our model that levels of environmental NAD+ precursors are important in determining the expression levels of EPA6, and possibly of other genes during UTI. Furthermore, from the above UTI experiments, when comparing the number of mice that were colonized by yeast cells in their bladders and kidneys, only 6 out of 10 mice infected with the tna1Δ tnr1Δ tnr2Δ EPA6::FLP1 reporter strain were colonized, whereas all 10 mice infected with the wild-type reporter strain were colonized. However, these data did not reach significance (P = 0.13). In addition, for those mice that were colonized, the numbers of CFU recovered from the kidneys did not significantly differ between the wild-type reporter strain (2.7 × 104) and the tna1Δ tnr1Δ tnr2Δ mutant reporter strain (1.7 × 104) (P = 0.16) (data not shown).
FIG. 4.

Flp1 recombinase reporter measuring expression of EPA6 during UTI. The Hygs phenotype (expressed as a percentage of the total number of cells recovered) of two C. glabrata strains, 1697 (wild type [wt]) and 1700 (tna1 tnr1 tnr2 mutant), is shown. Organisms were recovered from kidney tissue 7 days after transurethral inoculation. Individual diamonds correspond to cells recovered from one animal; each the bar represents the mean.
Gene induction under conditions of niacin limitation in C. glabrata is primarily mediated by relief of Hst1-mediated repression.
In C. glabrata, our model of gene induction by niacin limitation is that the limiting environmental supply of niacin lowers the intracellular [NAD+] and thus reduces the activities of the Sir2 family of NAD+-dependent histone deacetylases. In S. cerevisiae, three members of this family, Sir2, Hst1, and Hst2, are known to be involved in transcriptional repression of gene expression. We reasoned that in C. glabrata, the induction of many genes in response to niacin limitation might result from the derepression of Sir2-, Hst1-, or Hst2-dependent transcriptional regulation. To identify the target genes of Sir2-, Hst1-, and Hst2-dependent regulation, we determined the transcription profile of C. glabrata sir2Δ (BG1216), sir4Δ (BG1217), hst1Δ (BG1218), and hst2Δ (BG1219) mutant strains. All of the experiments were done with log-phase cells grown in YPD medium. Consistent with their functions in transcription repression, this analysis identified only a list of genes that were highly expressed in each of the deletion strains relative to wild-type cells. In brief, the transcripts levels of 235, 15, 30, and 37 ORFs are more than 2.5-fold higher in the hst1Δ, hst2Δ, sir2Δ, and sir4Δ mutant cells than in wild-type cells, respectively. Primary microarray data and the complete lists of these ORFs have been deposited at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE6626. Several features can be deduced from these lists. First, all 30 ORFs that showed high expression levels due to the deletion of SIR2 are also highly expressed in the sir4Δ mutant strain, consistent with our expectations that Sir2 and Sir4 function in the same pathway. Twenty-nine of these ORFs are located within 20 kb of the chromosome ends in the sequenced C. glabrata genome, indicating their association with telomeres. These data strongly suggest that C. glabrata Sir2 and Sir4 function primarily in telomeric gene silencing. Second, there is only one ORF exhibiting elevated expression in both hst1Δ and sir2Δ mutant strains and three ORFs exhibiting elevated expression in both hst1Δ and hst2Δ mutant strains. No overlap was found between genes induced in the hst2Δ and sir2Δ mutant strains. These data suggest that C. glabrata Hst1, Hst2, and Sir2 regulate different sets of genes. Finally, the ORFs that are highly expressed in the hst1Δ and hst2Δ mutant strains are distributed throughout the C. glabrata genome and not concentrated in any defined regions. Specifically, of the 235 ORFs that are highly expressed in the hst1Δ mutant, only 7 are located within 20 kb of the sequenced chromosome ends, with the remaining 228 ORFs dispersed among 13 chromosomes. None of the 15 ORFs that showed elevated expression in the hst2Δ mutant strain are located near the chromosome ends. This suggests that both Hst1 and Hst2, unlike Sir2, act as gene-specific regulators, similar to their homologues in S. cerevisiae.
We next compared the list of 315 ORFs that are upregulated more than 2.5-fold by niacin limitation (from cells grown in SC medium containing 0.016 versus 3.25 μM NA) with the lists of ORFs that showed significantly higher expression in the hst1Δ, hst2Δ, and sir2Δ mutant backgrounds. There are 103 ORFs that produce elevated expression both under niacin limitation and in the hst1Δ mutant background. By contrast, there are only three common ORFs that produce elevated expression both under niacin limitation and in the sir2Δ mutant background; two ORFs exhibited elevated expression both under niacin limitation and in the hst2Δ mutant background. These data strongly suggest that the transcriptional response to niacin limitation in C. glabrata is primarily due to derepression of Hst1-repressed genes and, to a lesser extent, to derepression of Sir2- and Hst2-repressed genes.
Transcription of TNA1, TNR1, and TNR2 is regulated by Hst1.
The three genes, TNA1, TNR1, and TNR2, which are most highly induced by niacin limitation are also among the most highly expressed genes in the hst1Δ mutant background but not in the sir2Δ and hst2Δ mutant backgrounds. This result was confirmed by an S1 nuclease protection assay detecting steady-state transcript levels (Fig. 5A). Additionally, the S1 nuclease protection assay was performed with the tnr1Δ or tnr2Δ mutant strain, and in each case, elevated transcript levels from the remaining TNR were observed only in the hst1Δ mutant background, confirming that both TNR genes are regulated by Hst1 (data not shown). Additionally, we restored HST1 on a self-replicating plasmid in the hst1Δ mutant strain. As expected, the ectopic expression of HST1 repressed the transcription of TNA1, TNR1, and TNR2 in SC medium but not in SC-NA medium (data not shown). In S. cerevisiae, the Sum1 transcription factor is known to bind Hst1 and recruit Hst1 to its target genes (2, 25). Hence, we tested if the deletion of the C. glabrata orthologue of SUM1 (ORF CAGL0J10956g) resulted in the elevated expression of TNA1, TNR1, and TNR2. As shown in Fig. 5B, the transcription profile of these three genes in the sum1Δ mutant strain was similar to that in the hst1Δ mutant strain. All of these data strongly suggest that the transcription of TNA1, TNR1, and TNR2 is regulated by Hst1, probably via the Sum1 transcription factor but independently of Sir2 and Sir4. The three ORFs that are most highly expressed both under niacin limitation and in the sir2Δ mutant background include EPA6 and EPA7, conforming to our previously published results. The derepression of EPA6 expression in the sir2Δ and sir4Δ mutant backgrounds was confirmed by the experiment shown in Fig. 5A.
FIG. 5.

(A) Sirtuin regulation of TNA1, TNR1, and TNR2. S1 nuclease analysis of TNA1, TNR1/TNR2, and ACT1 transcript levels. RNA was prepared from five strains, the wild type (WT; BG2) and the hst1 (BG1218), hst2 (BG1219), sir2 (BG1216), sir4 (BG1217) mutants. RNA was prepared from cells grown in SC medium containing 3.25 μM NA to an OD600 of 0.5. (B) S1 nuclease analysis of TNA1, TNR1/TNR2, and ACT1 transcript levels. RNA was prepared from three strains, the wild type (BG2) and the hst1 (BG1218) and sum1 (BG1462) mutants, growing in SC medium containing 3.25 μM (lanes 1, 3, and 5) or 16 nM (lanes 2, 4, and 6) NA. The OD600 of all of the cultures was 0.5. (C) S1 nuclease analysis of EPA6, TNR1/TNR2, and ACT1 transcript levels. Strain BG2 was grown in SC medium containing the indicated initial concentrations of NA; all cultures were grown to an OD600 of 0.5.
Since, in C. glabrata, genes whose expression is responsive to the NAD+ status of the cell are subject to Hst1- or Sir2-mediated repression, we tested whether Hst1- and Sir2-regulated genes might respond differentially to the perturbation of intracellular [NAD+]. Since, in C. glabrata, TNR1 and TNR2 are regulated exclusively by Hst1 and EPA6 is regulated exclusively by Sir2, we tested whether these Hst1- and Sir2-regulated genes were derepressed at different levels of niacin limitation. We carried out two separate experiments. First, as shown in Fig. 5C, wild-type C. glabrata cells were grown to a constant cell density (OD600 = 0.5) in SC medium containing a decreasing levels of NA. Full transcriptional induction of TNR1/TNR2 was observed in cultures supplemented with 32 nM or less NA, while for EPA6, full induction was observed in cultures supplemented with 16 nM or less NA. Thus, there might be a modest difference in the induction of EPA6 relative to that of the TNA1, TNR1, and TNR2 genes. In the second experiment, we transferred cells from SC medium to fresh SC or SC with low NA (0.016 μM) and grew them for 24 h, collecting cells at various points in the time course (Fig. 6A). As shown in Fig. 6B, when cells were grown in SC, EPA6 was not substantially induced at any time point, whereas TNA1 and TNR1/2 were induced only at later time points (11 and 23.5 h). In low-NA medium, TNA1 and TNR1/2 showed similar induction kinetics over the time course and reached half-maximal induction by h 6. Again, EPA6 showed slightly delayed induction kinetics, reaching half-maximal induction at h 11.
FIG. 6.

Analysis of C. glabrata grown in SC medium containing 3.25 or 0.016 μM NA. (A) The values are time points for the collection of cells used in quantitative reverse transcription-PCR and for analysis of intracellular NAD+ and NADH concentrations. (B) Quantitative reverse transcription-PCR analysis of TNA1, TNR1/2, and EPA6 transcript levels. RNA was prepared from wild-type C. glabrata grown in SC medium containing 3.25 or 0.016 μM NA. The indicated time points correspond to those in panel A. The transcript levels of TNR1/TNR2, TNA1, and EPA6 were normalized to levels of HHT2, a gene with constant expression levels under normal and low-NA conditions based on our microarray analysis. (C) Intracellular NAD+ and NADH concentrations. Cells grown in SC medium containing 0.016 μM NA have very low levels of intracellular NAD+ and NADH compared with those of cells grown in SC containing 3.25 μM NA. Error bars represent the standard error of the mean.
To demonstrate formally that growth in low-NA medium results in a drop in intracellular NAD+ concentrations, we monitored intracellular NAD+ and NADH at each time point. In Fig. 6C, we show that the levels of NAD+ and NADH do not substantially change in SC medium until the last two time points as the cell culture reaches saturation. In SC, the notable drops in intracellular NAD+ and NADH levels coincide with the induction of TNA1 and TNR1/2. For cells in SC-NA medium, we documented a dramatic drop in total intracellular NAD+ and NADH as the time course progressed, resulting by h 6 in levels below 3 amol NAD+/cell (compared to 58 amol NAD+/cell when grown in SC medium). Coincident with this drop in intracellular NAD+, transcription of TNA1, TNR1, and TNR2 was induced strongly by h 6. This correlation in both SC and SC-NA media between a drop in NAD+/NADH levels and the transcriptional induction of TNA1, TNR1, and TNR2 is consistent with our hypothesis that induction occurs in response to decreasing intracellular NAD+ levels.
To further explore the potential for Hst1 and Sir2 to differentially respond to intracellular NAD+ levels, we measured the apparent Km of purified rSir2 and Hst1. One report on S. cerevisiae has shown that the apparent Km of purified rHst1 for NAD+ is higher than that for rSir2 during HDAC reactions carried out in vitro (2), suggesting that at intermediate concentrations of NAD+, S. cerevisiae Hst1 might be relatively inactive compared to Sir2. We tested whether in C. glabrata a similar difference in affinity for NAD+ might contribute to the modest difference in the derepression of EPA6, TNA1, and TNR1/2 at different physiological [NAD+]. We expressed C. glabrata Hst1 and Sir2 in E. coli and measured the apparent Kms of these two purified recombinant enzymes for NAD+. The HDAC reactions were carried out by monitoring the release of 3H label from the [3H]acetyl histone H4 peptide (amino acids 2 to 24). As shown in Fig. 7, we determined that the apparent Km of rHst1 is approximately 118 ± 19 μM, while the apparent Km of rSir2 is approximately 339 ± 40 μM, which is well above that of rHst1.
FIG. 7.
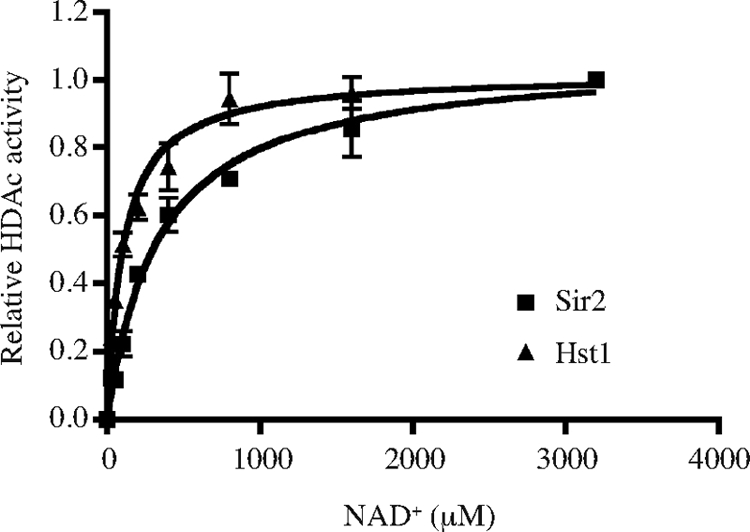
Apparent Kms of C. glabrata Hst1 and Sir2. Relative HDAC activities of rHst1 (squares) and rSir2 (triangles) are shown. Bacterially expressed and purified Hst1 and Sir2 were incubated with [3H]acetylated histone H4 peptide (40,000 cpm/reaction mixture) in the presence of different concentrations of NAD+. The plots are from assays carried out in triplicate. The measured Km of rHst1 is approximately 118 ± 19 μM, and that of rSir2 is approximately 339 ± 40 μM.
DISCUSSION
In living cells, NAD+ and its phosphorylated derivative NADP+, together with their reduced forms, NADH and NADPH, are central coenzymes in many redox reactions. Additionally, NAD+ serves as a substrate for enzymatic reactions that do not involve oxidative chemistry. Through the latter class of reactions, NAD+ is consumed and converted into NAM and ADP-ribose derivatives (reviewed in reference 3). In S. cerevisiae and C. glabrata, these reactions are catalyzed by the Sir2 family of histone deacetylases, whose functions are involved in many important metabolic events, including transcriptional control, longevity control, and cell cycle regulation. Thus, it is essential to balance the synthesis and consumption of NAD+ and keep its level at homeostasis for normal cellular functions. Unlike S. cerevisiae, C. glabrata lacks the ability to synthesize NAD+ de novo from tryptophan. Therefore, its growth depends on the environmental supply of NAD+ precursors, such as NA, NAM, and NR. When encountering a limitation of NAD+ precursors in its growth environment, C. glabrata upregulates a group of genes that potentially facilitate the restoration of NAD+ homeostasis in the cell. Among these genes are TNA1, TNR1, and TNR2, which encode high-affinity transporters of various NAD+ precursors. The functions of these three genes are essential for C. glabrata to grow in media, including human urine, which contain low levels of NAD+ precursors.
Besides these tightly regulated high-affinity transporters of NAD+ precursors, C. glabrata apparently possesses low-affinity uptake systems which permit the cells to grow independently of the functions of Tna1, Tnr1, and Tnr2 when the environmental supply is abundant. Under the extreme condition in which C. glabrata is starved for niacin and cell growth is arrested, TNA1, TNR1, and TNR2 are required to rapidly restore the growth of starved C. glabrata cells after supplementation with NAD+ precursors. In niacin-starved tna1Δ tnr1Δ tnr2Δ mutant cells, a very extended lag phase in the recovery of niacin-starved cells is seen. This lag phase is seen even when the cells are grown in the presence of enough niacin to totally suppress the log-phase growth defect of the transporter mutants. Thus, we provide evidence for two separable physiological functions of the high-affinity transporters: on the one hand, at moderately low levels of niacin, they are required to support maximal growth; on the other, for starved cells, they are central to reestablishing NAD+ homeostasis prior to growth. One possibility for this extended lag phase is that the putative low-affinity uptake systems for NAD+ precursors (which clearly can support robust growth once the cells are grown) are compromised in such niacin-starved cells, either directly because of NAD+ depletion or indirectly because of perturbation of energy metabolism, for example. Other C. glabrata genes that are significantly upregulated by niacin limitation include EPA6 and EPA7, which encode adhesins. The functional importance of altering adherence in response to niacin limitation is not known. One possibility that remains to be tested is that adherent yeast cells can access intracellular host NAD+ precursors, in which case the EPA genes could also play a role in maintaining NAD+ homeostasis.
In S. cerevisiae, environmental niacin limitation results not in cessation of growth but rather in derepression of the Hst1-regulated BNA genes, which encode enzymes of the de novo NAD+ biosynthetic pathway that act to restore intracellular NAD+ levels (2). In C. glabrata, the response to niacin limitation does not include transcriptional induction of the kynurenine pathway genes, which are not encoded in the genome, but rather is characterized by induction of the Hst1-regulated TNA1, TNR1, and TNR2 genes. In both species, therefore, Hst1 mediates the primary transcriptional response to NAD+ limitation by regulating genes central to restoring NAD+ homeostasis.
How does niacin limitation affect sirtuin-regulated gene expression? In S. cerevisiae, growth in the absence of niacin results in the induction of Hst1-regulated genes, probably as a result of loss of Hst1 activity as the cellular NAD+ concentration drops (2). Sirtuin function has also been postulated to respond to the NAD+/NADH ratio (15, 16) or, alternatively, to the intracellular concentration of NAM, a competitive inhibitor of sirtuins (1, 23). We considered whether intracellular NAM levels in the cell might contribute to the derepression of Hst1- or Sir2-regulated genes upon niacin limitation. From our microarray studies, the PNC1 gene, which encodes nicotinamidase, is strongly upregulated upon niacin limitation. Induction of PNC1 would have the effect of reducing NAM levels and thereby reducing the competitive inhibition of sirtuins. We consider it unlikely, therefore, that NAM inhibition of sirtuin function contributes to derepression of Hst1- or Sir2-regulated genes upon niacin limitation in C. glabrata. Rather, in C. glabrata, we have shown that niacin limitation results in a decrease in intracellular NAD+ and we suggest that this is directly responsible for loss of sirtuin function and derepression of sirtuin-regulated genes. In this regard, we found that TNA1 and the TNR genes were transcriptionally induced when intracellular NAD+ levels fell below 12 amol/cell (approximately 240 to 400 μM), with full induction apparent in cells containing 2 amol/cell (approximately 40 to 70 μM). C. glabrata cells are small (3 to 5 μm in diameter), and for these calculations we estimate the cell volume to be 30 to 50 fl. Thus, intracellular NAD+ (only a fraction of which is free) appears to be in a sensitive range for regulation of sirtuin activity, based on our in vitro measurements of Sir2 and Hst1 Kms.
As intracellular NAD+ levels fall, are Hst1 and Sir2 functions differentially affected? In support of this possibility, we noticed modest differences in the degree of niacin limitation leading to induction of the Sir2-regulated EPA6 gene relative to the Hst1-regulated TNA1, TNR1, and TNR2 genes: in both Fig. 5C and 6B, as NAD+ levels decrease, induction of TNA1, TNR1, and TNR2 precedes induction of EPA6. This potentially suggested that Hst1, as in S. cerevisiae, might have a lower binding affinity for NAD+ and consequently be more sensitive to drops in the cellular NAD+ concentration. However, our Km measurements suggested that Hst1 has a higher affinity for NAD+ than does Sir2 and our experiments do not, therefore, support a model in which differential innate affinity of Hst1 and Sir2 for NAD+ plays a role in the preferential induction of Hst1-regulated genes. That said, our data also do not exclude this model, since our experiments are subject to the caveats that we are measuring an apparent Km in HDAC reactions where the acetylated peptide substrate is not present at saturating concentrations and that the substrate for the Sir2- or Hst1-mediated deacetylation in vivo is the full-length histone in the context of chromatin rather than a peptide in solution. A further consideration is that Hst1-mediated repression and Sir2-mediated silencing may have inherently different stabilities since Hst1 mediates local deacetylation of histones at the promoters of repressed genes, while Sir2 mediates regional deacetylation of histones across contiguous silenced regions. We speculate that the effect on transcription of a drop in sirtuin activity may be quite different in these two situations. In this regard, it will be informative to assess the Sir complex association and histone acetylation status of the TNA1, TNR, and EPA6 loci as a function of the intracellular NAD+ concentration. Lastly, the regulation of Sir2- and Hst1-regulated genes is not solely at the level of sirtuin function; rather, transcription of EPA6, TNA1, and TNR1/2 is also regulated positively by factors that act independently of intracellular NAD+ concentrations and this complexity of regulation may also contribute to the differential regulation of Sir2- and Hst1-regulated genes.
Is NAD+ regulation of gene expression important during infection? C. glabrata colonizes the gastrointestinal and genitourinary tracts. On mucosal surfaces, C. glabrata must compete with other microbes for nutrients, which likely results in nutrient limitation, including limitation of NAD+ precursors; niacin regulation of TNA1, TNR1, TNR2, EPA6, and EPA7 may, therefore, be important quite generally in the adaptation of C. glabrata to the host mucosal environment. In the urinary tract, our data suggest that NAD+ levels directly influence C. glabrata gene expression. The measured levels of excreted available niacin in urine samples were on the order of 100 nM, and we have shown that growth in urine samples or in SC medium supplemented with this level of niacin results in expression of the TNA1, TNR1, and TNR2 genes. Separately, we know that expression of the transporters is required for growth in urine since the tna1 tnr1 tnr2 triple mutant completely fails to grow in urine. These data suggest that NAD+ limitation is an important signal governing C. glabrata gene regulation during urinary tract colonization or infection. We propose that further characterization of the response of C. glabrata to niacin limitation may shed light on the adaptation of C. glabrata to growth in the urinary tract and potentially in other mucosal niches.
Supplementary Material
Acknowledgments
We thank Jeff Corden and Rachel Green for help in purifying rHst1 and rSir2. We thank Hani Zaher for help in preparing Fig. 3 and 7. We thank Brian Green and members of the Cormack lab for careful reading of the manuscript. We are grateful to Rupinder Kaur and Alejandro De Las Peñas for gifts of plasmids and strains.
This work was supported by NIH grants AI046223 and DK075732 and the National Research Council of Canada Genomics and Health Initiative.
Footnotes
Published ahead of print on 18 May 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
National Research Council of Canada publication NRC 50666.
REFERENCES
- 1.Anderson, R. M., K. J. Bitterman, J. G. Wood, O. Medvedik, H. Cohen, S. S. Lin, J. K. Manchester, J. I. Gordon, and D. A. Sinclair. 2002. Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. J. Biol. Chem. 27718881-18890. [DOI] [PubMed] [Google Scholar]
- 2.Bedalov, A., M. Hirao, J. Posakony, M. Nelson, and J. A. Simon. 2003. NAD+-dependent deacetylase Hst1p controls biosynthesis and cellular NAD+ levels in Saccharomyces cerevisiae. Mol. Cell. Biol. 237044-7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belenky, P., K. L. Bogan, and C. Brenner. 2007. NAD+ metabolism in health and disease. Trends Biochem. Sci. 3212-19. [DOI] [PubMed] [Google Scholar]
- 4.Belenky, P. A., T. G. Moga, and C. Brenner. 2008. Saccharomyces cerevisiae YOR071C encodes the high affinity nicotinamide riboside transporter Nrt1. J. Biol. Chem. 2838075-8079. [DOI] [PubMed] [Google Scholar]
- 5.Bieganowski, P., and C. Brenner. 2004. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117495-502. [DOI] [PubMed] [Google Scholar]
- 6.Castaño, I., S. J. Pan, M. Zupancic, C. Hennequin, B. Dujon, and B. P. Cormack. 2005. Telomere length control and transcriptional regulation of subtelomeric adhesins in Candida glabrata. Mol. Microbiol. 551246-1258. [DOI] [PubMed] [Google Scholar]
- 7.Celic, I., H. Masumoto, W. P. Griffith, P. Meluh, R. J. Cotter, J. D. Boeke, and A. Verreault. 2006. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr. Biol. 161280-1289. [DOI] [PubMed] [Google Scholar]
- 8.Cormack, B. P., and S. Falkow. 1999. Efficient homologous and illegitimate recombination in the opportunistic yeast pathogen Candida glabrata. Genetics 151979-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cormack, B. P., N. Ghori, and S. Falkow. 1999. An adhesin of the yeast pathogen Candida glabrata mediating adherence to human epithelial cells. Science 285578-582. [DOI] [PubMed] [Google Scholar]
- 10.De Las Peñas, A., S. J. Pan, I. Castaño, J. Alder, R. Cregg, and B. P. Cormack. 2003. Virulence-related surface glycoproteins in the yeast pathogen Candida glabrata are encoded in subtelomeric clusters and subject to RAP1- and SIR-dependent transcriptional silencing. Genes Dev. 172245-2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Domergue, R., I. Castaño, A. De Las Peñas, M. Zupancic, V. Lockatell, J. R. Hebel, D. Johnson, and B. P. Cormack. 2005. Nicotinic acid limitation regulates silencing of Candida adhesins during UTI. Science 308866-870. [DOI] [PubMed] [Google Scholar]
- 12.Halme, A., S. Bumgarner, C. Styles, and G. R. Fink. 2004. Genetic and epigenetic regulation of the FLO gene family generates cell-surface variation in yeast. Cell 116405-415. [DOI] [PubMed] [Google Scholar]
- 13.Iraqui, I., S. Garcia-Sanchez, S. Aubert, F. Dromer, J. M. Ghigo, C. d'Enfert, and G. Janbon. 2005. The Yak1p kinase controls expression of adhesins and biofilm formation in Candida glabrata in a Sir4p-dependent pathway. Mol. Microbiol. 551259-1271. [DOI] [PubMed] [Google Scholar]
- 14.Kilby, N. J., M. R. Snaith, and J. A. Murray. 1993. Site-specific recombinases: tools for genome engineering. Trends Genet. 9413-421. [DOI] [PubMed] [Google Scholar]
- 15.Lin, S. J., E. Ford, M. Haigis, G. Liszt, and L. Guarente. 2004. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 1812-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin, S. J., M. Kaeberlein, A. A. Andalis, L. A. Sturtz, P. A. Defossez, V. C. Culotta, G. R. Fink, and L. Guarente. 2002. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418344-348. [DOI] [PubMed] [Google Scholar]
- 17.Lin, S. S., J. K. Manchester, and J. I. Gordon. 2001. Enhanced gluconeogenesis and increased energy storage as hallmarks of aging in Saccharomyces cerevisiae. J. Biol. Chem. 27636000-36007. [DOI] [PubMed] [Google Scholar]
- 18.Llorente, B., and B. Dujon. 2000. Transcriptional regulation of the Saccharomyces cerevisiae DAL5 gene family and identification of the high affinity nicotinic acid permease TNA1 (YGR260w). FEBS Lett. 475237-241. [DOI] [PubMed] [Google Scholar]
- 19.Ma, B., S. J. Pan, M. L. Zupancic, and B. P. Cormack. 2007. Assimilation of NAD+ precursors in Candida glabrata. Mol. Microbiol. 6614-25. [DOI] [PubMed] [Google Scholar]
- 20.Maas, N. L., K. M. Miller, L. G. DeFazio, and D. P. Toczyski. 2006. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol. Cell 23109-119. [DOI] [PubMed] [Google Scholar]
- 21.Panozzo, C., M. Nawara, C. Suski, R. Kucharczyka, M. Skoneczny, A. M. Becam, J. Rytka, and C. J. Herbert. 2002. Aerobic and anaerobic NAD+ metabolism in Saccharomyces cerevisiae. FEBS Lett. 51797-102. [DOI] [PubMed] [Google Scholar]
- 22.Rusche, L. N., A. L. Kirchmaier, and J. Rine. 2003. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu. Rev. Biochem. 72481-516. [DOI] [PubMed] [Google Scholar]
- 23.Sandmeier, J. J., I. Celic, J. D. Boeke, and J. S. Smith. 2002. Telomeric and rDNA silencing in Saccharomyces cerevisiae are dependent on a nuclear NAD+ salvage pathway. Genetics 160877-889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah, G. M., D. Poirier, C. Duchaine, G. Brochu, S. Desnoyers, J. Lagueux, A. Verreault, J. C. Hoflack, J. B. Kirkland, and G. G. Poirier. 1995. Methods for biochemical study of poly(ADP-ribose) metabolism in vitro and in vivo. Anal. Biochem. 2271-13. [DOI] [PubMed] [Google Scholar]
- 25.Xie, J., M. Pierce, V. Gailus-Durner, M. Wagner, E. Winter, and A. K. Vershon. 1999. Sum1 and Hst1 repress middle sporulation-specific gene expression during mitosis in Saccharomyces cerevisiae. EMBO J. 186448-6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.