

Abstract
Poly(ADP-ribose) polymerase 1 (PARP1) and SIRT1 deacetylase are two NAD-dependent enzymes which play major roles in the decision of a cell to live or to die in a stress situation. Because of the dependence of both enzymes on NAD, cross talk between them has been suggested. Here, we show that PARP1 is acetylated after stress of cardiomyocytes, resulting in the activation of PARP1, which is independent of DNA damage. SIRT1 physically binds to and deacetylates PARP1. Increased acetylation of PARP1 was also detected in hearts of SIRT1−/− mice, compared to that detected in the hearts of SIRT1+/+ mice, confirming a role of SIRT1 in regulating the PARP1 acetylation in vivo. SIRT1-dependent deacetylation blocks PARP1 activity, and it protects cells from PARP1-mediated cell death. We also show that SIRT1 negatively regulates the activity of the PARP1 gene promoter, thus suggesting that the deacetylase controls the PARP1 activity at the transcriptional level as well. These data demonstrate that the activity of PARP1 is under the control of SIRT1, which is necessary for survival of cells under stress conditions.
During cellular stress, proteins undergo a variety of posttranslational modifications that result in their increased or decreased activity. One such modification is poly(ADP-ribosyl)ation, which is catalyzed by a family of enzymes called poly(ADP-ribose) polymerases (PARPs). This is initiated by transfer of an ADP-ribose unit from NAD to glutamate or aspartate residues of the target protein, and it proceeds with successive additions of many ADP-ribose units to the substrate, resulting in the synthesis of a large chain of branched ADP-ribose polymers, which are subsequently degraded by poly(ADP-ribose) glycohydrolase (30).
PARP1 (116 kDa) is a prototype member of the PARP family of enzymes. It is ubiquitously expressed and accounts for most of the poly(ADP-ribosyl)ation of proteins in vivo (30). PARP1 is located in the nucleus as well as in the mitochondria, and it plays an important role in the DNA repair process and in the maintenance of genome stability. The enzyme consists of a characteristic three-domain structure: a DNA binding domain at the amino terminus, a catalytic domain at the carboxy terminus, and an automodification domain in the middle, which is poly(ADP-ribosyl)ated by itself (30). PARP1 is activated in response to DNA damage, such as single-strand breaks, which could develop as a response to various pathological conditions, such as inflammatory diseases, diabetes, reperfusion injury, or oxidative stress. PARP1 is also known to be activated by processes independent of DNA damage, including phosphorylation, and high levels of Mg2+, Ca2+, and polyamines (31). While basal activation of PARP1 is needed for maintenance of normal homeostasis of the cell, overactivation of PARP1 consumes NAD and results in cell death due to depletion of intracellular NAD stores (10). This characteristic makes it important for PARP1 activity to be tightly regulated for survival of the cell.
One group of factors which are most affected by changes in intracellular levels of NAD are the class III histone deacetylases (HDACs), also called sirtuins or SIRTs. SIRT1 is a prototype member of the sirtuin family, which is considered a nuclear sensor of the redox state of the cell (29). SIRT1 has been implicated in transcriptional silencing, genetic control of aging, cell metabolism, and calorie restriction-mediated longevity of the organism (11). Mutation of ySir2 shortens the life span of Saccharomyces cerevisiae by 40%, whereas overexpression of the protein extends the life span in multiple experimental models, including those of yeast, Caenorhabditis elegans, and Drosophila (15, 28, 32). Increased cellular NAD levels have been shown to activate SIRT1, whereas high nicotinamide and/or NADH levels inhibit its activity (29).
Because both PARP1 and SIRT1 use NAD for their activity and are capable of operating many common pathways, cross talk between these proteins has been suggested (36). It has been thought that the increased activity of one molecule might interfere with the activity of the other. In fact, PARP1 has been shown to enhance the transcription activity of NF-κB, whereas SIRT1 was found to inhibit NF-κB activity (12, 34). The functional activity of P53 has also been shown to be regulated oppositely by PARP1 and SIRT1 (20, 21). We have previously shown that PARP1 overactivation suppresses the activity of SIRT1 by depleting cellular NAD levels (23). Additionally, SIRT1 activation was found to be capable of blocking apoptosis-inducing factor (AIF) release from mitochondria resulting from the overactivation of PARP1, thus suggesting that these two proteins might be able to counterbalance each other's activity to control the balance between cell survival and death (18). However, the mechanism by which SIRT1 cancels out PARP1 activity is not yet known.
Here, we show that PARP1 is acetylated under stress conditions and that this enhances its enzymatic activity. SIRT1 is a strong deacetylase for PARP1. SIRT1-mediated deacetylation inhibits the enzymatic activity of PARP1. In addition, we show that SIRT1 negatively regulates the activity of the PARP1 gene promoter, leading to repressed synthesis of the PARP1 protein. Thus, under stress conditions, SIRT1 is capable of regulating PARP1 activity at both the transcription and posttranslation levels.
MATERIALS AND METHODS
Antibodies used.
The following antibodies and conjugates were used in this study: rabbit anti-PARP1 (sc-7150; Santa Cruz); mouse anti-PARP1 (sc-8007; Santa Cruz); rabbit SIRT1 (07-131; Upstate); rabbit anti-PCAF (sc-8999; Santa Cruz); rabbit anti-Flag (ab1162; Abcam); rabbit anti-acetyl-lysine (06-933; Upstate); mouse anti-acetyl-lysine (Ac-K-103; Cell Signaling); rabbit anti-acetyl-lysine (9441; Cell Signaling); mouse anti-poly(ADP-ribose) [anti-P(ADP)R] (Alx-804-220; Alexis); rabbit anti-P(ADP)R (Alx-210-890; Alexis); mouse antihistones (MAB052; Chemicon); goat anti-RNA polymerase II (sc-5943; Santa Cruz); anti-α-tubulin (sc8035; Santa Cruz); mouse anti-GST (sc-138; Santa Cruz); rabbit anti-cMyc (sc-789; Santa Cruz); goat antiactin (sc-1616; Santa Cruz); goat anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GADPH; sc20357; Santa Cruz), anti-Flag M2 affinity gel (A2220; Sigma); goat anti-rabbit immunoglobulin G-horseradish peroxide (IgG-HRP) (sc-2054; Santa Cruz); donkey anti-mouse IgG-HRP (sc-2096; Santa Cruz); donkey anti-goat IgG-HRP (sc-2056; Santa Cruz).
Plasmid constructs.
The luciferase reporter plasmid with multiple Gal4 DNA binding sites in the promoter region and the expression plasmids encoding the Gal4 DNA binding domain (Gal4DBD), Gal4DBD fused to the SIRT1 catalytic core domain (Gal4DBD-mCORE), and Gal4DBD fused to the catalytic core domain with a mutation that eliminates deacetylase activity [Gal4DBD-mCORE(H355A)] have been described previously (27). The PARPproCAT gene, a chloramphenicol acetyltransferase (CAT) reporter gene fused to DNA fragments from the recombinant PARP1 gene promoter (−237 bp upstream promoter), was provided by S. Guerin (Oncology and Molecular Endocrinology Research Center, Laval University, Quebec, Canada) (35). Glutathione S-transferase (GST)-PARP1 full-length expression vector, GST fusion expression vectors for different domains of PARP1, and cytomegalovirus expression vectors for Myc- and Flag-tagged PARP1 have been described previously (13). The wild-type SIRT1 and mutant SIRT1 (H355A) expression plasmids were provided by W. Gu (Department of Pathology, College of Physician and Surgeons, Columbia University, New York, NY). The PCAF and HDAC1 expression plasmids were obtained from Addgene.
Cell culture, mechanical stretch, transfection, and adenovirus infection.
All animal protocols were reviewed and approved by the University of Chicago Institutional Animal Care and Use Committee. Primary cultures of 2-day-old neonatal rat heart myocytes were carried out using an established procedure described previously (23). For stress experiments, cells were seeded at a density of 0.5 × 106/ml on Bioflex plates and subjected to cyclic mechanical stretch after 24 h of plating. The frequency of cyclic stretch was 0.2 Hz, with pulsation of 10% elongation 12 times/minute. Twenty-four to thirty hours after seeding, cells were used for adenovirus infection. For all the adenoviral experiments, viruses at a multiplicity of infection (MOI) of 10 were used unless mentioned otherwise. Cardiomyocytes were exposed to 20 μM phenylephrine (PE) or 5 μM angiotensin II for 48 h when required. HeLa or Cos7 cells were maintained in Dulbecco's modified Eagle's medium supplemented with penicillin-streptomycin and 10% fetal bovine serum (complete growth medium). For transfections, 1.2 × 106 cells were grown in 10-cm plates or 2 × 105 in a six-well plate and transfected with appropriate plasmids using the Superfect transfection reagent (Qiagen) or Lipofectamine (Invitrogen) according to the manufacturer's protocol. For cotransfection studies, β-galactosidase (β-Gal) plasmid was used as the control. Luciferase and β-Gal activities were measured as described previously (23).
Subcellular fractionation, immunoprecipitation, and Western analyses.
Subcellular protein fractions of mouse hearts were prepared using the NE-PER nuclear and cytoplasmic extraction kit (Pierce) according to the manufacturer's protocol. Western blotting and immunoprecipitation experiments were done using a standard protocol as described elsewhere (7).
In vitro protein binding assay with and without NAD.
In vitro synthesized-Flag-SIRT1 was immunoprecipitated using Flag beads and incubated with [35S]methionine-labeled PARP1 in a protein binding (radioimmunoprecipitation assay [RIPA]) buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM Na2-EDTA, 1% Triton X-100, 0.1% NP-40, 1 mM phenylmethylsulfonyl fluoride, and 10 μl/ml protease inhibitor cocktail) (Sigma) with different concentrations of NAD (10 μM, 100 μM, 300 μM, 1 mM) or without NAD at 4°C on a rotator overnight. Beads were separated and then washed thrice with RIPA buffer, with a final wash of phosphate-buffered saline (PBS). Bound complexes were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and detected by autoradiography. In another set of experiments, Flag-SIRT1 on beads was initially incubated with NAD (1 mM) in RIPA buffer for 1 h at 4°C on a rotator. The NAD-saturated bead-bound SIRT1 was washed thrice with PBS, washed once with binding buffer, and then incubated with [35S]methionine-labeled PARP1 not bound to NAD in the protein binding buffer overnight at 4°C. The next morning, beads were separated, washed thrice with RIPA buffer, washed once with PBS, and then resolved by SDS-PAGE, and coprecipitation of proteins was detected by autoradiography. For the GST pulldown assay, 4 μg of GST or GST fusion protein containing different segments of PARP1, purified using by glutathione-Sepharose 4B beads (GE Healthcare), was incubated for 2 h at 4°C on a rotator with the in vitro-translated [35S]methionine-labeled Flag-SIRT1 protein. Beads were washed thrice with a buffer containing 200 mM NaCl, 50 mM Tris-HCl (pH 7.5), 0.5% Nonidet P-40, 1 mM dithiothreitol [DTT], protease inhibitor cocktail (Sigma), and 1% bovine serum albumin and were finally rinsed once with PBS. Bound proteins were resolved by SDS-PAGE and detected by autoradiography.
In vitro acetylation and deacetylation assay.
GST-PARP amino acids (aa) 1 to 214, GST-PARP aa 477 to 524, and Flag-PARP1 were used as substrates for in vitro acetylation. Briefly, 2 μg of substrate protein bound to the appropriate beads was resuspended in 1× histone acetyltransferase (HAT) buffer (Upstate Biotechnology). A typical acetylation reaction mixture was comprised of 1 μg of the active PCAF enzyme (GST-PCAF; Upstate Biotechnology), 0.5 μCi [14C]acetyl coenzyme A ([14C]Ac-CoA) (MC 269; Moravek Biochemicals) or 0.3 mM Ac-CoA (Sigma), 30 μM trichostatin A (TSA) and 50 mM nicotinamide (NAM) in 1× HAT buffer (50 mM Tris-HCl [pH 8.0], 10% glycerol, 0.1 mM EDTA, 1 mM DTT). Reaction mixtures were incubated at 30°C for 30 to 60 min on a rotator. Reactions were terminated by adding SDS sample buffer and resolved on a 12% SDS-polyacrylamide gel. Proteins were transferred to a Hybond-P membrane (GE Healthcare) and detected by autoradiography. In situations where cold Ac-CoA was used, protein acetylation was detected by Western analysis with anti-Ac-K antibody. The same membrane was also probed with anti-GST antibody or subjected to Coomassie staining to demonstrate equal loadings of proteins in each lane. For the deacetylation assay, acetylated PARP fusion proteins bound to beads were washed as described above and resuspended in 1× HDAC buffer (50 mM Tris-HCl [pH 8.0], 4 mM MgCl2, 0.2 mM DTT). The acetylated PARP proteins were either incubated in just 1× HDAC buffer (control) or with recombinant immunoprecipitated HDAC1, HDAC4, or SIRT1 (500 ng/reaction). For SIRT1-mediated deacetylation, 100 μM of NAD was included in the HDAC buffer. The reaction mixture was incubated for 1.5 h at 30°C on a Nutator. Proteins were resolved by SDS-PAGE and analyzed by either autoradiography or Western blotting with anti-Ac-K antibody.
In vitro poly(ADP-ribosyl)ation assay.
Acetylated or nonacetylated Flag-PARP1 (100 ng/sample) was incubated with [32P]NAD (800 Ci/mmol; 1 μl/sample) in 50 μl of reaction volume containing 100 mM Tris-HCl (pH 8.0), 20 mM MgCl2, and 1 mM DDT for 20 min at 30°C. The reaction was terminated by adding Laemmli sample buffer. The denatured proteins were resolved by SDS-PAGE, transferred to polyvinylidene difluoride membrane and detected by either autoradiography or Western blotting with the appropriate antibodies. In experiments where the effect of DNA was examined, activated DNA (Trevigen) or genomic DNA (1 μg/sample) was added to the buffer before incubating PARP1 with radiolabeled NAD.
Cell death assay.
HeLa cells were cultured in six-well plates at 70% confluence in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and were treated with 500 μM of N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) for 15 min in serum-free medium. Subsequently, the MNNG-containing medium was removed and replaced with complete growth medium and maintained for 3 h. In another set of experiments, cells were overexpressed with the SIRT1 wild type and/or pretreated with 4 mM 3-aminobenzamide (3AB) and/or 500 nM TSA. Cell death was analyzed by fluorescence-activated cell sorter analysis using propidium iodide staining or annexin V staining. The cell death results were also confirmed by a trypan blue cell viability assay (1). Results were analyzed using Flojo software.
Real-time quantitative PCR analyses.
Total cellular RNA was isolated from cardiomyocytes, using the Trizol reagent (Invitrogen, CA). Residual genomic DNA was digested by incubating the RNA preparation with 0.5 units of RNase-free DNase I per μg of total RNA in 1× reaction buffer for 15 min at room temperature, followed by heat inactivation at 90°C for 5 min. The quality of DNase I-treated RNA was tested by formaldehyde-agarose gel electrophoresis. Two micrograms of DNase-treated RNA was reverse transcribed using the Superscript III kit (Invitrogen). The resultant cDNA was diluted 10-fold prior to PCR amplification. A reverse transcriptase-negative reaction served as a negative control.
The nucleotide sequences of the PCR primers used were as follows: PARP1, 5′-CCCTCACCGAATCTCCTTAG (forward) and 5′-CCCAACTTTCCCTCTACTGC (reverse); ß-actin, 5′-CAAGATCATTGCTCCTCCTG (forward) and 5′-TCATCGTACTCCTGCTTGCT (reverse); CAT, 5′-TTAAACGTGGCCAATATGGA (forward) and 5′-GACATGGAAGCCATCACAGA (reverse); β-Gal, TTGAACTGCCTGAACTACCG (forward) and 5′-TATTACCCAGCTCGATGCAA (reverse). A real-time quantitative PCR was performed with the ABI 7900HT sequence detection system (Applied Biosystems) using a SYBR green endoplasmic reticulum quantitative PCR kit for the ABI Prism system (Invitrogen).
RNA interference analysis.
HeLa cells were transfected with 100 nM ON-TARGETplus small interfering RNA (siRNA) specific for human PCAF using DharmaFect transfection reagents per the manufacturer's instructions. After 48 h of transfection, cells were washed with PBS and treated with MNNG (100 μM) for 15 min in a serum-free medium. Cells were harvested, and lysates were prepared using high-salt (300 mM NaCl) RIPA buffer. PARP1 was immunoprecipitated from these lysates and analyzed by SDS-PAGE and then by Western blotting.
NAD estimation.
Cellular NAD content was measured according to the method described by Jacobson and Jacobson (14) with slight modifications. Briefly, cells (1 × 104) were harvested in PBS and pelleted by centrifugation, and the pellet was suspended in 100 μl of 0.5 M perchloric acid. The cell lysate was neutralized with equal volumes of 1.0 M KOH and 0.33 M KH2PO4-K2HPO4 (pH 7.5) and centrifuged to remove the KClO4 precipitate. The supernatant (50 μl) was mixed with 200 μl of reaction buffer [600 mM ethanol, 0.5 mM 3-(4,5 dimethylthiazol)-2,5 diphenyl-tetrazolium bromide, 2 mM phenazine ethosulfate, 5 mM EDTA, 1.0 mg/ml bovine serum albumin, and 120 mM bicine (pH 7.8)] and incubated for 5 min at 37°C. For a NAD standard curve, a known amount of NAD was added in place of the supernatant. The reaction was initiated by adding 25 μl of alcohol dehydrogenase (0.5 mg/ml in 100 mM bicine [pH 7.8]) and incubating the mixture for 20 min at 37°C. Afterwards, the reaction was quenched by adding 250 μl of 12 mM iodoacetate, and the optical density was determined at 570 nm wavelength. The NAD content is determined from the standard curve and normalized to the protein content of the extract.
Statistical analysis.
Student's t test was used to analyze statistical significance between two groups. All P values corresponded to two-tailed tests, and a P value of <0.05 was considered statistically significant.
RESULTS
PARP1 is acetylated under stress conditions.
PARP1 is known to be activated in hemodynamically overloaded hearts (24). To examine whether PARP1 is responsive to physical stress of cardiomyocytes, we subjected rat heart myocytes to cyclic mechanical stretch (10%) for 4 h. Subsequently, the cell lysate was analyzed for acetylation and poly(ADP-ribosyl)ation of cardiac proteins by Western analyses using anti-Ac-K and anti-P(ADP)R antibodies. We found robust poly(ADP-ribosyl)ation and acetylation of many cellular proteins (Fig. 1a and b). One protein that was running close to a molecular mass of 100 kDa was consistently acetylated in stretched cells. By probing the same membrane with anti-PARP1 antibody, we found that this acetylated protein was in fact identical to the protein recognized by the PARP1 antibody, suggesting that PARP1 might be acetylated under stretch conditions (Fig. 1c). To demonstrate that the increased poly(ADP-ribosyl)ation and acetylation of proteins had resulted from stress of cells, and was not related to any hidden deformity of cells due to mechanical stretch, we performed a similar experiment in which cells were treated with hypertrophy agonist phenylephrine (PE) or angiotensin II, both known inducers of stress of cardiomyocytes. Both agonists caused acetylation of PARP1, and that was associated with the robust poly(ADP-ribosyl)- ation of cellular proteins (Fig. 1d). These experiments indicated that, under stress conditions, PARP1 is acetylated in cardiomyocytes and this is coupled with its increased enzymatic activity.
FIG. 1.
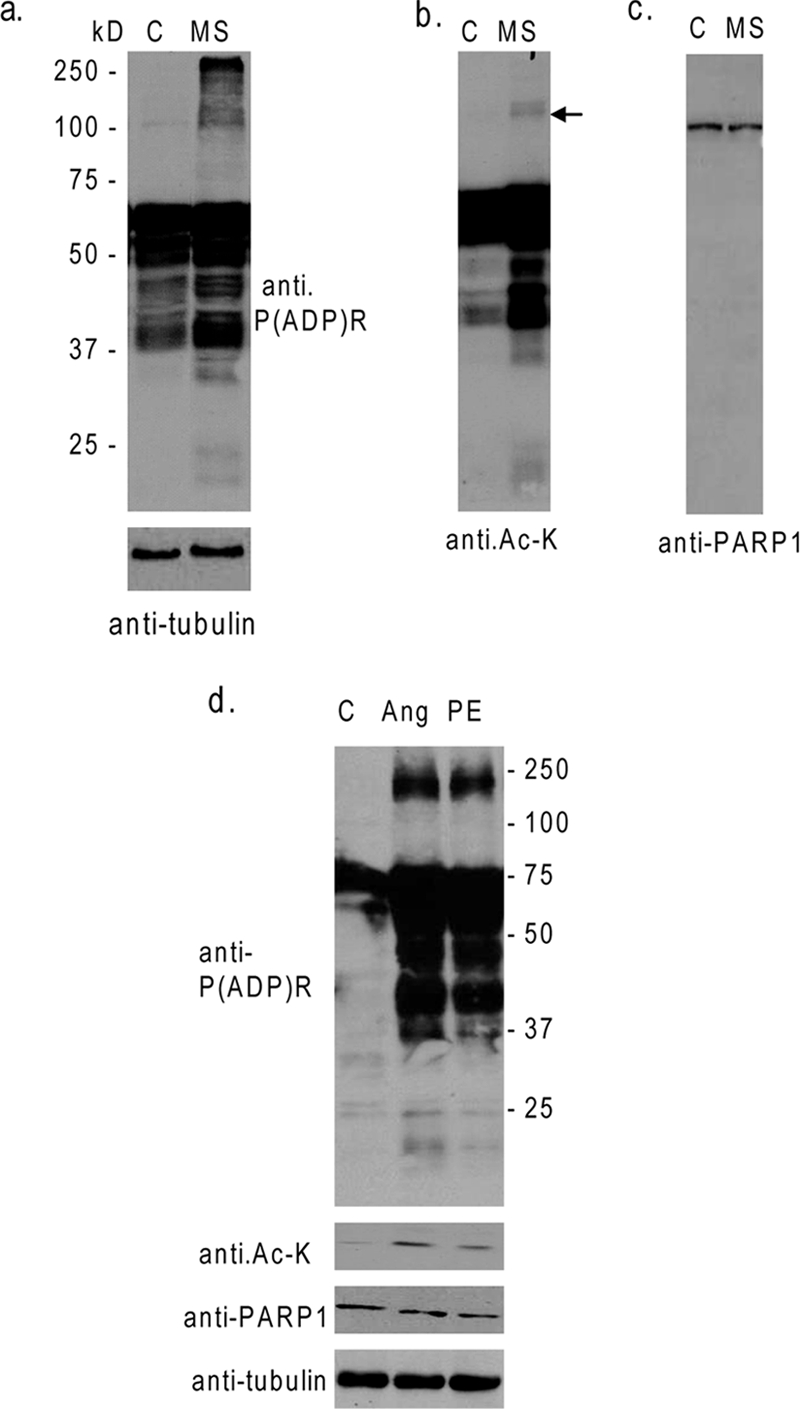
PARP1 is acetylated and activated after stress of cardiomyocytes. (a) Cardiomyocytes were subjected to cyclic mechanical stretch (MS) of 10% elongation for 4 h. Subsequently, cell lysates were analyzed by Western analysis, with use of anti-P(ADP)R and anti-tubulin antibodies. (b and c) The same membrane as that in panel a was probed with anti-Ac-K and anti-PARP1 antibodies. Note that increased acetylation of PARP1 was associated with increased poly(ADP-ribosyl)ation of cardiac proteins. (d) The cell lysate of cardiomyocytes treated with PE (20 μM) or angiotensin II (Ang; 5 μM) for 48 h was analyzed by Western blotting with different antibodies, as indicated. C, control.
Acetylation activates PARP1, independent of DNA damage.
The activity of the HATs p300/CBP and PCAF is known to be induced during stress of cardiomyocytes. We therefore tested the acetylation of PARP1 by both p300 and PCAF. Results indicated that PCAF alone was sufficient to acetylate PARP1 and that the addition of p300 and PCAF together had no additional effect. We subsequently used PCAF for the acetylation of PARP1 in all future experiments. In vitro-synthesized recombinant PARP1 (Flag-PARP1) was subjected to acetylation with GST-PCAF and Ac-CoA. After completion of the acetylation reaction, Flag-PARP1 was precipitated on beads and incubated with [32P]NAD in a Tris buffer to analyze auto-poly(ADP-ribosyl)ation of PARP1. As shown in Fig. 2a, PARP1 was substantially acetylated by PCAF, and the addition of Ac-CoA to the reaction mixture further increased the acetylation of PARP1. The ability of PCAF to acetylate PARP1 in the absence of exogenous Ac-CoA is likely to be related to Ac-CoA inherently bound with the enzyme, as PCAF was self-acetylated in these reactions without adding Ac-CoA (Fig. 2a, bottom panels). When PARP1 was incubated with [32P]NAD, we found acetylation-dependent poly([32P]ADP-ribosyl)ation of PARP1, thus again suggesting a role of protein acetylation in enhanced activity of the enzyme (Fig. 2a, top panel). To substantiate these findings, we tested three different preparations of PARP1, one obtained from commercial sources (Sigma) and two synthesized in our laboratory as Myc- and Flag-tagged PARP1 proteins, for their response to PCAF-mediated acetylation and found identical results. In one experiments, we also treated PCAF with DNase I to exclude the possibility of contaminated DNA as a cause of PARP1 activation; however, our results were same as those with the untreated PCAF (data not shown).
FIG. 2.

PCAF-dependent acetylation activates PARP1. (a) In vitro-synthesized recombinant PARP1 (Flag-PARP1) was subjected to acetylation with GST-PCAF with or without Ac-CoA and then incubated with [32P]NAD in a buffer. Poly(ADP-ribosyl)ation of PARP1 was analyzed by autoradiography. Before addition of radiolabeled NAD, a portion (20%) of the reaction mixture was removed and analyzed by Western analysis by use of anti-Ac-K, anti-Flag, and anti-GST antibodies. (b) HeLa cells were overexpressed with PCAF-specific siRNA or scrambled siRNA for 48 h. PCAF knockdown was determined by Western blotting. In another set of experiment, PCAF-targeted cells were treated with MNNG (100 μM) for 15 min. Afterwards, PARP1 was immunoprecipitated, and acetylation of protein was analyzed by Western blotting. (c) The histone H1 was incubated with nonacetylated or acetylated Flag-PARP1 in the presence of [32P]NAD. Poly(ADP-ribosyl)ation of H1 was analyzed by autoradiography. To confirm equal loadings of protein in each lane, a portion of the reaction mixture was removed before adding NAD and was subjected to Western analysis with anti-Ac-K, anti-Flag, and anti-H1 antibodies. +, present; −, absent.
To demonstrate PCAF-dependent acetylation of PARP1 in vivo, we knocked out PCAF in HeLa cells by using PCAF-specific siRNA and then examined PARP1 acetylation under stress conditions (MNNG treatment). As shown in Fig. 2b, there was a notably reduced level of PARP1 acetylation in cells in which PCAF was knocked out, compared to that in cells that received scrambled siRNA, thus suggesting that PCAF is involved in the acetylation of PARP1 under stress conditions.
To test whether acetylation also enhanced the activity of PARP1 for other substrates (trans-poly[ADP-ribosyl]ation), we tested poly(ADP-ribosyl)ation of H1 histones, a known substrate of PARP1. We found that poly([32P]ADP-ribosyl)- ation of H1 was also notably enhanced following acetylation of PARP1 with PCAF, albeit to a lesser extent than that observed for the self-poly(ADP-ribosyl)ation of PARP1 (Fig. 2c). These results thus demonstrated that acetylation of PARP1 enhanced its activity for auto-poly(ADP-ribosyl)ation as well as for transferring ADP-ribose moieties to trans substrates.
PARP1 is a DNA binding protein, and it has been shown to be highly activated by binding to nicked DNA. We, therefore, examined the effect of exogenous DNA on acetylation-mediated activation of PARP1 (data not shown). Recombinant PARP1 was acetylated with PCAF, incubated with nicked DNA or intact genomic DNA, and then tested for its ability for poly([32P]ADP-ribosyl)ation. Acetylation of PARP1 resulted in enhanced self-poly([32P]ADP-ribosyl)ation in the absence of DNA. Incubation of PARP1 with intact genomic DNA had no noticeable effect on the basal activity of PARP1, and the acetylation-mediated auto-poly(ADP-ribosyl)ation of PARP1 was again significantly increased in the presence of genomic DNA. When PARP1 was incubated with nicked DNA, there was robust activation of the enzyme, as expected. PARP1 acetylation in the presence of nicked DNA showed no further activation of the enzyme over that of the nonacetylated PARP1. These results thus demonstrated that (i) acetylation induces PARP1 activation independent of DNA damage and (ii) DNA causes maximal activation of PARP1, and in this situation, acetylation-mediated activation of the enzyme cannot be observed (data not shown).
SIRT1 binds to PARP1 and deacetylates it.
To begin to understand the interplay between PARP1 and SIRT1, we examined first the ability of SIRT1 to bind to PARP1 in vivo. Cos7 cells were overexpressed with Flag-SIRT1 and Myc-PARP1; subsequently, the cell lysate was prepared and subjected to immunoprecipitation and then Western analysis with the appropriate antibodies. Nonspecific IgG was used as a negative control for immunoprecipitation. As shown in Fig. 3a, we found that PARP1 was coprecipitated with SIRT1 and vice versa. To demonstrate interaction between endogenous PARP1 and SIRT1, cardiomyocytes stimulated with PE were subjected to immunoprecipitation with SIRT1 antibody, and the resulting beads were analyzed by Western blotting using anti-PARP1 antibody. Results of this experiment demonstrated that PARP1 was coprecipitated with SIRT1, but not with the nonspecific IgG control, thus indicating PARP1-SIRT1 interaction in vivo (Fig. 3b). We then carried out GST pulldown assay to identify short regions of PARP1 targeted by SIRT1. We found that out of six GST-PARP fusion proteins analyzed, only two, containing aa 1 to 214 and aa 477 to 524 segments of PARP1 successfully pulled down [35S]methionine-labeled SIRT1, indicating that these two regions of PARP1 are directly interacting with SIRT1. We also noticed that between these two segments, the N-terminal aa 1 to 214 region of PARP1 had notably higher affinity to bind to SIRT1 than did the other region (data not shown).
FIG. 3.

SIRT1 binds to and deacetylates PARP1 in vitro and in vivo. (a) Cos7 cells were overexpressed with Flag-SIRT1 and Myc-PARP1. The cell lysate was subjected to immunoprecipitation (IP) with anti-Flag, anti-Myc, or nonspecific-IgG-conjugated agarose beads. Precipitated beads were analyzed by Western blotting with anti-Flag or anti-Myc antibodies. (b) Endogenous SIRT1 interacts with PARP1 under stress conditions. The cell lysate obtained from PE-treated cardiomyocytes was subjected to immunoprecipitation with either nonspecific IgG or specific anti-SIRT1 antibody. The resulting beads were analyzed by Western blotting with anti-PARP1 antibody. (c) Full-length Flag-PARP1 was subjected to acetylation with PCAF and [14C]Ac-CoA. The acetylated protein was immunoprecipitated with anti-Flag agarose beads and incubated with beads containing in vivo-synthesized SIRT1, HDAC1, or HDAC4 under the appropriate buffer conditions. The acetylation status of PARP1 was determined by SDS-PAGE and then by autoradiography. Equal loadings of proteins were confirmed by staining the gel with Coomassie dye. (d) Cos7 cells were transfected with plasmids encoding Myc-PARP1, PCAF, SIRT1, HDAC1, or HDAC4 in different combinations and treated with TSA or NAM for 24 h as indicated. Myc-PARP1 was immunoprecipitated, and the level of acetylation was analyzed by Western blotting with anti-Ac-K and anti-Myc antibodies. (e) Quantification of PARP1 deacetylation by SIRT1 in vivo. Values are means of the results for three experiments. (f) Hearts of SIRT1+/+ and SIRT1−/− mice were subjected to nuclear and cytoplasmic protein fractionations. Fractions were characterized by Western blotting with antibodies against fraction-specific proteins. PARP1 was immunoprecipitated from nuclear fractions and then analyzed by Western blotting with anti-Ac-K and anti-PARP1 antibodies. (g) Quantification of PARP1 acetylation in nuclear fractions of SIRT1 wild-type and knockout hearts. +, present; −, absent.
We then tested the ability of SIRT1 to deacetylate PARP1. In vitro-synthesized Flag-PARP1 was incubated with PCAF and [14C]Ac-CoA in HAT buffer. The acetylated Flag-PARP1 was immunoprecipitated and then incubated with beads containing in vivo-synthesized SIRT1 in HDAC buffer containing NAD plus TSA or NAM. In this experiment, we also incubated acetylated PARP1 with HDAC1 and HDAC4 in the appropriate buffers, which served as positive and negative controls, respectively (13). As shown in Fig. 3c, beads containing SIRT1 or HDAC1 had the ability to deacetylate PARP1 substantially, but not the beads with HDAC4. Previously, Hassa et al. have reported that aa 1 to 214 and aa 373 to 524 segments of PARP1 are acetylated (13). These regions include two SIRT1 binding domains of PARP1, the aa 1 to 214 and aa 477 to 524 domains (data not shown). We therefore asked whether these segments of PARP1 could be deacetylated by SIRT1. We found that SIRT1 was capable of deacetylating both the aa 1 to 214 and aa 477 to 524 segments of PARP1, whereas HDAC1 was effective for the deacetylation of the aa 477 to 524 segment, but not the aa 1 to 214 segment of PARP1 (data not shown). HDAC4 was incapable of deacetylating either PARP1 segment, consistent with previous results (13). These studies thus demonstrated that while the N-terminal aa 1 to 214 segment of PARP1 is specifically deacetylated by SIRT1, the automodification domain of PARP1 comprising the aa 477 to 524 region is targeted by both HDAC1 and SIRT1.
To demonstrate deacetylation of PARP1 by SIRT1 in vivo, Cos7 cells were overexpressed with Myc-PARP1 and PCAF together with SIRT1, HDAC1, or HDAC4. Cells were also treated with TSA or NAM to inhibit endogenous class I and II HDACs or class III HDACs, respectively. Subsequently, the cell lysate was prepared, and PARP1 was immunoprecipitated and analyzed by Western blotting using anti-Ac-K and anti-Myc antibodies. As shown in Fig. 3d and e, overexpression of PCAF led to acetylation of PARP1 in vivo, which was substantially reduced by overexpression of SIRT1 or HDAC1 but not HDAC4. These results thus indicated that SIRT1 and HDAC1 were capable of deacetylating PARP1 under in vivo conditions.
To confirm these findings, we examined the acetylation status of PARP1 in SIRT1 knockout mice. We prepared nuclear and cytoplasmic fractions of heart tissue obtained from SIRT1+/+ and SIRT1−/− mice. Both fractions were characterized by using fraction-specific protein antibodies. As shown in Fig. 3f, RNA polymerase II was present only in the nuclear fraction, and GAPDH was present only in cytoplasmic fraction, whereas tubulin was expressed both in the cytoplasmic and nuclear fractions, as expected. Expression of PARP1 was enriched in the nuclear faction, though a tiny portion of PARP1 was also detected in the cytoplasmic fraction. PARP1 was immunoprecipitated from nuclear fractions and then analyzed by Western analysis with anti-Ac-K antibody. The results of this experiment revealed that PARP1 was nearly threefold more acetylated in the nuclear fraction of SIRT1−/− than in the nuclear fraction of SIRT1+/+ mice (Fig. 3g). These results strongly indicated that SIRT1 plays a major role in regulating acetylation of PARP1 in vivo.
SIRT1-mediated deacetylation blocks PARP1 enzymatic activity.
To examine the effect of SIRT1 on the enzymatic activity of PARP1, we tested the poly(ADP-ribosyl)ation ability of acetylated and deacetylated enzymes. In vitro-synthesized Flag-PARP1 was acetylated with PCAF and then deacetylated with the SIRT1 wild type or the mutant (H355A) lacking deacetylase activity. Both acetylated and deacetylated PARP1s were then incubated with [32P]NAD in Tris buffer for auto-poly(ADP-ribosyl)ation of the enzyme. As shown in Fig. 4a, acetylated PARP1 was highly poly([32P]ADP-ribosyl)ated, compared to PARP1 not subjected to acetylation (compare lanes 1 and 2). Incubation of acetylated PARP1 with the SIRT1 wild type, but not the mutant, reduced the poly([32P]ADP-ribosyl)ation of PARP1 to nearly zero (lane 4), and that correlated with deacetylation status of the enzyme, thus suggesting that SIRT1 deactivates PARP1 by deacetylation. A similar experiment was also done with HDAC1 to test its ability to deactivate PARP1. As shown in Fig. 4b, HDAC1 was also capable of deactivating PARP1, but HDAC4, which served as a negative control, was not. HDAC1-mediated deactivation of PARP1 was again correlated with deacetylation of the enzyme, thus further confirming a role of reversible acetylation in the regulation of PARP1 activity. To demonstrate this effect in vivo, we infected cardiomyocytes with adenovirus vectors synthesizing the SIRT1 wild type or the mutant. After overnight infection, cardiomyocytes were subjected to mechanical stretch for 4 h. Afterwards, cells were harvested, and the cell lysate was analyzed by Western blotting using anti-P(ADP)R antibody. The results of this experiment again demonstrated that SIRT1 overexpression was capable of blocking the PARP1 activity mediated by cell stress (Fig. 4c).
FIG. 4.

Deacetylation by SIRT1 and HDAC1 blocks the PARP1 enzymatic activity. (a and b) Flag-PARP1 was acetylated by PCAF and Ac-CoA. Beads with acetylated Flag-PARP1 were separated and incubated with the SIRT1 wild type (wt), the SIRT1 mutant (mut), HDAC1, or HDAC4 in the appropriate deacetylase buffers. Subsequently, the beads with Flag-PARP1 were removed from the deacetylase buffer and suspended in ribosylation buffer containing [32P]NAD. Poly(ADP-ribosyl)ation of PARP1 was determined by autoradiography. Acetylation of PARP1 was determined by Western blotting with anti-Ac-K and anti-Flag antibodies. (c) Cardiomyocytes were infected with adenovirus vectors synthesizing wild-type or mutant SIRT1. Cells were then subjected to mechanical stretch for 4 h as described for Fig. 1. The cell lysate was analyzed by Western blotting with antibodies against P(ADP)R, PARP1, and tubulin. (d) Cardiomyocytes plated onto serum-free medium were treated with PE (20 μM), splitomicin (60 μM), and/or resveratrol (50 μM) for 24 h. PARP1 was immunoprecipitated and analyzed by Western blotting with the use of anti-Ac-K and anti-PARP1 antibodies. +, present; −, absent.
To confirm that endogenous SIRT1 was capable of deacetylating PARP1, we treated cardiomyocytes with a SIRT1 inhibitor, splitomicin, or an activator, resveratrol, and then stimulated cells with PE. Afterwards, PARP1 was immunoprecipitated from the cell lysate and analyzed for acetylation by Western blotting. As shown in Fig. 4d, PARP1 was acetylated by PE treatment of cells, as expected. Addition of splitomicin to these cultures further enhanced the PARP1 acetylation, while the resveratrol treatment did the opposite. These results thus demonstrated that endogenous SIRT1 was capable of regulating PARP1 acetylation in vivo.
Cellular NAD levels dictate SIRT1 and PARP1 interaction.
PARP1 activation is known to deplete cellular NAD levels. We therefore hypothesized that SIRT1 may deactivate PARP1 to preserve cellular NAD levels and thus retain its own function. If this is right, then cellular NAD levels are likely to play a role in the regulation of SIRT1-PARP1 interaction. To test this hypothesis, we examined the SIRT1 and PARP1 interaction in vitro in the absence or in the presence of gradually increasing concentrations of NAD in the buffer (Fig. 5a). Results of this experiment showed that SIRT1 strongly binds to PARP1 in the absence of NAD, and this binding gradually diminishes as the concentration of NAD in the buffer increases, being completely abolished at a 1 mM concentration of NAD in the buffer. Although the 1 mM concentration of NAD is nearly three times higher than the intracellular levels of NAD reported, the data obtained from this in vitro experiment (where protein concentrations used are also likely to be beyond the physiological range) suggested that NAD levels do play a role in controlling the SIRT1 and PARP1 interaction.
FIG. 5.

NAD levels dictate PARP1-SIRT1 interaction. (a) Beads containing Flag-SIRT1 were incubated with [35S]methionine-labeled PARP1 ([35S]PARP1) in a protein binding buffer containing either no NAD or gradually increasing concentrations of NAD. Beads were then separated and washed, and proteins bound to beads were analyzed by SDS-PAGE and then by autoradiography. (b) Flag-SIRT1 or Flag alone (negative control) was incubated with NAD (1 mM) in separate tubes. The NAD-bound Flag-SIRT1 or Flag was then incubated with [35S]methionine-labeled PARP1 not bound to NAD. Beads were separated, and coprecipitation of [35S]methionine-labeled PARP1 with Flag beads was analyzed by SDS-PAGE and then by autoradiography. (c) HeLa cells were treated with 100 μM of MNNG for 15 min. PARP1 was immunoprecipitated from the lysate, and the resulting beads were analyzed by Western analysis with anti-SIRT1 and anti-PARP1 antibodies. (d) Relative NAD levels in the same cell lysate (n = 4). (e) PARP1 and SIRT1 counteract each other's activity. Cos7 cells were cotransfected with the reporter plasmid (0.5 μg) and different combinations of expression plasmids (described in Materials and Methods) as shown below each bar diagram. Forty-eight hours following transfection, cells were harvested, and the luciferase activity was measured. The expression of β-Gal was used as a reference control. Values are means ± the standard errors (SE) of the results for five experiments. (f) NAD content levels in selected transfectants (n = 5). +, present; −, absent.
Since SIRT1 needs NAD to deacetylate PARP1, we asked next whether SIRT1 prebound with NAD is capable of associating with PARP1. For this experiment, in vitro-synthesized SIRT1 was saturated with NAD and then tested for its ability to coprecipitate in vitro-synthesized PARP1 which was not bound to NAD. As shown in Fig. 5b, SIRT1 bound with NAD was capable of coprecipitating PARP1 unbound to NAD (lane 3). These results thus indicated that NAD-bound SIRT1 retains its ability to bind to PARP1, which may be necessary to deactivate PARP1 by deacetylation in stress situations.
To confirm a role of NAD levels for SIRT1-PARP1 interaction in vivo, we treated Cos7 cells with 100 μM MNNG for 15 min, which was insufficient to induce cell death. Subsequently, the cell lysate was prepared, PARP1 was immunoprecipitated, and the resulting immune complex was analyzed for coprecipitation of SIRT1 by Western blotting. In the same lysate, NAD levels were also measured. The results of this experiment demonstrated that MNNG treatment for 15 min led to a decline in cellular NAD levels by nearly 30%. At this time point, a notable amount of SIRT1 was seen coprecipitated with PARP1, compared to controls not exposed to MNNG treatment, thus again suggesting that cellular NAD levels, in part, dictate SIRT1-PARP1 interaction (Fig. 5c and d).
To further confirm a role of cellular NAD levels in SIRT1-PARP1 interplay, we utilized a previously characterized reporter gene-based transcription assay, in which a Gal4DBD fusion system is used to measure the activity of both SIRT1 and PARP1 (23). In this assay system, the expression plasmid encodes a fusion protein with GAL4DBD fused to the SIRT1 catalytic core domain (GAL4DBD-mCORE), and the reporter plasmid has a luciferase reporter gene that is driven by the thymidine kinase minimum promoter and four tandem repeats of GAL4 binding sites. Cos7 cells were cotransfected with different combinations of plasmids, and 48 hours following transfection, cells were harvested and the reporter gene activity and the cell NAD levels measured. As shown in Fig. 5e, the reporter gene activity was substantially repressed by overexpression of the GAL4DBD-mCORE plasmid, compared to expression of the GAL4DBD control, but not by the mCORE mutant plasmid (Fig. 5e, lanes 1, 2, and 3), as expected. When cells were cotransfected with a higher ratio of PARP1 plasmids to the GAL4DBD-mCORE plasmid, the transcription repression activity of mCORE was blocked by PARP1 overexpression, and this was accompanied with a nearly 50% decline in cellular NAD levels (Fig. 5e and f, lanes 4), suggesting that PARP1-mediated depletion of cellular NAD levels hampers the activity of SIRT1 catalytic core domain, consistent with previous results (23).
Because our earlier experiments demonstrated that the acetylation of PARP1 by PCAF activates PARP1 enzymatic activity, we overexpressed cells with suboptimal concentrations of PARP1, which was incapable of blocking the activity of the SIRT1 core domain, together with increasing concentrations of PCAF or the corresponding mutant (Fig. 5e, lanes 6 to 9). The results of this experiment demonstrated that, while PCAF alone had no effect on the activity of the mCORE plasmid (Fig. 5e, lane 5), in combination with PARP1, it eliminated the activity of the SIRT1 core domain in a concentration-dependent manner (Fig. 5e, lanes 7 to 9). Measurement of the NAD levels of these transfectants indicated that reduced activity of the mCORE plasmid was again accompanied by significantly decreased levels of cellular NAD (Fig. 5e and f, compare lanes 6 and 9). Since SIRT1 is capable of deactivating PARP1, we next asked whether overexpression of the mCORE plasmid can neutralize the effect of PARP-PCAF combination in this assay system. We transfected cells with a threefold larger amount of mCORE plasmid together with the same amounts of PARP1 and PCAF plasmids as before. As shown in Fig. 5e, lane 10, larger amounts of the Gal4DBD-mCORE plasmid completely blocked the effect of PARP1-PCAF combination, and the reporter gene silencing activity of the SIRT1 core domain was restored back to near control levels. Measurement of the NAD levels of these cells revealed that the SIRT1 (GAL4DBD-mCORE) overexpression prevented the PARP1-PCAF-mediated decline in cellular NAD levels (Fig. 5f, compare lanes 9 and 10). These results together further confirmed that (i) PARP activity is regulated in vivo by PCAF and SIRT1 in opposite fashions and (ii) cellular NAD levels, in part, play a role in the interplay between PARP1 and SIRT1.
SIRT1 inhibits the transcription activity of the PARP1 gene.
The results mentioned above demonstrate that SIRT1 controls PARP1 activity at the posttranslational level. In our earlier experiments, we noticed that during hypertrophy of cardiomyocytes, PARP1 is not only activated, but its proteins levels are also highly elevated (24). We therefore hypothesized that for SIRT1 to control PARP1 activity, it may be necessary for the deacetylase to modulate the PARP1 gene expression as well. To test this hypothesis we overexpressed cardiomyocytes with wild-type SIRT1 or its mutant by use of adenovirus vectors. Cells were then treated with the hypertrophy agonist PE for 48 h. Subsequently, the cell lysate was analyzed by Western blotting for the expression of endogenous PARP1 levels. We found consistently reduced levels of PARP1 in cells overexpressed with wild-type SIRT1, but not in those overexpressed with the mutant, whereas the expression levels of β-actin and β-Gal, which was used to monitor transfection efficiency, remained unaltered, thus suggesting the specificity of the effect of SIRT1 on PARP1 expression (Fig. 6a and b). Reduced expression level of a protein could be related to increased degradation or to reduced synthesis of the protein. To determine whether increased degradation of protein had contributed to decreased levels of PARP1, we examined the effect of two protein degradation blockers, MG132 and leupeptin, which prevent protein degradation by inhibiting proteasomes and lysosomes, respectively. We found, however, no increase in PARP1 levels in SIRT1-expressing cells by using either inhibitor (data not shown). We then examined the steady-state levels of PARP1 mRNA in different groups of cells by real-time PCR analysis (Fig. 6c). The results of this experiment demonstrated that SIRT1 overexpression significantly reduced the levels of endogenous PARP1 mRNA transcripts, and that correlated with the reduced protein levels observed by the Western analysis, suggesting that SIRT1 may negatively regulate the PARP1 gene expression. To obtain direct evidence for the role of SIRT1 in regulating PARP1 gene expression, we analyzed the effect of the deacetylase on the promoter activity of the PARP1 gene. Cells were cotransfected with a reporter plasmid that has the CAT reporter gene driven by the −237 bp upstream promoter region of PARP1 gene, together with expression plasmids synthesizing the SIRT1 wild type or the mutant. As shown in Fig. 6d and e, the CAT reporter gene expression was dramatically reduced in cells overexpressed with the SIRT1 wild type, but not in those overexpressed with the mutant, suggesting that SIRT1 negatively regulates the activity of the PARP1 gene promoter. Based on these results, we conclude that SIRT1 is capable of regulating the activity of PARP1 at the transcriptional as well as at the posttranslational levels.
FIG. 6.

SIRT1 negatively regulates PARP1 gene expression. (a) Cardiomyocytes were overexpressed with the SIRT1 wild type (10 and 20 MOI) or the mutant (20 MOI) by use of adenovirus vectors and were then stimulated by PE (20 μM) for 48 h. Cells were also infected with β-Gal adenovirus (10 MOI), which served as the negative control. The cell lysate was analyzed by Western blotting with anti-PARP1, antiactin, and β-Gal antibodies. In the same cell lysate, DNA content was also measured. (b) Quantification of PARP1 content in different groups of cardiomyocytes. (c) Endogenous levels of PARP1 mRNA in different groups of cardiomyocytes, measured by real-time PCR. (d) Cos7 cells were cotransfected with the CAT reporter plasmid, in which the CAT gene was driven by the −237 bp upstream promoter region of the PARP1 gene (PARPPro-CAT), and the expression plasmids encoding either wild-type (wt) or mutant (mut) SIRT1. The gel image shows the amplification of CAT transcripts from different groups of cells under identical conditions. The actin transcript was used as a reference control. (e) CAT mRNA levels in different groups of transfections, measured by real-time PCR and normalized to β-Gal mRNA. All values are means ± SE of the results for four to six different experiments. +, present; −, absent.
To further understand the role of NAD in the interplay between PARP1 and SIRT1, we measured the affinity constant (Km) of both enzymes by utilizing the Michaelis-Menten formulation. The estimated Km of SIRT1 for deacetylation of PARP1 is 1.2 μM, which is consistent with a Km (1.2 μM) of SIRT1 measured for another peptide (CISBIO) (data not shown). The Km of PCAF-acetylated PARP1 is 995 nM, which is close to the Km value of 1.1 μM reported previously for pERK-activated PARP1 (4) (data not shown). These similar Km values for SIRT1 and Ac-PARP1 indicate that both enzymes might be equally active at a low concentration of NAD. It should be noted, however, that these values might differ for endogenous reactions of stressed cells.
Deacetylation of PARP1 by SIRT1 prevents cell death.
In order to demonstrate functional significance of PARP-SIRT1 interplay, we examined the effect of SIRT1 on PARP-mediated cell death (Fig. 7a and b). HeLa cells were treated with 3AB (4 mM), a PARP1 inhibitor, or MNNG (500 μM), a PARP1 activator, for 3 h in the presence or absence of TSA, a class I and II HDAC inhibitor. We found that MNNG treatment alone induced cell death in ∼70% of cells, whereas a combination of MNNG and TSA led to nearly 100% cell death. When cells were treated with 3AB, we found that, while it had no effect on control cells, the cell death induced by the combination of MNNG and TSA was reduced dramatically by 3AB treatment, suggesting involvement of PARP1 activation in MNNG-plus-TSA-mediated cell death. We then examined the effect of SIRT1 on this type of cell death. Cells were overexpressed with the SIRT1 wild type or the mutant and then treated with MNNG either alone or together with TSA. The results demonstrated that overexpression of the SIRT1 wild type, but not that of the mutant, markedly reduced cell death induced by MNNG alone as well as by that induced by the combination of MNNG and TSA. These results thus strongly indicated that SIRT1 was capable of blocking cell death induced by PARP1 activation.
FIG. 7.

SIRT1 overexpression blocks cell death induced by acetylated PARP1. HeLa cells were overexpressed with the SIRT1 wild type or the mutant protein. The next day, cells were treated for 4 h with 3AB with TSA overnight and/or for 3 h with MNNG in different combinations. (a) The extent of cell death was measured by estimating the percentage of annexin V-positive cells by fluorescence-activated cell sorter analysis. (b) Quantification of cell death in different treatment groups. Values are means ± SE of the results for three experiments. Asterisks indicate that the rate of cell death is significantly (P < 0.01) lower than that for the MNNG-plus-TSA-treated group. (c) Schematic model illustrating interplay between PARP1 and SIRT1. Cell stress induces acetylation of PARP1 leading to its activation. In cardiomyocytes, stress also induces synthesis of new PARP1 that again feeds into PARP1 activation. Activated PARP1 promotes poly(ADP-ribosyl)ation of itself as well as that of other proteins by adding multiple ADP-ribose units from NAD to the target protein. This reaction consumes cellular NAD stores and generates NAM as a by-product. Reduction of cellular NAD levels promotes interaction between SIRT1 and PARP1, resulting in deactivation of PARP1. SIRT1 is also capable of suppressing the activity of the PARP1 gene promoter, leading to decreased PARP1 protein synthesis. Both these mechanisms help cells to survive under mild stress conditions. Under severe stress conditions, however, depletion of NAD levels beyond certain critical levels inhibits the activity of SIRT1, leading to hyperacetylation of target proteins, such as P53, Ku70 and PARP1, which eventually proceeds to cell death.
DISCUSSION
The interplay of PARP1 and SIRT1 was predicted before because of the dependence of both enzymes on the same substrate, NAD (36). Previous reports from this laboratory have demonstrated that during stress, PARP1 overexpression depletes cellular NAD levels and that this leads to suppression of the deacetylase activity of SIRT1 (23). In this study, we provided evidence for the other arm of the interplay. We demonstrate a new mode of PARP1 activation by PCAF-mediated acetylation, which is independent of DNA damage. SIRT1 has the ability to physically bind to PARP1 and deacetylate it, and that results in suppression of PARP1 enzymatic activity. In addition, we show that SIRT1 is also capable of negatively regulating expression of the PARP1 gene under stress conditions. These data (and the data reported previously) provide strong evidence for the existence of a functional interplay between PARP1 and SIRT1 and give novel insights into the cell survival/death pathways modulated by SIRT1 during stress.
Acetylation of PARP1 enhances its enzymatic activity.
PARP1 is abundantly expressed in cardiomyocytes, and it was shown to be activated during hypertrophy of myocytes where no evidence of DNA damage could be detected (23). Several lines of evidence presented in this study show that PARP1 is activated by acetylation during stress of cardiomyocytes. Notably, (i) mechanical stretch of myocytes for 4 h, when no DNA damage or cell death could be detected, was sufficient to cause massive protein poly(ADP-ribosyl)ation; (ii) increased poly (ADP-ribosyl)ation of cardiac proteins was associated with acetylation of PARP1; and (iii) increased poly(ADP-ribosyl)- ation of proteins was blocked by overexpression of SIRT1, thus indicating that acetylation of PARP1 had indeed contributed to the increased PARP1 activity. These results are consistent with a previous report in which angiotensin II treatment, a known inducer of oxidative stress, was shown to produce hypertrophy only in wild-type mice, but not in PARP1-deficient mice (25). There are other reports showing that increased protein acetylation contributes to hypertrophy of myocytes during stress situations (8, 9). Data presented in this study demonstrate that PARP1 might be one of the downstream targets of stress stimuli causing induction of hypertrophy and death of cardiomyocytes.
More direct evidence for acetylation-mediated activation of PARP1 was obtained by in vitro studies, in which incubation with PCAF was found to be sufficient to increase the formation of P(ADP)R polymers. PARP1 has been shown before to be acetylated by p300/CBP, and that promotes its ability to regulate NF-κB-dependent gene transcription (12, 13). Hassa et al. reported that the PARP1 domain corresponding to aa 373 to 525 is highly acetylated by p300/CBP (13). Within this region, they have identified five different lysine residues acetylated by p300/CBP (13). These lysine residues fall within one of the PARP1 fragments (aa 477 to 525 segment) that we identified as a target of PCAF-mediated acetylation. In addition, we have found that the N-terminal aa 1 to 214 segment of PARP1 is also highly acetylated by PCAF. Acetylation of this (aa 1 to 214) segment of PARP1 by p300/CBP was also noticed before, but it was acetylated to a much lesser extent than were the other segments (13). In our experiments, we found that the aa 1 to 214 segment of PARP1 is as good a substrate as the aa 477 to 525 segment is for PCAF-mediated acetylation. These results suggest that PCAF and p300/CBP may have different preferences for different lysine residues of PARP1. Since PCAF and p300/CBP have been shown to be activated by common as well as by different stimuli at different stages of development, it is not unreasonable to anticipate that they may differently control PARP1 activity in different situations (6, 26, 33).
Results obtained with the use of DNA indicated that activation of PARP1 by acetylation was independent of DNA damage. Similar results were reported previously for pERK2-mediated activation of PARP1 (4). These findings revealed participation of PARP1 activation in events that are not necessarily related to the repair of DNA damage. These events may include recruitment of PARP1 by transcription factors, such as AP2, TEF1, or NF-κB, at the specific gene-regulatory sites in the DNA as well as binding of PARP1 to histones for chromatin remodeling (2, 12, 19). Therefore, it appears that besides the DNA machinery, there are alternative mechanisms of PARP1 activation; these include acetylation (this study) and phosphorylation (17), as well as interaction of PARP1 with other proteins as reported before (4).
SIRT1 physically binds to PARP1 and regulates its enzymatic activity.
With respect to deacetylation of PARP1, it has been reported that the members of class I HDACs, HDAC1, -2, and -3, are capable of deacetylating PARP1, but HDAC8 and class II HDACs, HDAC4 and -5, are not (13). In this study, we found that SIRT1 was also capable of deacetylating PARP1. A single point mutation in the catalytic core domain of SIRT1, which eliminated its deacetylase activity, was capable of binding to PARP1 (not shown); however, it failed to suppress the synthesis of P(ADP)R polymers, suggesting that the catalytic activity, and not the protein binding ability of SIRT1, was necessary for blocking the PARP1 activity. Because the conserved core domain (mCORE) of SIRT1 was sufficient to cancel the PARP1 effect, it is likely that other sirtuin analogues which carry deacetylase activity will also have potential to regulate the activity of PARP1. In our experiments, HDAC1 and SIRT1 both were capable of deacetylating the aa 477 to 525 segment of PARP1, and both were able to block its catalytic activity, suggesting that there could be redundancy between these two deacetylases in regulating certain modes of PARP1 activation. We also noticed that SIRT1 has a unique ability to deacetylate the aa 1 to 214 segment of the PARP1. Although the functional significance of this effect of SIRT1 is not known to us at present, deacetylation of full-length PARP1 by SIRT1 was notably higher than that which occurred with HDAC1, suggesting that this may be related to the ability of SIRT1 to deacetylate the aa 1 to 214 region of PARP1. Because this region of PARP1 is involved in DNA binding, it is likely that SIRT1 also plays a role in controlling the DNA binding ability of the enzyme (30). Studies are currently under way in our laboratory to understand this effect.
Functional significance of PARP1-SIRT1 interaction.
In many instances, PARP1 has been shown to exert an effect opposite to that elicited by SIRT1 for the same target. For example, poly(ADP-ribosyl)ation of P53 promotes its nuclear accumulation and increases its transcriptional activity, whereas SIRT1 deacetylates P53 and decreases its transcriptional activity (16, 21). The same is true for other common targets of PARP1 and SIRT1, such as NF-κB, FOXO, and Ku70 (3, 21, 22, 34). Therefore, in order for cells to proceed with one enzyme, it may be necessary to control the activity of the other. The results presented here demonstrate that both PARP1 and SIRT1 have the ability to counterbalance each other's activity. In Fig. 7, a scheme is presented showing how they might regulate each other's activity. During stress situations, PARP1 is activated by acetylation leading to poly(ADP-ribosyl)ation of the target proteins. Mild activation of PARP1 can regulate various physiologic functions by modulating different cellular mechanisms, e.g., chromatin structure, gene transcription, and cell metabolism (30). In situations of severe stress, overactivation of PARP1 can deplete cellular NAD stores, leading to repression of the activity of SIRT1 and other NAD-dependent pathways (18, 23). Cellular NAD levels also control the transcription of the SIRT1 gene (37). It is therefore likely that reduced NAD levels after PARP1 activation suppress the SIRT1 activity, not only by reducing the availability of NAD for the deacetylase reaction, but also by suppressing synthesis of the SIRT1 protein. This argument is consistent with a previous study in which exogenous addition of NAD was found to be capable of activating SIRT1 and blocking the detrimental effects of PARP1 (23, 25).
To check this damaging mechanism, the cell has designed a clever mechanism to block the activity of PARP1 by SIRT1, before it becomes itself inert due to decreases in NAD levels. SIRT1 blocks the PARP1 activity by deacetylation, and that helps to maintain cellular NAD levels. It should be noted, however, that SIRT1 could potentially also block the activity of PARP1 by deacetylating and inhibiting the activity of acetyltransferases. Both p300/CBP and PCAF had been shown to be targeted by SIRT1 to inhibit the acetylation status of these enzymes (3, 5, 22). In our studies, because SIRT1 was capable of blocking the PARP1 activity in in vitro assays, it was clear that the deacetylase has a direct effect on regulating the activity of PARP1. From the viewpoint of enzyme kinetics, the calculated Km of SIRT1 is 1.2 μM, which is nearly same as the Km of 995 nM of acetylated PARP1. These values indicate that during stress, both enzymes will be functionally active, even in subcellular compartments (such as the nucleus) where NAD levels are relatively low (compared to those in the mitochondria). Because of such a low Km value of acetylated PARP1, it may be necessary to block the activity of PARP1, not only at the posttranslational level but also at the transcriptional level. We have found that SIRT1 is capable of regulating the activity of PARP1 at both levels.
In summary, our data show for the first time that PARP1 activity is regulated by acetylation of the protein. SIRT1 is capable of deacetylating and deactivating PARP1, whereas PARP1 regulates SIRT1 activity by depleting cellular NAD levels. It should be noted, however, that PARP1 may also directly control the activity of SIRT1 by poly(ADP-ribosyl) ation of the deacetylase, a possibility which needs to be explored. Because PARP1 and SIRT1 have many common targets, it is likely that the interplay between these two molecules is one of the important regulatory mechanisms that determine the cellular decision between life and death. These findings have deep implications for various pathological conditions and may pave the way for designing future therapeutic strategies.
Acknowledgments
We thank the following investigators for their generous gift of plasmids used in this study: S. Guerin, H. Boulares, W. Gu, and S. Imai. We also thank M.W. McBurney for providing SIRT1 knockout mice.
This study was partially supported by NIH grants RO1 HL-77788, HL-83423, HL087823, and PO1 HL058064.
Footnotes
Published ahead of print on 26 May 2009.
REFERENCES
- 1.Altman, S. A., L. Randers, and G. Rao. 1993. Comparison of trypan blue dye exclusion and fluorometric assays for mammalian cell viability determinations. Biotechnol. Prog. 9671-674. [DOI] [PubMed] [Google Scholar]
- 2.Butler, A. J., and C. P. Ordahl. 1999. Poly(ADP-ribose) polymerase binds with transcription enhancer factor 1 to MCAT1 elements to regulate muscle-specific transcription. Mol. Cell. Biol. 19296-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen, H. Y., S. Lavu, K. J. Bitterman, B. Hekking, T. A. Imahiyerobo, C. Miller, R. Frye, H. Ploegh, B. M. Kessler, and D. A. Sinclair. 2004. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell 13627-638. [DOI] [PubMed] [Google Scholar]
- 4.Cohen-Armon, M., L. Visochek, D. Rozensal, A. Kalal, I. Geistrikh, R. Klein, S. Bendetz-Nezer, Z. Yao, and R. Seger. 2007. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol. Cell 25297-308. [DOI] [PubMed] [Google Scholar]
- 5.Fulco, M., R. L. Schiltz, S. Iezzi, M. T. King, P. Zhao, Y. Kashiwaya, E. Hoffman, R. L. Veech, and V. Sartorelli. 2003. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol. Cell 1251-62. [DOI] [PubMed] [Google Scholar]
- 6.Goodman, R. H., and S. Smolik. 2000. CBP/p300 in cell growth, transformation, and development. Genes Dev. 141553-1577. [PubMed] [Google Scholar]
- 7.Gupta, M., S. Samant, S. Smith, and S. Shroff. 2008. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J. Biol. Chem. 28310135-10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gusterson, R., B. Brar, D. Faulkes, A. Giordano, J. Chrivia, and D. Latchman. 2002. The transcriptional co-activators CBP and p300 are activated via phenylephrine through the p42/p44 MAPK cascade. J. Biol. Chem. 2772517-2524. [DOI] [PubMed] [Google Scholar]
- 9.Gusterson, R. J., E. Jazrawi, I. M. Adcock, and D. S. Latchman. 2003. The transcriptional co-activators CREB-binding protein (CBP) and p300 play a critical role in cardiac hypertrophy that is dependent on their histone acetyltransferase activity. J. Biol. Chem. 2786838-6847. [DOI] [PubMed] [Google Scholar]
- 10.Ha, H. C., and S. H. Snyder. 1999. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 9613978-13982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haigis, M. C., and L. P. Guarente. 2006. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev. 202913-2921. [DOI] [PubMed] [Google Scholar]
- 12.Hassa, P. O., C. Buerki, C. Lombardi, R. Imhof, and M. O. Hottiger. 2003. Transcriptional coactivation of nuclear factor-kappaB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. J. Biol. Chem. 27845145-45153. [DOI] [PubMed] [Google Scholar]
- 13.Hassa, P. O., S. S. Haenni, C. Buerki, N. I. Meier, W. S. Lane, H. Owen, M. Gersbach, R. Imhof, and M. O. Hottiger. 2005. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J. Biol. Chem. 28040450-40464. [DOI] [PubMed] [Google Scholar]
- 14.Jacobson, E. L., and M. K. Jacobson. 1976. Pyridine nucleotide levels as a function of growth in normal and transformed 3T3 cells. Arch. Biochem. Biophys. 175627-634. [DOI] [PubMed] [Google Scholar]
- 15.Kaeberlein, M., M. McVey, and L. Guarente. 1999. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 132570-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanai, M., K. Hanashiro, S. H. Kim, S. Hanai, A. H. Boulares, M. Miwa, and K. Fukasawa. 2007. Inhibition of Crm1-p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat. Cell Biol. 91175-1183. [DOI] [PubMed] [Google Scholar]
- 17.Kauppinen, T. M., W. Y. Chan, S. W. Suh, A. K. Wiggins, E. J. Huang, and R. A. Swanson. 2006. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc. Natl. Acad. Sci. USA 1037136-7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolthur-Seetharam, U., F. Dantzer, M. W. McBurney, G. de Murcia, and P. Sassone-Corsi. 2006. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle 5873-877. [DOI] [PubMed] [Google Scholar]
- 19.Kraus, W. L., and J. T. Lis. 2003. PARP goes transcription. Cell 113677-683. [DOI] [PubMed] [Google Scholar]
- 20.Langley, E., M. Pearson, M. Faretta, U. M. Bauer, R. A. Frye, S. Minucci, P. G. Pelicci, and T. Kouzarides. 2002. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 212383-2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo, J., A. Y. Nikolaev, S. Imai, D. Chen, F. Su, A. Shiloh, L. Guarente, and W. Gu. 2001. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107137-148. [DOI] [PubMed] [Google Scholar]
- 22.Motta, M. C., N. Divecha, M. Lemieux, C. Kamel, D. Chen, W. Gu, Y. Bultsma, M. McBurney, and L. Guarente. 2004. Mammalian SIRT1 represses forkhead transcription factors. Cell 116551-563. [DOI] [PubMed] [Google Scholar]
- 23.Pillai, J. B., A. Isbatan, S. Imai, and M. P. Gupta. 2005. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J. Biol. Chem. 28043121-43130. [DOI] [PubMed] [Google Scholar]
- 24.Pillai, J. B., H. M. Russell, J. Raman, V. Jeevanandam, and M. P. Gupta. 2005. Increased expression of poly(ADP-ribose) polymerase-1 contributes to caspase-independent myocyte cell death during heart failure. Am. J. Physiol. Heart Circ. Physiol. 288H486-H496. [DOI] [PubMed] [Google Scholar]
- 25.Pillai, J. B., M Gupta, S. B. Rajamohan, R. Lang, J. Raman, and M. P. Gupta. 2006. Poly(ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 291H1545-H1553. [DOI] [PubMed] [Google Scholar]
- 26.Puri, P. L., V. Sartorelli, X. J. Yang, Y. Hamamori, V. V. Ogryzko, B. H. Howard, L. Kedes, J. Y. Wang, A. Graessmann, Y. Nakatani, and M. Levrero. 1997. Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol. Cell 135-45. [DOI] [PubMed] [Google Scholar]
- 27.Revollo, J. R., A. A. Grimm, and S. Imai. 2004. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 27950754-50763. [DOI] [PubMed] [Google Scholar]
- 28.Rogina, B., and S. L. Helfand. 2004. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA 10115998-16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saunders, L. R., and E. Verdin. 2007. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene 265489-5504. [DOI] [PubMed] [Google Scholar]
- 30.Schreiber, V., F. Dantzer, J. C. Ame, and G. de Murcia. 2006. Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 7517-528. [DOI] [PubMed] [Google Scholar]
- 31.Szabo, C., P. Pacher, and R. A. Swanson. 2006. Novel modulators of poly(ADP-ribose) polymerase. Trends Pharmacol. Sci. 27626-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tissenbaum, H. A., and L. Guarente. 2001. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410227-230. [DOI] [PubMed] [Google Scholar]
- 33.Wong, K., J. Zhang, S. Awasthi, A. Sharma, L. Rogers, E. F. Matlock, C. Van Lint, T. Karpova, J. McNally, and R. Harrod. 2004. Nerve growth factor receptor signaling induces histone acetyltransferase domain-dependent nuclear translocation of p300/CREB-binding protein-associated factor and hGCN5 acetyltransferases. J. Biol. Chem. 27955667-55674. [DOI] [PubMed] [Google Scholar]
- 34.Yeung, F., J. E. Hoberg, C. S. Ramsey, M. D. Keller, D. R. Jones, R. A. Frye, and M. W. Mayo. 2004. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 232369-2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaniolo, K., A Rufiange, S. Leclerc, S. Desnoyers, and S. L. Guerin. 2005. Regulation of the poly(ADP-ribose) polymerase-1 gene expression by the transcription factors Sp1 and Sp3 is under the influence of cell density in primary cultured cells. Biochem. J. 389423-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang, J. 2003. Are poly(ADP-ribosyl)ation by PARP-1 and deacetylation by Sir2 linked? Bioessays 25808-814. [DOI] [PubMed] [Google Scholar]
- 37.Zhang, Q., S. Y. Wang, C. Fleuriel, D. Leprince, J. V. Rocheleau, D. W. Piston, and R. H. Goodman. 2007. Metabolic regulation of SIRT1 transcription via a HIC1:CtBP corepressor complex. Proc. Natl. Acad. Sci. USA 104829-833. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]