

Metal-sensitive fluorescent probes have been shown to accurately and rapidly report metal complexation through a change in the fluorophore's spectral properties1-3. Previous studies have focused on quasi-static metal-fluorophore complexes, where a single metal of interest complexes with the fluorophore, altering its emission in some discernable fashion. The resultant emission could be further modified by changing the ligands attached to metals or altering the metal oxidation states. Neither experimental nor theoretical studies have been conducted regarding dynamic metal-fluorophore complexes in which the nature of the bound metal itself is variable. Such changes, for example, may transform a non-fluorescent complex into a fluorescent system if a quencher metal could convert into a non-quencher metal. The difficulty in achieving this transformation arises from the high stability of metals. However, a select group of metals such as radionuclides, is ideally suited for this application because spontaneous radioactivity decay of an unstable parent metal could transform it to a different, more stable metal.
Herein we propose the first molecular system that demonstrates the feasibility of altering the fluorescence properties of an organic dye as a function of radionuclide decay. Previous studies have shown that 64Cu decays to 64Zn and 64Ni through two pathways, as shown in the following equation:
We found a clear differentiation in fluorescence between Cu2+ and its decay products, with Cu2+ acting as a strong quencher, Zn2+ as an enhancer, and Ni2+ having no net effect on the dye fluorescence. Correlation of the radionuclide decay with the dye fluorescence enhancement validates the hypothesized metal effect and suggests a realm of possible applications for complementing fluorescence with radioactivity.
To study the influence of inter-metal conversion through radioactive decay on the fluorescence properties of organic dyes, we prepared LS479, a novel near-infrared fluorescent dye with a covalently linked metal chelating group (Scheme 1). LS479 is structurally similar to a known dye 1,1′,3,3,3′,3′-hexamethylindotricarbocyanine (HITC), which is widely used in analytical chemistry and biological optical imaging studies4. The chelating group is derived from diethylenetriamine pentaacetic acid (DTPA). The synthesis of LS479 (Scheme 1) involves the methylation of 5-nitro-2,3,3-trimethylindolenine to provide the indole analog 2 in a 60% yield. Reduction of the nitro group, followed by N-alkylation using N,N-bis[(tert-butyloxycarbonyl) methyl]-2-bromoethylamine 1 afforded the indole derivative 4 in an overall yield of 65%. Treatment of 1,2,3,3-tetramethylindolinium iodide with glutacondialdehyde dianil, acetyl chloride, and acetic anhydride produced the hemicyanine intermediate 5 in an 87% yield. Reaction of 5 with the indole derivative 4 under reflux in anhydrous ethanol with anhydrous sodium acetate afforded Boc-protected intermediate 6, which was isolated in good yield (74.5%). The hydrolysis of 6 to afford LS479 was accomplished with TFA and the desired product was purified by preparative HPLC.
Scheme 1.
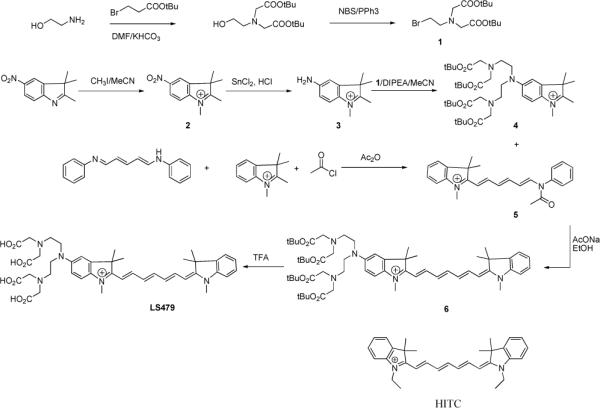
Synthesis of LS479 and structure of HITC
We used absorption studies to establish that metal complexation would not be lost upon radioactive decay of 64Cu2+ and that the parent and daughter nuclides remain bound to LS479. A solution of HITC or LS479 in water was titrated with Cu2+, Ni2+, and Zn2+ chlorides and the absorption and emission spectra were recorded. HITC, lacking the chelating group, did not show any isosbestic points, and the shape of the absorption spectra upon metal titration remained the same (see SI Fig. S1-S3), suggesting the absence of metal chelation to the dye molecule. Similarly, we did not observe significant changes in the fluorescence of HITC upon addition of metal salts (see SI Fig. S4). In contrast, transformations in the absorption spectra (see SI Fig. S5-S7) of LS479 showed that all the metals remained bound to the dye. In all cases, clear isosbestic points were observed, especially for Zn2+ and Ni2+.
Analysis of the fluorescence spectra showed that all three metals displayed different effects on emission of LS479 (Fig. 1). Cu had a strong quenching ability, achieving maximum quenching at a 1:1 molar ratio to the dye. The fluorescence intensity of Ni2+-LS479 complex remained the same as that of LS479 at all molar ratios examined. Furthermore, Zn significantly enhanced the fluorescence of LS479, with the emission of a 1:1 mol ratio of the Zn2+-LS479 complex approximately 5 times higher than the intensity of LS479. The metal effects on fluorescence and absorption of LS479 were concentration dependent. The metal effects on the dye fluorescence were mostly linear until approximately 1:1 ratio of metal to dye was established. Obviously, at this point, all the dye molecules were chelated to the metal and no free dye existed in solution. Further addition of metal resulted in minimal alteration of the spectra.
Figure 1.

Left panel: titration emission spectra of LS479 with CuCl2 (top), NiCl2 (middle), ZnCl2 in water (excitation 675 nm). Arrows indicate the increase of metal concentration. Copper was shown to quench, zinc enhanced fluorescence, and nickel had no net effect. Right panel: fluorescence intensity at the maximum emission (760 nm) as a function of metal to dye ratio. (last statement is removed)
Having demonstrated that Cu2+ was a fluorescence quencher of LS479, Ni2+ did not have an effect, and Zn2+ significantly enhanced fluorescence, we hypothesized that a solution of completely decayed 64CuCl2 would possess similar effects on the fluorescence spectra of LS479 as a result of the daughter nuclides Ni2+ and Zn2+. Considering that the half-life of 64Cu is 12.7 h, we used a completely decayed solution of 64CuCl2 in water (> 12 half-lives), which ensured that the solution contained 61% 64Ni2+ and 39% 64Zn2+ in addition to any other incipient metals5. The remaining amount of 64Cu2+ was virtually zero. As expected, the emission spectra were drastically different from “cold” Cu2+ titration spectra (Fig. 2). The spectral changes induced by decayed 64Cu2+ on LS479 were similar to those caused by a model daughter isotope mixture (Ni:Zn ratio ~1.5) of 64Cu2+ decay (see SI Fig. S8-S9 for absorption spectra).
Figure 2.
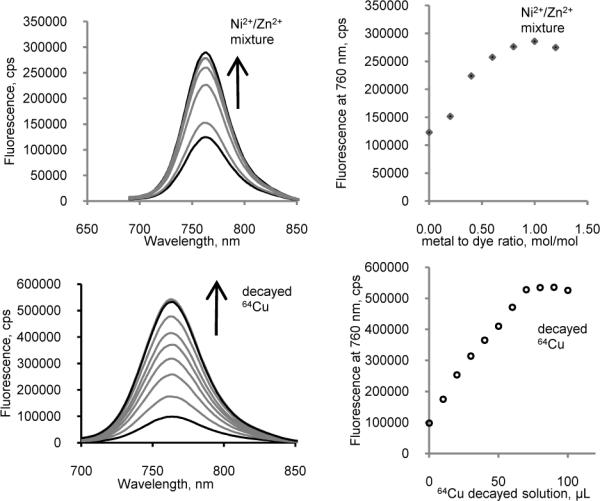
Left panel: titration emission spectra of LS479 with a mixture [61% natNi2+ + 39% natZn2+] (top) and decayed 64Cu2+ (bottom) (ex. 675 nm). Right panel: fluorescence intensity at 760 nm as a function of decayed 64Cu addition, where the saturation point apparently corresponds to a 1:1 metal to dye ratio. Addition of either solution to LS479 resulted in marked fluorescence enhancement.
The above results using “cold” metal models suggest that “hot” Cu would quench the fluorescence of LS479. Indeed, addition of 64Cu2+ to a solution of LS479 rapidly quenched the dye fluorescence with a concomitant increase in fluorescence over time (Fig. 3).There were no changes in the absorbance spectra of LS479 which indicates that the observed fluorescence increases were not diminished or altered by radiolysis. The resultant fluorescence enhancement reflected the transformation of 64Cu2+ into 64Ni2+ and 64Zn2+. As 64Cu further decayed to 64Ni2+ and 64Zn2+, the quenching effect decreased and the fluorescence increased to 130% relative to the emission of unquenched LS479 for a net enhancement of fluorescence. After 72 h, nearly all of the 64Cu decayed into stable 64Ni2+ and 64Zn2+ (2% of initial activity remaining). Consequently, there were no changes in fluorescence intensity thereafter.
Figure 3.
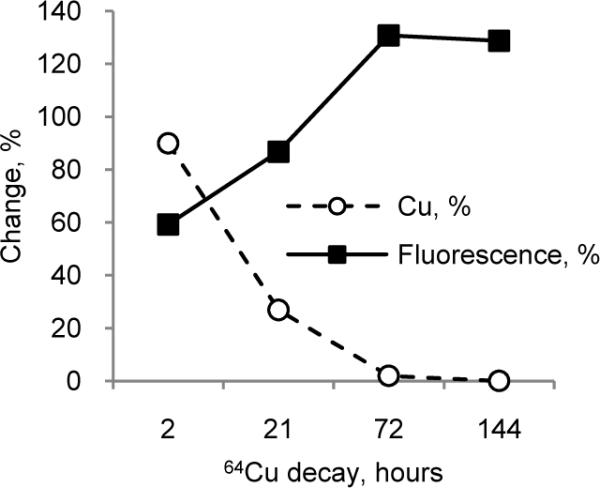
Change in 64Cu2+ (%) concentration as a result of decay (calculated from exponential decay) and change in relative fluorescence of Cu-LS479 (%) solution as a function of time. Unquenched LS479 (0.5 μ-M) corresponded to a hypothetical time-zero point (F0t) for fluorescence. Relative fluorescence was measured as an area under the emission curve after addition of 64Cu solution divided by the area before the addition (ex. 675 nm). An overall increase in fluorescence intensity was seen as a function of time.
The fluorescence quenching by Cu2+ is a general phenomenon with few exceptions6, spanning from organic molecules2 to quantum dots7 and fluorescent proteins8. However, the mechanism of fluorescence quenching still remains obscure3. Quenching has been attributed to spheres of action static quenching mechanism8, paramagnetic nature of the metal9, heavy-atom effect10, energy transfer from the dye (donor) to Cu2+ (acceptor)11, and charge transfer from π* to d-orbitals12. We ruled out the first mechanism, spheres of action static quenching, because it considers a certain probability of collision quenching13 which is not the case in Cu2+-LS479 complex where the probability reaches 100%, with maximum quenching occurring at 1:1 metal-dye ratio. Paramagnetism is also an inadequate explanation because another paramagnetic complex, octahedral Ni2+14, did not exhibit any quenching. Heavy-atom effect was discounted on the ground that the heaviest Zn2+ enhanced the fluorescence of LS479 instead of quenching it.
To delineate the contribution of metal chelation to the observed fluorescence properties of LS479 metal chelates, we prepared and compared the absorption spectra of Cu2+-, Ni2+- and Zn2+-DTPA complexes and the emission spectra of free LS479 (Fig. 4). The result showed a large spectral overlap between the dye and Cu-DTPA complex and insignificant overlap for Ni2+ and Zn2+ complexes, thus supporting that energy transfer mechanism is responsible for the strong quenching by Cu2+ and the neutrality by Ni2+ and Zn2+. Similarly, quenching could be explained by an excited state charge transfer, where the excited electron travels from the π* orbital of a dye to a d-orbital of similar energy level of Cu2+, where the absorbed energy non-radiatively dissipates. The higher energy of d-orbitals in Ni2+ compared to π* precludes this transfer, allowing the fluorescence of LS479 to occur naturally from π*→ π. Indeed, the absorbance max in the visible spectra of Cu-DTPA was 740 nm, while Ni-DTPA was shorter at 610 nm. A similar mechanism of quenching might occur in porphyrin, which in its free form emits at around 600 nm but both Cu2+ and Ni2+ cause fluorescence quenching15.
Figure 4.
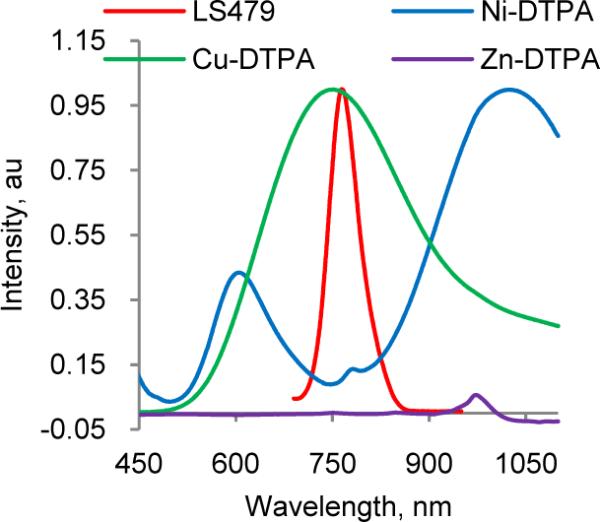
Normalized emission (ex. 690 nm) of LS479 and absorption spectra of Cu-DTPA and Ni-DTPA and Zn-DTPA. (Abs. max of Zn-DTPA complex was <450 nm). Substantial overlap between LS479 and copper suggests that energy transfer is a possible explanation for quenching. Note the lack of overlap with zinc and nickel.
The fluorescence enhancement of LS479 observed upon complexation with Zn suggests that the lack of vacant d-orbital precludes metal-ligand charge transfer and the absence of absorption bands prevents the energy transfer. A distinguishing characteristic of LS479 compared to HITC was the presence of a lone electron pair on the conjugated amine group. Lone electron pairs typically act as highly efficient fluorescence quenchers via an excited state charge transfer mechanism (ESCT)16. Indeed, the quantum yield of LS479 (Φ=0.018) in unbuffered water was ~6 times lower than that of HITC (Φ=0.11). In addition, the fluorescence lifetime of LS479 (τ =0.35 ns) was shorter than HITC (τ =0.41 ns), further indicating the presence of a fluorescence quenching process.
The addition of 64CuCl2 also resulted in fluorescence quenching. The net fluorescence enhancement upon 64Cu2+ decay strongly supports our model of synchronized fluorescence enhancement with radioactive decay. This new method of monitoring fluorescence enhancement through the decay of radionuclides has a variety of potential applications. For example, it may open new possibilities in studying fundamental molecular transformations such as determining how quickly a molecule readjusts from one state to another after radioactive decay. Furthermore, combining radionuclide decay with fluorescence enhancement could provide a unique complementary signaling mechanism for radionuclear-optical multimodal biological imaging. In fact, the new radiolabeled LS479 represents a paradigm shifting strategy in multimodal molecular imaging because 64Cu2+ and near infrared fluorescent dyes such as LS479 are routinely used in positron emission tomography 17 and optical imaging 18 of living organisms, respectively. Moreover, the strong correlation between fluorescence quenching and the presence of 64Cu2+ chelation provides a dynamic method to study the in vivo stability of the radionuclide chelates, where fluorescence enhancement reports demetallation of the radionuclide. Finally, the new molecular construct and mechanism could be used to develop visible radioactivity decay devices for environmental and nuclear forensic applications. Future work will be focused on these potential applications.
Supplementary Material
Acknowledgment
This study was support in part by the NIH extramural grants (R01 CA109754, R33 CA 123537, R33 CA100972, U54 CA136398 and U54 CA119342), the NIH Roadmap for Medical Research as funding source for the IPDC, and the Intramural Research Program of Eunice Shriver NICHD
Footnotes
Supporting Information Available. Synthesis, experimental procedures, calculations and optical spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Domaille DW, Que EL, Chang CJ. Nat. Chem. Biol. 2008;4(3):168–75. doi: 10.1038/nchembio.69. [DOI] [PubMed] [Google Scholar]; Kimura E, Koike T. Chem. Soc. Rev. 1998;27(3):179–184. [Google Scholar]
- 2.Fabbrizzi L, Licchelli M, Pallavicini P, Perotti A, Taglietti A, Sacchi D. Chem. Eur. J. 1996;2(1):75–82. [Google Scholar]
- 3.Rurack K. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2001;57(11):2161–95. doi: 10.1016/s1386-1425(01)00492-9. [DOI] [PubMed] [Google Scholar]
- 4.Kanofsky JR, Sima PD. Photochem. Photobiol. Sci. 2000;71(4):361–368. doi: 10.1562/0031-8655(2000)071<0361:saerfq>2.0.co;2. [DOI] [PubMed] [Google Scholar]; Loura LM, Fedorov A, Prieto M. Biophys. J. 1996;71(4):1823–36. doi: 10.1016/S0006-3495(96)79383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lowenthal G, Airey P. Practical Applications of Radioactivity and Nuclear Radiations. Cambridge University Press; Cambridge: 2001. [Google Scholar]
- 6.Yang L, McRae R, Henary MM, Patel R, Lai B, Vogt S, Fahrni CJ. Proc. Natl. Acad. Sci. U.S.A. 2005;102(32):11179–84. doi: 10.1073/pnas.0406547102. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zeng L, Miller EW, Pralle A, Isacoff EY, Chang CJ. J. Am. Chem. Soc. 2006;128(1):10–1. doi: 10.1021/ja055064u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Rosenzweig Z. Anal. Chem. 2002;74(19):5132–8. doi: 10.1021/ac0258251. [DOI] [PubMed] [Google Scholar]
- 8.Rahimi Y, Goulding A, Shrestha S, Mirpuri S, Deo SK. Biochem. Biophys. Res. Commun. 2008;370(1):57–61. doi: 10.1016/j.bbrc.2008.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chien JCW, Conner WP. J. Am. Chem. Soc. 1968;90(4):1001–1006. [Google Scholar]
- 10.Rae M, Fedorov A, Berberan-Santos MN. J. Chem. Phys. 2003;119(4):2223–2231. [Google Scholar]
- 11.Meallet-Renault R, Herault A, Vachon JJ, Pansu RB, Amigoni-Gerbier S, Larpent C. Photochem. Photobiol. Sci. 2006;5(3):300–10. doi: 10.1039/b513215k. [DOI] [PubMed] [Google Scholar]
- 12.Antipas A, Dolphin D, Gouterman M, Johnson EC. J. Am. Chem. Soc. 1978;100(24):7705–9. [Google Scholar]
- 13.Lakowicz JR. Principles of fluorescence spectroscopy. 3rd ed. Springer; New York: 2006. p. 954 S. [Google Scholar]
- 14.Cotton FA, Wilkinson G, Murillo CA, Bochmann M. Advanced Inorganic Chemistry. 6th ed. Wiley; New York: 1999. p. xv.p. 1355. [Google Scholar]
- 15.Dolphin D. The Porphyrins. Vol. 3. Academic Press; New York: 1978. p. 571. [Google Scholar]
- 16.Atwood JL, Steed JW. Encyclopedia of Supramolecular Chemistry. CRC Press; Boca Raton: 2004. [Google Scholar]
- 17.Anderson C, Connett J, SW S, PA R, LW G, GW P, KR Z, CF M, CF MJ, CF W. J. Nucl. Med. 1992;33(9):1685–1691. [PubMed] [Google Scholar]
- 18.Citrin D, Lee AK, Scott T, Sproull M, Menard C, Tofilon PJ, Camphausen K. Mol. Cancer Ther. 2004;3(4):481–8. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.