

Abstract
Cytolytic T cell-centric active specific and adoptive immunotherapeutic approaches might benefit from the simultaneous engagement of CD4+ T cells. Considering the difficulties in simultaneously engaging CD4+ and CD8+ T cells in tumor immunotherapy -- especially in an antigen specific manner -- “redirecting” CD4+ T cells to MHC class I-restricted epitopes through engineered expression of MHC class I-restricted epitope specific T cell receptors (TCR) in CD4+ T cells has emerged as a strategic consideration. Such TCR engineered CD4+ T cells have been shown to be capable of synthesizing cytokines as well as lysing target cells. We have carried out a critical examination of functional characteristics of CD4+ T cells engineered to express the α and β chains of a high functional avidity TCR specific for the melanoma epitope, MART-127–35 (M1), as a prototypic human tumor antigen system. We found that unpolarized CD4+CD25− T cells engineered to express the M1 TCR selectively synthesize Th1 cytokines and exhibit a potent antigen-specific lytic granule exocytosis-mediated cytolytic effector function of comparable efficacy to that of CD8+ CTL. Such TCR engineered CD4+ T cells, therefore, might be useful in clinical immunotherapy.
Introduction
``Cancer vaccines`` and adoptive immunotherapy for cancer have achieved impressive clinical responses in some patients (1, 2) but the overall result of such treatments has been somewhat modest. Intense efforts are underway to improve the outcome of these forms of therapy. Most ``cancer vaccines`` and adoptive immunotherapeutic strategies seek to achieve a robust and long-lived cytolytic T lymphocyte (CTL) response as most cancer cells express only MHC class I restricted epitopes. The task of generating a robust and long-lived CTL response to tumor-associated antigens – most of which are “self” antigens – faces many constraints. Among these is the lack of an effective strategy to engage CD4+ T helper (Th) cells.
Clinical trials with various forms of non-cognate help (universal class II epitope, tetanus toxoid, KLH, etc.) have been undertaken without clear evidence of benefit. A limited number of studies with MHC class II determined helper epitopes – as “cognate help”— has also been initiated. Unfortunately, only a few class II restricted determinants have so far been defined and only a limited number of patients can be matched for the appropriate MHC class I and class II alleles for which epitopes are available. The task of engaging cognate help targeting MHC class II restricted epitopes in anti-tumor immunity, therefore, has turned out to be difficult. Lately, “redirecting” CD4+ T cells to MHC class I-restricted epitopes through engineered expression of class I-restricted epitope specific T cell receptors (TCR) in CD4+ T cells has emerged as a strategic consideration. It has been shown that CD4+ T cells engineered to express relevant MHC class I-restricted epitope specific T cell receptors (TCR) can function as effector cells (3–7). They usually exhibit a mixed (i.e., Th1 as well as Th2/3) cytokine synthesis profile (3, 5, 6) unless prior to transduction, they are polarized to Th1 phenotype (7). It has also been shown that MHC class I-restricted epitope specific TCR engineered CD4+ T cells provide “help” toward the generation of memory CTL response in animal models (8, 9). These observations and the pressing need for developing an effective way that would engage “cognate help” in tumor immunotherapy in humans prompted us to undertake a critical examination of the biology of CD4+ T cells engineered to express a relevant set of MHC class I-restricted human tumor epitope specific TCR. Here we show that human CD4+CD25− T cells engineered to express a TCR specific for the HLA-A2.1-restricted melanoma epitope, Melan-A/MART-127–35 (hereafter to be referred to as M1 epitope) synthesize only Th1 cytokines IFN-γ and IL-2 and no IL-4 or IL-10. They also exhibit MHC class I-restricted and granule-mediated cytolytic activity in recognition of the M1 epitope presented on HLA-A2.1 positive surrogate target cells as well as melanoma cells naturally displaying the M1 epitope.
Materials and Methods
Study population, cell lines, culture medium and reagents
HLA-A2-positive melanoma patients and healthy donors were included in the study with informed consent. Culture medium, and other reagents used to generate M1 epitope specific T cells have been described previously (10). T2 cells, human melanoma cell line A375 was a gift of Steven A. Rosenberg, Surgery Branch, National Cancer Institute, Bethesda, Maryland. A375 cells engineered to express Melan-A/MART-1, A375-M have been described before (11). The melanoma cell lines PT-M and JL-M were established from two HLA-A2.1 positive melanoma patients.
Construction of recombinant lentivirus expressing MART-127–35TCR
A MART-127–35 antigenic epitope specific oligoclonal CTL line was generated from a limiting dilution micro-well seeded with three MART-127–35 peptide pulsed DC-activated CD8+ T cells generated as described before (10, 11). Total RNA was extracted from the CTL line using a Qiagen RNeasy Kit, and was used to clone out the TCR genes using the BD SMART™ RACE kit following the manufacturer’s instructions. In brief, the first-strand cDNA was synthesized in two steps: step 1 constituted the first-strand synthesis coupled with (dC) tailing by RT; step 2 constituted the template switching and extension by RT, resulting in the 5’-RACE-Ready cDNA capped with a common primer at the 5’ end. The cDNA was then used to run 5’-RACE PCR to clone the TCR genes. From the oligoclonal CTL line, 3 productively rearranged TCR alpha chain genes and three productively rearranged TCR beta chain genes were cloned. Using a TCR pairing assay, two TCR pairs, α3β15 and α2β14, were determined to be specific for the MART127–35 epitope based on the HLA-A2/MART-1 tetramer (Beckman Coulter) staining. To construct the FUW-M1-TCR/sr39tk lentivector, the alpha chain and beta chains of M1 TCR and the sr39tk imaging/suicide gene were linked with F2A and P2A elements (12) using assembly PCR, and were then cloned into the FUW lentivector (13).
The lentiviral vector was packaged by transient transfection of 293T cells with the vector plasmid, a plasmid expressing the HIV-1 gag and pol proteins (p8.9) and a plasmid encoding the VSV-G protein, as described before (14). The vector supernatant was concentrated approximately 500-fold by ultrafiltration (Centricon plus 70 ultrafiltration filters; Millipore, Bedford, MA) followed by ultracentrifugation at 500 × g for 2 h. Concentrated vector supernatants were resuspended in X-Vivo15 serum-free medium and stored at −70° C until used. Titers were determined by transduction of sub-confluent HT29 cells with serial dilutions of vector supernatant followed by extraction of genomic DNA after one week and quantification of integrated vector copy number by quantitative polymerase chain reaction (qPCR) using primers to the HIV-1 leader region (15). The numbers of transducing units per milliliter (TU/mL) were calculated by multiplying the vector copy number, which was extrapolated from a standard curve of known vector copy number, the number of cells at the time of supernatant addition, and the supernatant dilution.
Transduction of CD4+ T cells with lentiviral vector
Purified CD4+CD25− subsets of CD4+ T cells were isolated from CD4+ T cells by magnetic beads purification method (Invitrogen Inc., USA) from Ficoll-Hypaque density gradients purified human peripheral blood lymphocytes as reported before (10, 11). Purified CD4+CD25− T cells were then activated with 5µg plate-bound anti-CD3 antibody and cultured in the presence of 100 u/ml IL-2 for 5 days. Activated CD4 T cells (0.5–1 ×106 cells) were transduced with the recombinant virus at 10 MOI. Transduction was carried out for 120 min. by slow shaking (∼100rpm), on a bench top mixer at room temperature. The next day, cells were transduced again under identical conditions and then cultured for 5 days in the presence of 1000 u/ml IL-15. Transduction efficiency was quantified by staining transduced cells with M1 epitope specific tetramer (Beckman Coulter, USA). When needed, the TCR transduced populations were expanded by in-vitro stimulation with M1 peptide pulsed matured DC, once or twice, as reported earlier (10, 11).
Cytokine synthesis by the transduced T cells
The ability of the TCR transduced CD4+ T cells to produce cytokines was determined in cytokine synthesis assay as previously described (10, 11). Briefly, they were stimulated by T2 cells pulsed with the cognate peptide (MART-127–35) or the control peptide (MAGE-3271–279) at an effector T cells to target ratio of 10 for 16h. Supernatants were then analyzed for various cytokines. Melanoma cells were also used to stimulate the transduced cells to determine if they would respond to them.
Cytotoxicity assay
The cytolytic ability of T cells was examined by the chromium release microcytotoxicity assay was done as described previously (16).
ELISA
IL-2, IL-4, IL-10 and IFN-gamma cytokines were measured by sandwich ELISA kit (Immunotech, Marseilles, France) according to the manufacturer’s instruction. The details have been published (10).
Results
An oligoclonal MART-127–35 epitope specific CTL line was generated by limiting dilution culture from the in vitro activated/expanded CD8+ T cells obtained from a patient who exhibited a remarkable high frequency of the M1 epitope specific TCR positive T cells in circulation (Fig. 1A). The M1 epitope specific TCR positive T cells in circulation of the donor were predominantly V β14 positive (Fig. 1B). Co-expression of the M1 epitope specific TCR -α and -β chains, derived from the M1 epitope specific CTL line led to the identification of two M1 epitope specific functional TCRs (Fig. 2A). Both of these TCR contained the Vβ14 region and Vα 2.1 and 2.2 regions respectively (Fig. 2A). Based on preliminary comparative studies, we elected to use the Vα 2.2/ Vβ14 TCR (to be referred as M14.2 TCR) to study the biology of TCR transduced CD4 T cells. A recombinant lentiviral vector encoding the M14.2 TCR was constructed (Fig. 2B). A human ubiquitin-C promoter was used to drive the expression of three transgenes MART-1 TCR alpha, MART-1 TCR beta, and suicide/imaging gene sr39tk. The suicide/imaging gene sr39tk was incorporated in our constructs for in-vivo imaging studies in a future clinical trial. Two “self-cleavage” 2A-like liners (F2A, derived from food-and-mouth disease virus; P2A, derived from Porcine teschovirus) (12) were used to link the three transgenes to achieve the optimal stoichiometric expression of these three proteins (Fig. 2C). The functional integrity of the two TCR chains was confirmed by transducing Jurkat cells (Fig 2D). A dose-dependent increase in M1 specific tetramer and anti-V β14 antibody stained Jurkat was observed in transduced populations (Fig 2D).
Figure. 1.
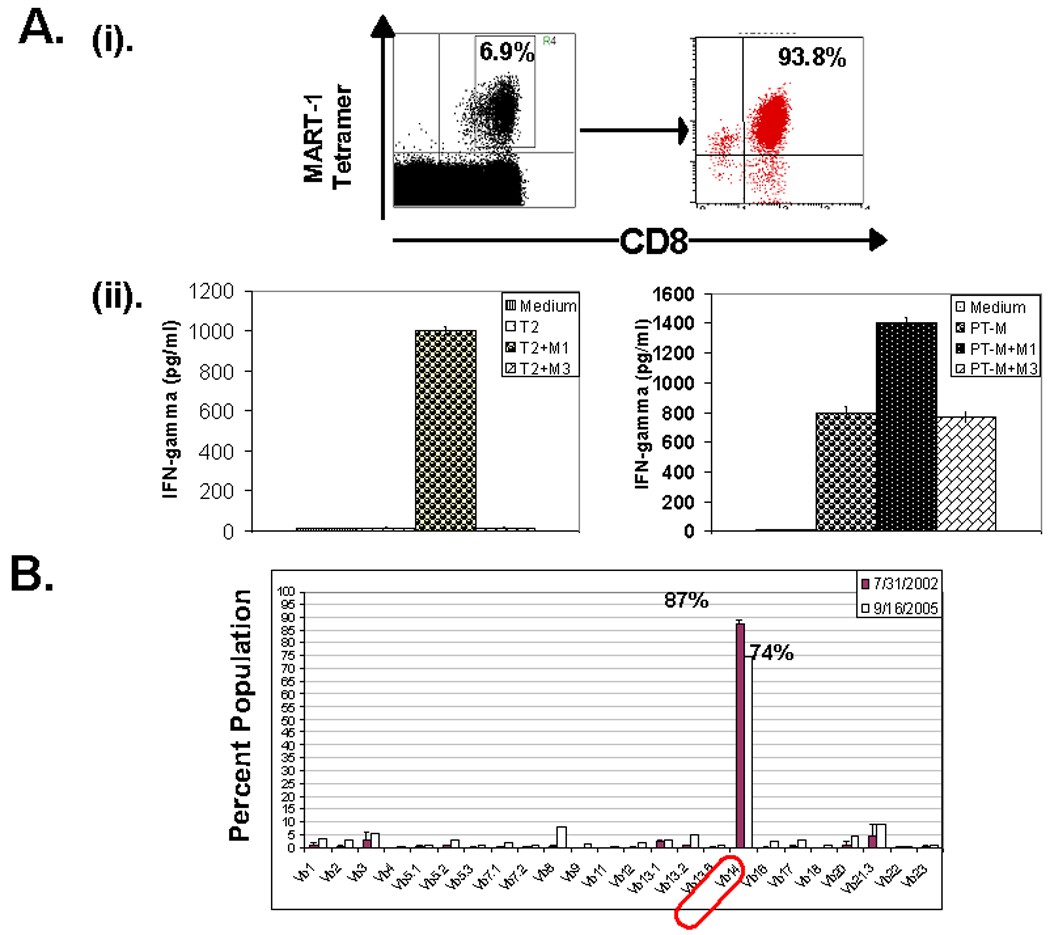
Generation of MART-127–35 epitope specific CTL line from the peripheral blood derived CD8 T cells of a melanoma patient. (A)– (i). An oligocolnal CTL line was generated from a melanoma patient’s peripheral blood derived CD8+ T cells, in a DC-T cell co-culture expansion protocol. (ii). The antigen specific effector function of the CTL line was tested by measuring IFN-γ release in the supernatant upon co-culture with either surrogate target cells, T2 (left panel) or MART-1 expressing human melanoma cells PT-M (right panel). CTL were co-cultured with target cells alone (T2/PT-M) or target cells pulsed with either cognate peptide, MART-127–35 (T2/PT-M +M1) or MAGE-3271–279 control peptide (T2/PT-M+M3) and cytokine released in supernatant was measured next day. (B). TCR V-β clonotypic analysis of patient derived MART-127–35 epitope specific CD8 T cells.
Figure. 2.
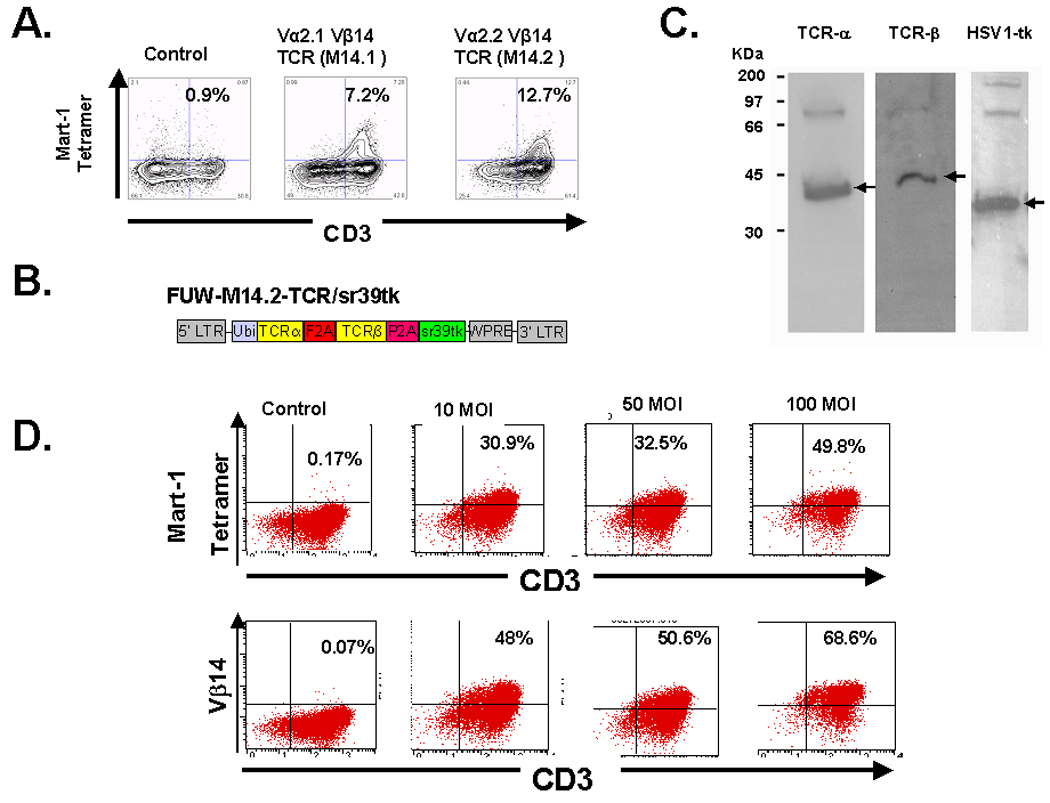
Construction of a recombinant lenti virus vector encoding MART-127–35 epitope specific TCR. (A). Co-expression of PCR amplified −α and −β chains led to the identification of two functional M1 epitope specific TCRs. Both of these TCR consisted of Vβ14 region and Vα2.1 and Vα2.2 regions respectively. The Vα2.1/ Vβ14 TCR was named as M14.1, while Vα2.2/ Vβ14 TCR was named as M14.2 TCR. M14.2 TCR was used in follow-up studies (B). Cloning of M14.2 TCR −α and −β chains in a FUW-M1-TCR/sr39tk lentivirus vector. (C). 293T cells were transduced with the lentiviral vector FUW-M14-TCR/sr39tk, lysed and immunoblotted. Specific antibodies were used to detect the constant region of the −α and −β chains of TCRs and the HSV1-tk. In the figure, arrows point to the expected size bands for each protein. Weaker high molecular weigh bands correspond to uncleaved products. The cleavage efficiency of the linkers was estimated to be > 90%. (D). Transduction of Jurkat cells with transgenic TCR.
We next examined the functional integrity of the transgene in CD4+ T cells and characterized the nature of antigen-specific cytokine response exhibited by the M14.2 TCR transduced CD4 T cells. Fig. 3A shows that 12% of the non-polarized but pre-activated CD4+25− T cells transduced with the FUW vector expressing the M14.2 TCR. A fraction of the M14.2 TCR positive cells expressed CD4 molecules at lower density. The reason for this under-expression of CD4 molecules in the TCR transduced population presently remains unclear. The M14.2 TCR engineered CD4+ T cells synthesized IFN-γ and IL-2 but no IL-4 or IL-10 in an epitope specific manner (Fig. 3B). Of note, this Th1 type bias was also seen with the M14.2 TCR engineered CD4+ T cells from three other donors in six separate experiments (Fig. 4). The TCR transduced CD4+ T cells also recognized M1 antigen on the HLA-A2.1 positive MART-1 transduced melanoma line, A375 as well as the HLA-A2.1 positive melanoma cell line, PT-M, naturally expressing the MART-1 antigen (Fig. 3C and Fig. 5B). In contrast, they did not recognize the melanoma line JL that was HLA-A2.1 positive but did not express the MART-1 protein (Fig. 3C and Fig. 5B). Fig. 3D shows the epitope recognition profile of autologous CD8+ CTL. As can be seen, CD8+ CTL, generated from the same donor in our standard in vitro CTL generation protocol (10), also exhibited essentially identical epitope recognition profile and comparable cytokine synthetic capacity. The M14.2 TCR transduced CD4+ T cells also exhibited Th1 type bias against the melanoma lines (Fig. 5A). Of note, the TCR transduced CD4+ T cells exhibited a comparable high functional avidity for the cognate peptide as displayed by the natural M1 epitope specific high affinity TCR expressing CD8+ CTL (Fig. 6). The functional avidity of TCR transduced CD4 T cells and CD8+ CTL was examined at different peptide doses and at different effector to target ratios (Fig. 6A–D).
Figure. 3.
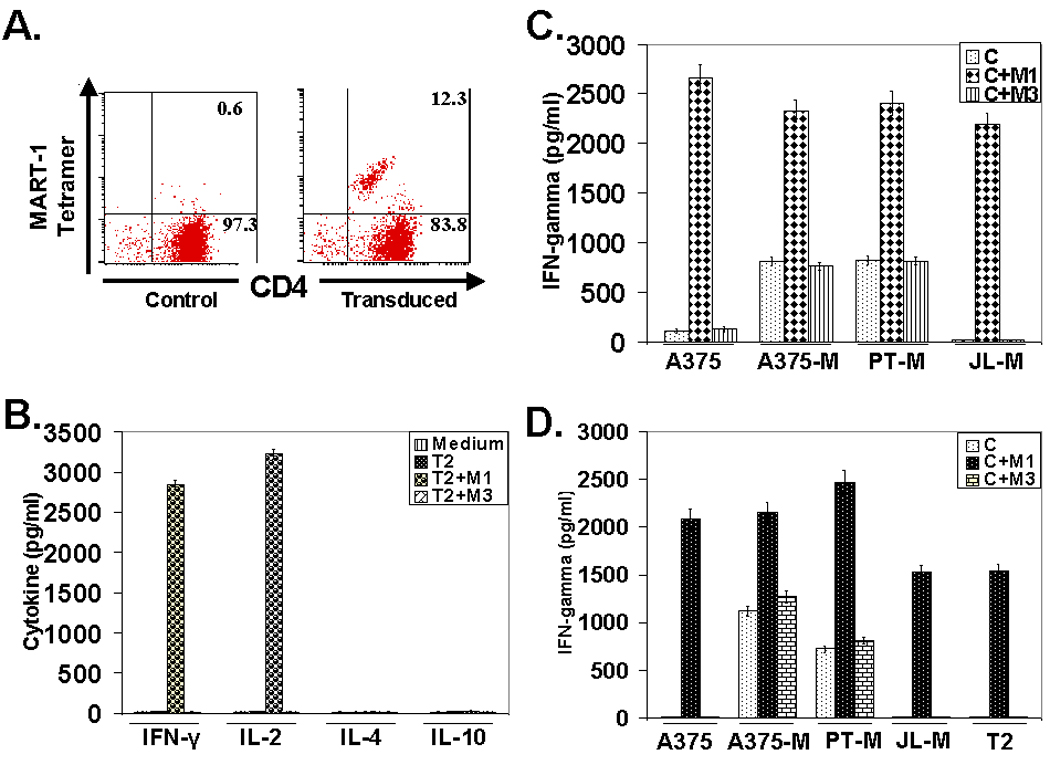
Transduction of T cells with transgenic TCR and analysis of the effector cytokine response (A). Transduction of activated human primary CD4 T cells with transgenic TCR. (B). Characterization of effector cytokine response of TCR engineered CD4 T cells. Transduced CD4 T cells were co-cultured with T2 cells alone or T2 pulsed with MART-127–35 peptide or MAGE-3271–279 control peptide and cytokines released in the supernatant were quantified 16 hr following co-culture. (C). Effector cytokine response of TCR engineered CD4 T cells against human melanoma cells. Transduced CD4 T cells were co-cultured with human melanoma cell lines, A375 wild type & MART-1 expressing A375 (A375-M), PT-M and JL-M. Co-culture was set up either with cells alone or cells pulsed with MART-127–35 peptide or MAGE-3271–279 control peptide and cytokines released in the supernatant were quantified 16 hr following co-culture. (D). Effector cytokine response analysis of human peripheral blood derived MART-127–35 epitope specific CD8+ CTL against surrogate target, T2 or human melanoma cells. CTL were co-cultured with T2 or melanoma cells alone or cells pulsed with cognate peptide (M1) or control peptide (M3) and IFN-γ released in the supernatant was measured 16 hr following co-culture, as done with TCR engineered CD4 T cells (Fig. 3C).
Figure. 4.
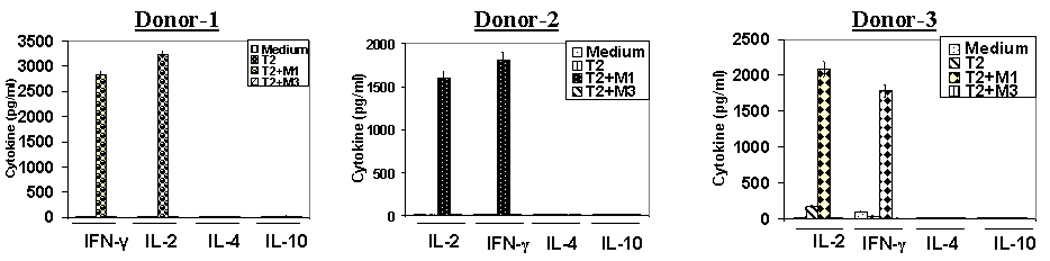
Characterization of cytokine response of TCR engineered CD4 T cells. CD4 T cells derived from three different donors were transduced with M14.2 TCR and co-cultured with T2 cells alone or T2 pulsed with MART-127–35 peptide or MAGE-3271–279 control peptide and cytokines released in the supernatant were quantified 16 hr following co-culture.
Figure. 5.
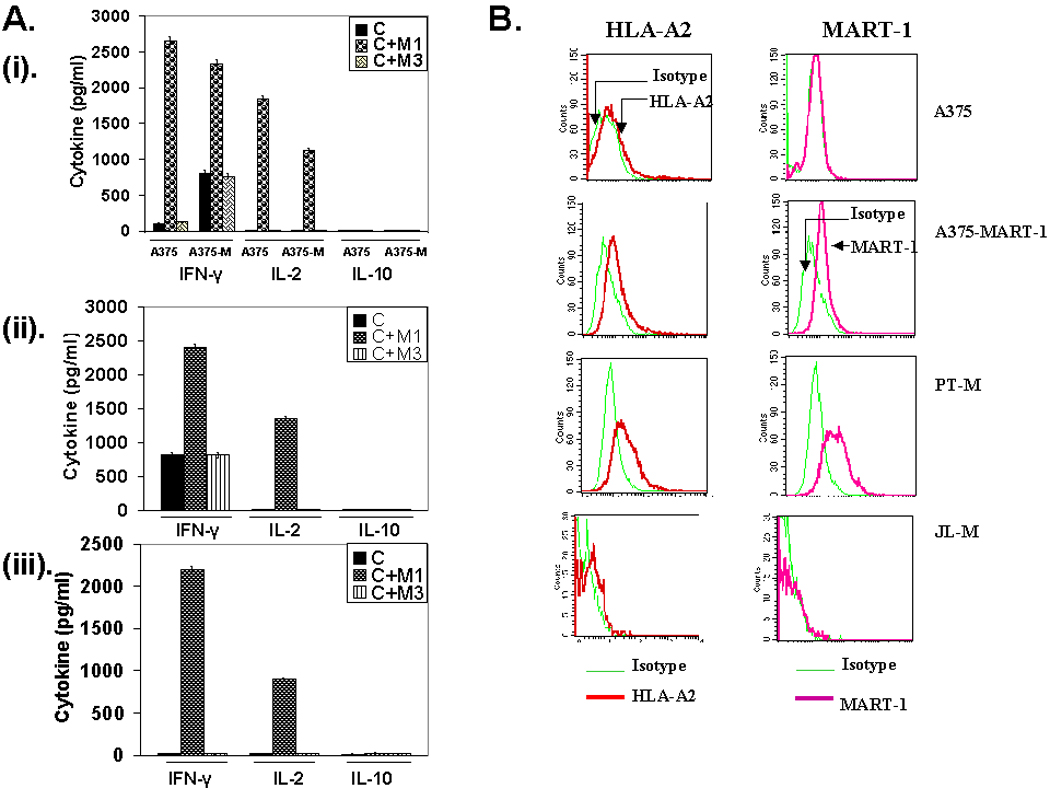
Characterization of cytokine response of TCR engineered CD4 T cells, in response to human melanoma cell lines. (A). M14.2 TCR transduced CD4 T cells were co-cultured with human melanoma cell lines, A375 wild type & MART-1 expressing A375 (i), PT-M (ii) and JL-M (iii). Co-culture was set up either with cells alone or cells pulsed with MART-127–35 peptide or MAGE-3271–279 control peptide and cytokines released in the supernatant were quantified 16 hr following co-culture. (B). Analysis of human melanoma cell lines, A375, A375-MART-1, PT-M and JL-M, for the expression of HLA-A2 molecules and MART-1 antigen.
Figure. 6.
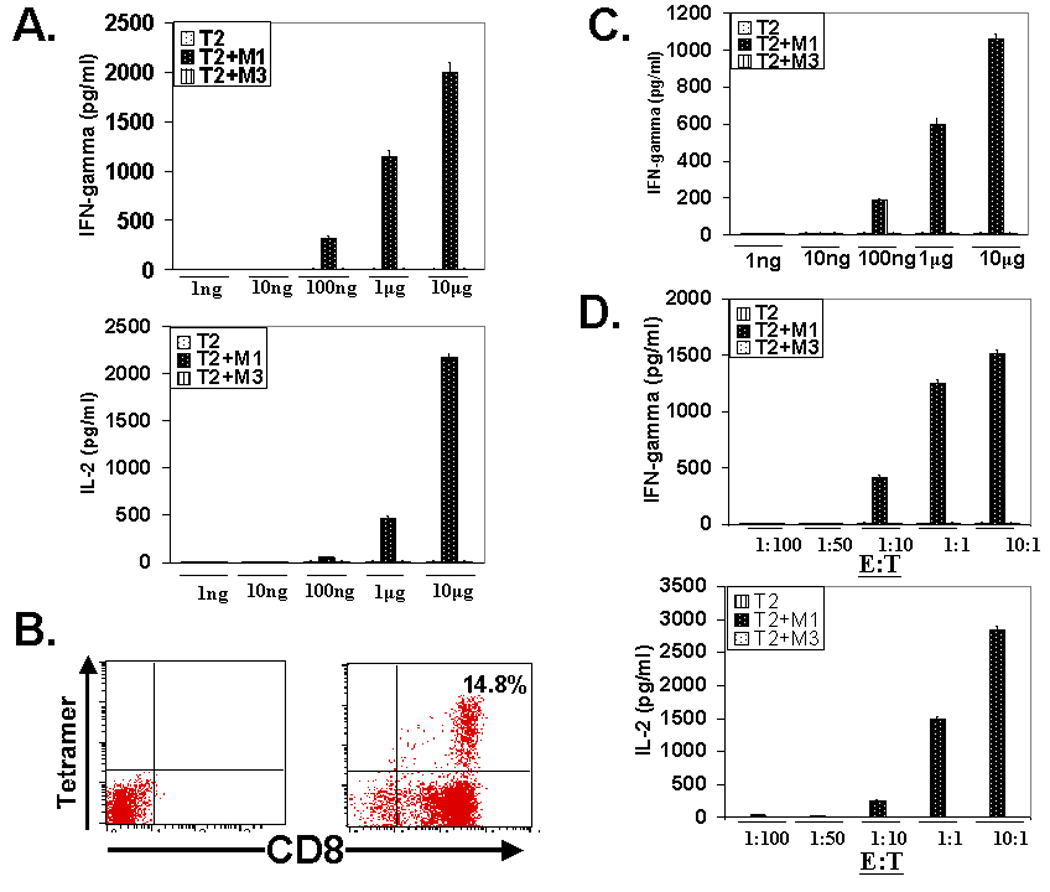
Transgenic TCR engineered CD4 T cells function as high avidity effector T cells. (A). Analysis of effector cytokine (IFN-γ and IL-2) release by transgenic TCR engineered CD4 T cells at a peptide dose range of 1ng-10µg. (B). Tetramer analyses of MART-127–35 antigenic epitope specific CTL population expanded from human peripheral blood, in a peptide pulsed DC-T cell co-culture expansion protocol. (C). Analysis of effector cytokine (IFN-γ) release by human peripheral blood derived CD8+ CTL precursor expanded MART-127–35 specific CTL population, at a peptide dose range of 1ng-10µg. (D). Analysis of effector cytokine (IFN-γ and IL-2) release by transgenic TCR engineered CD4 T cells at different effector to target ratios.
We next examined the cytolytic potential of transduced CD4 T cells (Fig. 7). As shown, the M14.2 TCR engineered CD4+ T cells lysed the M1 peptide loaded surrogate targets as well as lysed melanoma cells naturally expressing the epitope (Fig. 7A & 7B) just as efficiently as the in vitro generated natural CD8+ CTL (Fig. 7C & 7D).
Figure. 7.
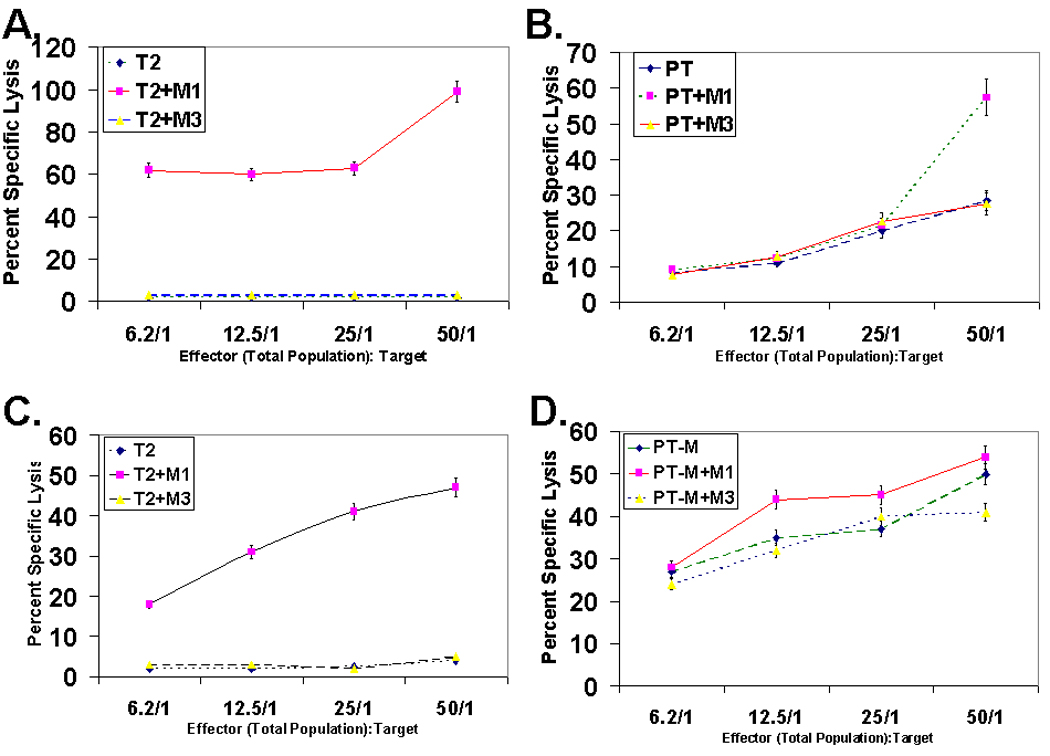
Cytolytic potential of MART-127–35 epitope specific TCR engineered CD4 T cells. (A & B). Cytolytic potential of TCR engineered CD4 T cells against peptide pulsed T2 cells (A) or MART-1 expressing melanoma cells, PT-M (B). Cytotoxicity assays were set up with target cells alone or pulsed with the MART-127–35 peptide or MAGE-3271–279 control peptide and Cr51 release was quantified 6 hr post co-culture. Of note, lysis of the PT-M cells were observed at all E:T ratios examined, the level of lysis was considerably higher at E:T of 50 when the PT-M cells were pulsed with the peptide (7B). (C & D). Cytolytic potential analysis of MART-127–35 epitope specific human peripheral blood derived CD8+ T cells against T2 (C) or PT-M melanoma cells (D). Cytotoxicity assay was done by Cr51 release assay with Cr51 loaded T2 or PT-M melanoma cells, as done for MART-127–35 epitope specific M14.2 TCR engineered CD4 T cells (Fig. 7A & 7B).
Next we compared the mechanism of the cytolytic effector function displayed by the transgenic TCR transduced CD4 T cells with the MART-127–35 epitope specific natural CD8+ CTL (Fig. 8). As shown in Fig. 8A, the cytolytic effector function exhibited by TCR engineered CD4 T cells was MHC class I restricted and the calcium chelant EGTA blocked this cytolytic activity. Since calcium is required for release of perforin and granzyme B leading to cytotoxic death of target cells, these data suggest that the lytic activity of these cells was mediated by the release of lytic granules (Fig. 8B). The M14.2 TCR engineered CD4+ T cell expressed cytolytic effector molecules, granzyme and perforin, and they underwent degranulation after encountering the cognate epitope (Fig. 8C), similar to the M1 epitope specific CD8+ CTL (Fig. 8D & 8E).
Figure. 8.
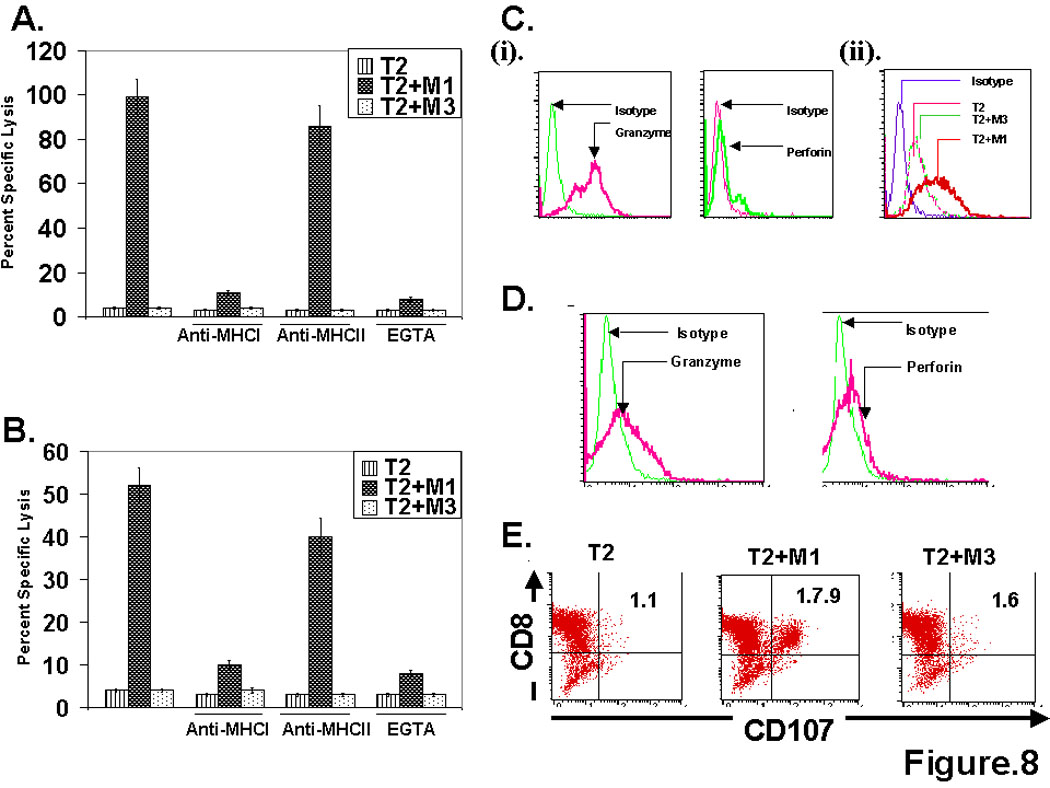
MART-127–35 epitope specific TCR engineered CD4 T cells mediate cytolytic effector function in a MHC class I restricted and cytolytic granule exocytosis dependent manner. (A & B). Analyses of cytolytic potential of MART-127–35 epitope specific M14.2 TCR engineered CD4 T cells (A) and MART-127–35 epitope specific human peripheral blood derived CD8+ CTL (B), in a Cr51 release assay, with Cr51 labeled T2 cells alone or T2 cells pulsed with MART-127–35 cognate peptide or MAGE-3271–279 control peptide. Cytotoxicity assay were set up either in the absence or presence of anti-MHC class I, anti-MHC class II antibodies. EGTA (4mM) pre-treatment was done to block the release of cytolytic granules. (C). M14.2 TCR engineered CD4 T cells express cytolytic effector molecules, granzyme and perforin (i), and undergo degranulation following antigen encounter with T2 cells pulsed with MART-127–35 peptide epitope (ii). Transgenic CD4 T cells were co-cultured either with T2 cells alone of T2 cells pulsed with MART-127–35 cognate peptide or MAGE-3271–279 control peptide and degranulation was measured by quantifying CD107 molecules, surface exposed upon antigen encounter. (D & E). Analyses of the expression of granzyme and perforin (D), and degranulation assay (E) were carried out with MART-127–35 epitope specific human peripheral blood derived CD8+ CTL, as done for TCR engineered CD4 T cells (Fig. 8C).
Discussion
Most active specific and adoptive immunotherapeutic approaches for cancer center around cytolytic T lymphocyte (CTL)-based responses generated through immunization or transferred adoptively with ex vivo generated CTL. CD4+ T cells might contribute in these strategies substantially (17–19), but the task of simultaneously engaging CD4+ T helper cells and CTL has turned out to quite difficult-- especially in an antigen specific manner. Accordingly, “redirecting” CD4+ T cells to recognize MHC class I-restricted epitopes through engineered expression of MHC class I-restricted epitope-specific TCR has gained attention. In animal models, it has indeed been shown that such TCR engineered CD4+ T cells can provide “help” (8, 9) as well as exhibit cytolytic effector function (20). In human systems, MHC class I restricted epitope specific TCR engineered CD4+ T cells have been shown to synthesize different cytokines in a co-receptor-dependent as well as co-receptor-independent manners (3–7). In general agreement with these studies, our results attest to the potential usefulness of such MHC class I-restricted epitope specific TCR engineered CD4+ T cells in tumor immunotherapy. Our studies also reveal a number of additional interesting aspects of their biology.
First, our studies clearly show that the MHC class I-restricted epitope specific TCR engineered CD4+ T cells can function in a co-receptor-independent manner. Co-receptor-dependent and -independent function by such TCR engineered CD4+ T cells has been described (21, 22). It is believed that a “high affinity/avidity” TCR might transmit productive signals without the participation of co-receptors. The affinity of the M14.2 TCR on CD4+ T cells was found to be in the nanomolar range and it was comparable to that of the naturally grown CD8+ CTL (Fig. 6). These transgenic TCR transduced CD4 T cells recognized the naturally expressed M1 epitope on melanoma cells, just as efficiently as the naturally grown CD8+ CTL (Fig. 3).
Second, in contrast to previous reports of MHC class I-restricted epitope specific TCR engineered CD4+ T cells synthesizing both type 1 and type 2/3 cytokines (3, 5, 6), the M14.2 TCR engineered CD4+ T cells synthesize only Th1 type cytokines (Fig. 3, Fig. 4 and Fig. 5). The reason for this Th1/Tc1 bias in the M14.2 TCR engineered CD4+ T cells remains unclear. It should be noted that we used only CD4+CD25− subsets. Additionally, we did not polarize them in Th1 type condition prior to transduction as was done in another study where Th1 type bias by this type of TCR engineered CD4+ T cells was reported (7). Given that CD4+ T cells from three different donors engineered to express the M14.2 TCR elaborated only Th1/Tc1 cytokine (Fig. 4), it does not appear to be an isolated incidence. Careful studies will be needed to understand if a given set of transgenic TCR dictates polarization or the functional polarization is an imprinted property acquired inherently during ontogeny or acquired in culture conditions. Whatever the explanation might turn out to be, one can envision a distinct advantage with the TCR engineered CD4+ T cells exhibiting Th1 bias from a translational viewpoint. TCR engineered CD4+ T cells exhibiting Th1 bias are likely to be true helper cells and more effective effector cells.
Third, the MHC class I-restricted and granule-mediated cytolytic property of the M14.2 TCR engineered CD4+ T cells provides a different understanding on the functional separation between a CD4+ T cell and a CD8+ T cell. Traditionally, CD8+ T cells are viewed as quintessential cytolytic T cells and CD4+ T cells as “helper” cells. Although, a role of CD4+ T cells in anti-tumor immunity is well documented in certain models (23–25), the mechanism by which CD4+ T cells exhibit anti-tumor immunity in these models has never been fully clarified. The M14.2 TCR engineered CD4+ T cells appear to be just as good killer cells as their CD8 counterparts, they use granule-mediated lytic machinery (Fig. 7 &Fig. 8), and they also elaborate a comparable amount of IFN-γ (Figs. 3C & 3D). The mechanism(s) that ``re-directs`` CD4+ T cells to perform as killer T cells presently remain unclear. Additional studies will be needed to address this issue. Although how CD4+ T cells are ``re-directed`` to function as cytolytic T cells by signals through the transgenic CTL derived TCR remains an open question, our data clearly show that the M14.2 TCR engineered CD4+ T cells can function as formidable effector cells. They could, therefore expand the effector response repertoire substantially in an active specific or adoptive immunotherapy schema.
Finally, it should be pointed out that engaging such MHC class I restricted epitope specific and co-receptor independent TCR engineered CD4+ T cells in tumor immunotherapy would have many advantages. For example, making CD4 T cells function through the same epitope that the CTLp also recognize resolves the problem in engaging the CTLp, Thp, and the APC presenting separate class I and class II epitopes simultaneously -- a conundrum in orchestrating help in a three cell model. Second, the strategy does not rely on recruiting a rare Thp. By introducing a fairly large number of TCR engineered CD4+ T cells, the scope for providing cognate help could be vastly improved. Additionally, such CD4+ T cells could do more than “help”. They could provide effector function of their own. The strategy can be employed for active specific as well as in adoptive therapy models. Most importantly, as a large number of MHC class I-restricted tumor associated epitopes and CD8+ T cells capable of recognizing them are defined, a large number of patients could be matched for administration of MHC class I-epitope specific TCR engineered CD4+ T cells. As such, the translational potential of MHC class I epitope engineered CD4+ T cells in the setting of active specific or adoptive immunotherapy will be considerable.
Acknowledgments
The work was supported by PHS grants CA 83130 (BM), CA 88059 (BM), CA129816 (JSE) and grants from the Dowling Foundation (BM), Samuel Waxman Foundation and WM Keck Foundation (JSE for UCLA-CALTECH-CHLA-USC-UCONN Consortium on Translational Program in Engineered Immunity).
References
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blattman JN, Greenberg PD. Cancer immunotherapy: a treatment for the masses. Science. 2004;305:200–205. doi: 10.1126/science.1100369. [DOI] [PubMed] [Google Scholar]
- 3.Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. 1999;163:507–513. [PubMed] [Google Scholar]
- 4.Morgan RA, Dudley ME, Yu YY, Zheng Z, Robbins PF, Theoret MR, Wunderlich JR, Hughes MS, Restifo NP, Rosenberg SA. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol. 2003;171:3287–3295. doi: 10.4049/jimmunol.171.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol. 2005;174:4415–4423. doi: 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roszkowski JJ, Lyons GE, Kast WM, Yee C, Van Besien K, Nishimura MI. Simultaneous generation of CD8+ and CD4+ melanoma-reactive T cells by retroviral-mediated transfer of a single T-cell receptor. Cancer Res. 2005;65:1570–1576. doi: 10.1158/0008-5472.CAN-04-2076. [DOI] [PubMed] [Google Scholar]
- 7.Tsuji T, Yasukawa M, Matsuzaki J, Ohkuri T, Chamoto K, Wakita D, Azuma T, Niiya H, Miyoshi H, Kuzushima K, Oka Y, Sugiyama H, Ikeda H, Nishimura T. Generation of tumor-specific, HLA class I-restricted human Th1 and Tc1 cells by cell engineering with tumor peptide-specific T-cell receptor genes. Blood. 2005;106:470–476. doi: 10.1182/blood-2004-09-3663. [DOI] [PubMed] [Google Scholar]
- 8.Morris EC, Tsallios A, Bendle GM, Xue SA, Stauss HJ. A critical role of T cell antigen receptor-transduced MHC class I-restricted helper T cells in tumor protection. Proc Natl Acad Sci U S A. 2005;102:7934–7939. doi: 10.1073/pnas.0500357102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kessels HW, Schepers K, van den Boom MD, Topham DJ, Schumacher TN. Generation of T cell help through a MHC class I-restricted TCR. J Immunol. 2006;177:976–982. doi: 10.4049/jimmunol.177.2.976. [DOI] [PubMed] [Google Scholar]
- 10.Mehrotra S, Chhabra A, Chattopadhyay S, Dorsky DI, Chakraborty NG, Mukherji B. Rescuing melanoma epitope-specific cytolytic T lymphocytes from activation-induced cell death, by SP600125, an inhibitor of JNK: implications in cancer immunotherapy. J Immunol. 2004;173:6017–6024. doi: 10.4049/jimmunol.173.10.6017. [DOI] [PubMed] [Google Scholar]
- 11.Chhabra A, Mehrotra S, Chakraborty NG, Mukherji B, Dorsky DI. Cross-presentation of a human tumor antigen delivered to dendritic cells by HSV VP22-mediated protein translocation. Eur J Immunol. 2004;34:2824–2833. doi: 10.1002/eji.200425192. [DOI] [PubMed] [Google Scholar]
- 12.Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 13.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 14.Haas DL, Case SS, Crooks GM, Kohn DB. Critical factors influencing stable transduction of human CD34(+) cells with HIV-1-derived lentiviral vectors. Mol Ther. 2000;2:71–80. doi: 10.1006/mthe.2000.0094. [DOI] [PubMed] [Google Scholar]
- 15.Sastry L, Johnson T, Hobson MJ, Smucker B, Cornetta K. Titering lentiviral vectors: comparison of DNA, RNA and marker expression methods. Gene Ther. 2002;9:1155–1162. doi: 10.1038/sj.gt.3301731. [DOI] [PubMed] [Google Scholar]
- 16.Chhabra A, Chakraborty NG, Mukherji B. Silencing of endogenous IL-10 in human dendritic cells leads to the generation of an improved CTL response against human melanoma associated antigenic epitope, MART-1 27–35. Clin Immunol. 2008;126:251–259. doi: 10.1016/j.clim.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keene JA, Forman J. Helper activity is required for the in vivo generation of cytotoxic T lymphocytes. J Exp Med. 1982;155:768–782. doi: 10.1084/jem.155.3.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 19.Bevan MJ. Helping the CD8(+) T-cell response. Nat Rev Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- 20.Kessels HW, Wolkers MC, van den Boom MD, van der Valk MA, Schumacher TN. Immunotherapy through TCR gene transfer. Nat Immunol. 2001;2:957–961. doi: 10.1038/ni1001-957. [DOI] [PubMed] [Google Scholar]
- 21.Roszkowski JJ, Yu DC, Rubinstein MP, McKee MD, Cole DJ, Nishimura MI. CD8-independent tumor cell recognition is a property of the T cell receptor and not the T cell. J Immunol. 2003;170:2582–2589. doi: 10.4049/jimmunol.170.5.2582. [DOI] [PubMed] [Google Scholar]
- 22.Willemsen R, Ronteltap C, Heuveling M, Debets R, Bolhuis R. Redirecting human CD4+ T lymphocytes to the MHC class I-restricted melanoma antigen MAGE-A1 by TCR alphabeta gene transfer requires CD8alpha. Gene Ther. 2005;12:140–146. doi: 10.1038/sj.gt.3302388. [DOI] [PubMed] [Google Scholar]
- 23.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med. 1998;188:2357–2368. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pardoll DM, Topalian SL. The role of CD4+ T cell responses in antitumor immunity. Curr Opin Immunol. 1998;10:588–594. doi: 10.1016/s0952-7915(98)80228-8. [DOI] [PubMed] [Google Scholar]
- 25.Mumberg D, Monach PA, Wanderling S, Philip M, Toledano AY, Schreiber RD, Schreiber H. CD4 (+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proc Natl Acad Sci U S A. 1999;96:8633–8638. doi: 10.1073/pnas.96.15.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]