

Abstract
Pneumonia caused by Streptococcus pneumoniae is a significant cause of morbidity and mortality during influenza virus epidemics. We had previously advanced the hypothesis that interactions of pneumococcus with the receptor for platelet activating factor (PAFR) in the lung were facilitated by antecedent influenza virus infection and play a major role in the pathogenesis of bacterial super-infections. Although influenza enhanced the adherence of pneumococci to respiratory epithelial cells in vitro, chemical or antibody-mediated blockade of the PAFR did not affectt adherence. In agreement with these data, mice lacking PAFR had similar bacterial loads within the lung compartment when compared to heterozygous littermates and were not protected from secondary pneumococcal pneumonia after influenza. Lack of support for this hypothesis and the observation of enhanced inflammation during secondary pneumococcal pneumonia in mice lacking PAFR may moderate enthusiasm for treatment strategies targeting the interaction of bacteria with PAFR.
Keywords: influenza virus, Streptococcus pneumoniae, pneumonia, platelet activating factor receptor
Introduction
The World Health Organization (WHO) estimates that annual influenza epidemics cause 3 − 5 million severe illnesses and 250,000 − 500,000 deaths each year in the developed world alone (1). The effect in the developing world is unknown and is likely to be even higher. Since only exceptionally virulent influenza viruses can cause death of the host from direct viral causes (2, 3), most mortality during seasonal influenza is either due to an increased physiologic load in a person with pre-existing medical problems or to secondary bacterial infections (4, 5). In typical epidemic years, about 25% of these deaths are estimated to be caused by secondary bacterial pneumonia, most frequently S. pneumoniae (6). Pneumococcus primarily affects young children, causing otitis media, sinusitis, and pneumonia, and the elderly, causing pneumonia. During annual epidemics, excess mortality following influenza is particularly common in elderly persons with comorbidities such as lung or heart disease (7, 8). Prevention or treatment of the secondary infections that follow influenza would be facilitated by an improved understanding of the mechanisms underlying the interaction between respiratory viruses and bacteria.
The mechanisms that underlie viral-bacterial synergism are poorly understood. It was dogma for many years that viral mediated damage to the lung epithelium exposed basement membrane and extra-cellular membrane elements to which bacteria could adhere. This was based on mouse models using highly virulent, mouse-adapted strains (9, 10), and on pathology studies done during the 1918 and 1957 pandemics (11-14). However, these outcomes are poorly representative of typical epidemic strains, which do not cause severe lung damage. Thus, it is likely that other, more subtle mechanisms are either partly or wholly responsible for the enhancement of bacterial infections seen during seasonal influenza.
We hypothesized in earlier reports that alterations of receptor interactions between pneumococcus and lung epithelia by uncovering, altering, or up-regulating potential receptors may facilitate super-infections (15-17). A particularly intriguing hypothesis, based on work from Cundell et al. demonstrating that inflammatory stimuli upregulate and activate the G-protein coupled receptor for the phospholipid mediator platelet activating factor (PAF) (18), suggested that the inflammatory response to influenza might make this receptor more available and more permissive in the lung, allowing better adherence and invasion of pneumococci (16). However, the results of our early attempt to test this hypothesis using a PAF receptor (PAFR) antagonist in an in vivo model of secondary bacterial pneumonia in mice were equivocal (16). The current study was conducted to answer this important question more authoritatively using mice deficient in expression of the PAFR.
Methods
Infectious agents
The Mount Sinai strain of mouse adapted influenza virus A/Puerto Rico/8/34 (H1N1), hereafter referred to as PR8, was grown in Madin-Darby canine kidney (MDCK) cells from stock from the influenza virus repository at St. Jude Children's Research Hospital. S. pneumoniae strains D39 (type 2), R6T (unencapsulated variant of the D39 strain used in animal experiments), A66.1 (type 3), and Norway T4 (type 4), were the kind gifts of Dr. Elaine Tuomanen at St. Jude Children's Research Hospital.
Mice
Six – to eight week old male and female mice lacking the PAFR were generated by the author PJM as described (19) and back-crossed at least 5 times onto a C57Bl/6 background prior to use in these studies. All experimental procedures were done under general anesthesia with inhaled isoflurane 2.5% (Baxter Healthcare Corporation, Deerfield, IL). Animals used in this study were cared for in accordance with the guidelines of the Committee on Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council) under an approved protocol from the Animal Care and Use Committee of St. Jude Children's Research Hospital in biosafety level 2 facilities.
Infectious model
Infectious agents were diluted in sterile PBS and administered intranasally in a volume of 100 μl (50 μl per nostril) to anesthetized mice held in an upright position. Since both male and female mice were used in experiments, care was taken to match groups for sex and weight prior to initiating experiments. Influenza virus was given at a dose of 100 units infectious for 50% of tissue culture wells (TCID50), and S. pneumoniae strains D39, A66.1, and T4 were given at doses of 1000, 100, and 10,000 CFU/ml, respectively, based on prior experience with these strains (16, 17, 20) and in an effort to achieve about 50% mortality following influenza virus infection so that either positive or negative effects of the absence of PAFR could be determined. Groups of 6 to 10 mice were weighed and followed at least daily for illness and mortality. Mice found to be moribund were euthanized and considered to have died on that day. Care was taken to insure all mice received equal anesthesia during the course of the experiments. For collection of lung homogenates, anesthetized mice were euthanized by inhalation of CO2. Lungs were removed under sterile conditions, washed three times in sterile PBS, and placed into 500 μl of sterile PBS. Quantitation of pneumococcal colony counts was then done by 10-fold serial dilutions on tryptic soy agar plates supplemented with 3% v/v sheep erythrocytes. For determination of cytokines, lung homogenates were centrifuged at 10,000 × g for 5 minutes and the supernatants frozen. The production of IL-1β, IL-6, IL-10, KC, MIP-1α and TNF-α was measured using the “mouse 17-plex” cytokine assay (Bio-Rad Laboratories, Hercules, CA) read on a Luminex 100 reader (Luminex Corp., Austin, TX) according to the manufacturer's instructions. Samples were diluted 1:4 and run in duplicate in all assays.
Adherence assay
Adherence assays were performed using standard methods as described previously (17) using pneumococcal strain R6T, which is an unencapsulated variant of the D39 strain used in animal experiments, following a 30 minute incubation of the lung epithelial cell line A549 with virus at 37 °C. Bacteria-only controls were treated identically, without the addition of virus. For inhibition of PAFR, 50 μg/ml anti-PAFR antibody (Alexis Biochemicals, San Diego, CA), 1μM ( ± ) trans-2,5,-bis (3,4,5-trimethoxyphenyl-1,3-dioxolane (active PAFR antagonist; Biomol International, Plymouth Meeting, PA), or 1μM ( ± ) cis-2,5,-bis (3,4,5-trimethoxyphenyl-1,3-dioxolane (inactive PAFR antagonist’ Biomol International) were added to the virus suspension, 30 min before incubation with monolayers. Intraperitoneal injection of antibody against PAFR prevented PAF-mediated cardiac arrest and death of mice following IV-injection (data not shown). The active (trans-) PAFR antagonist does not inhibit the growth of pneumococcus in vitro (data not shown).
Statistical analysis
Comparison of survival between groups of mice was done with the Log Rank chi-squared test on the Kaplan-Meier survival data. Comparisons of adherent bacteria, bacterial lung titers, and cytokine levels between groups were done using analysis of variance (ANOVA). A p-value of < 0.05 was considered significant for these comparisons. SigmaStat for Windows (SysStat Software, Inc., V 3.11) was utilized for all statistical analyses.
Results
Inhibition of PAFR does not alter pneumococcal adherence to lung epithelial cells
Inhibitors of PAFR receptor have been demonstrated to decrease adherence of pneumococcus to endothelial cells (19). To test whether this finding extended to lung epithelial cells, we pre-treated A549 cells with either an antibody that binds PAFR and inhibits the activity of PAF, or an active inhibitor of the PAFR, prior to performing a standard adherence assay. Neither antibody-mediated nor chemical inhibition of the PAFR affected adherence of pneumococcus to A549 cells (Fig 1A, B). Pre-incubation of monolayers with influenza virus significantly enhanced adherence 2−3 fold in the presence or absence of either antibody or the active inhibitor compared to the no-virus controls (Fig 1A, B). Thus, the enhanced in vitro adherence of pneumococci to respiratory epithelium seen following incubation with influenza virus is not mediated solely by PAFR.
Fig. 1.

Inhibition of PAFR does not decrease adherence of pneumococci to A549 cells. Adherence of S. pneumoniae strain R6 to A549 cell monolayers was assayed after a 30 minute pre-incubation with either PBS or influenza virus in (A) the presence or absence of antibody (Ab) against PAFR or (B) the presence of an active (trans) or inactive (cis) inhibitor of PAFR. The mean values of 4−6 independent experiments are presented with error bars indicating the standard deviation of the data. An asterisk indicates a significant difference in adherence by ANOVA (p < 0.05) compared to the corresponding control group that was not exposed to influenza virus.
PAFR is not responsible for the increased mortality from S. pneumoniae following influenza
To test our hypothesis that PAFR is involved in the pathogenesis of secondary bacterial infections following influenza in vivo, mice lacking PAFR were infected with influenza and challenged 7 days later with pneumococcus. If our hypothesis was correct, we would expect the mice lacking PAFR to survive bacterial challenge, while those with intact PAFR would succumb to pneumococcal lung infection. To insure that our findings were generalizable, 3 different pneumococcal strains known to be virulent for mice in this model were utilized (20). In all 3 experiments, mice lacking PAFR had lower rates of survival following bacterial super-infection than did age- and sex- matched heterozygous litter mates (Figure 2). These data indicate that the enhanced mortality due to pneumococcal pneumonia seen following influenza infection is not dependent on PAFR.
Fig. 2.

Lack of PAFR does not protect mice from secondary pneumococcal pneumonia following influenza. Groups of 10 mice lacking PAFR (KO) were infected with influenza virus then challenged with pneumococcus 7 days later and followed for mortality in comparison to age- and sex- matched heterozygous littermates (WT). Different strains of mouse-virulent S. pneumoniae were utilized, (A) D39 (type 2), (B) A66.1 (type 3), or (C) T4 (type 4). No significant differences in survival were demonstrated by the log rank test on the Kaplan-Meier survival data for D39 (p = 0.244), A66.1 (p = 0.111), or T4 (p = 0.146).
PAFR is not necessary for productive infection of the mouse lung following influenza
Since our primary hypothesis was that influenza virus infection was enhancing the adherence of pneumococci in the lungs leading to increased pneumonia and death, we measured bacterial lung titers in infected mice. Mice were infected with influenza virus, challenged with pneumococcus 7 days later, and lungs were removed 48 hours after pneumococcal challenge for determination of titers. All mice were productively infected with each of the 3 strains of pneumococcus, and no differences in mean titer were seen in any pairwise comparisons (Fig 3). Interestingly, 3 out of 6 control mice secondarily infected with pneumococcus strain T4, which is known to cause bacteremia, had positive blood cultures at 48 hours, compared to 0 of 8 mice lacking PAFR. No mouse infected with strain A66.1 had a positive blood culture, and mice infected with strain D39 were not assessed for bacteremia.
Fig. 3.
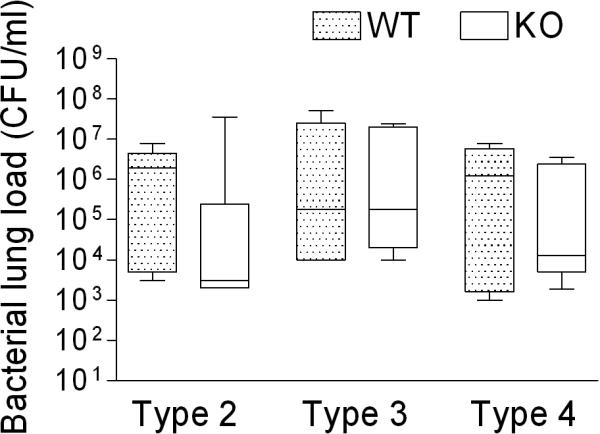
Post influenza pneumococcal lung titers are not altered by the absence of the PAFR. Groups of 6−9 mice lacking PAFR (KO) were infected with influenza virus then challenged with pneumococcus 7 days later and assayed 48 hours after pneumococcal challenge for bacterial lung load in comparison to age- and sex- matched heterozygous littermates (WT). Different strains of mouse-virulent S. pneumoniae were utilized, D39 (type 2), A66.1 (type 3), or T4 (type 4). The 25th – 75th percentiles are represented by the shaded box-plots with the horizontal bar indicating the mean value. Error bars indicate the standard deviation of the measurements. No significant differences in bacterial lung load were demonstrated by the ANOVA for any comparisons (p = 0.200).
Local cytokine production during pneumonia is increased in mice lacking PAFR
In our original study on viral-bacterial interactions and the PAFR, pharmacologic inhibition of PAFR in the mouse model of secondary bacterial pneumonia led to a bi-phasic response. Initially, mice appeared to be protected from secondary infection with lower bacterial lung titers and less weight loss 24 hours after secondary bacterial challenge. However, thereafter treated mice became rapidly ill compared to control mice and succumbed to infection between 48 and 72 hours (16). We speculated at the time, based on histopathologic study of the lungs, that an increased inflammatory response following blockade of PAFR may have contributed to mortality (16). Therefore, during the current study we assayed a set of 6 cytokines and chemokines shown to be elevated during secondary pneumococcal infections following influenza (20). Mice lacking PAFR had significantly higher amounts of the pro-inflammatory mediators IL-1β, TNFα, IL-6, KC, and MIP-1α in lung homogenates than did control mice 48 hours after secondary pneumococcal challenge with strain T4 (Figure 4). Thus, although the numbers of pneumococci were not elevated in the lungs, the host response to the bacteria was amplified. These data moderate enthusiasm for treatment strategies designed to block access of pathogens to PAFR.
Fig. 4.

Pro-inflammatory cytokines levels are enhanced in the lungs of mice lacking PAFR. Groups of 6 mice lacking PAFR (KO) were infected with influenza virus then challenged with pneumococcus strain T4 7 days later and assayed 48 hours after pneumococcal challenge for cytokines in lung homogenates in comparison to age- and sex- matched heterozygous littermates (WT). Mean values are presented with error bars indicating standard deviation. An asterisk (*) indicates a significant difference by ANOVA (p < 0.05) compared to WT.
Discussion
Development of strategies to prevent or treat secondary bacterial infections following influenza will require a better understanding of the mechanisms that underlie this medically important interaction. We have taken a reductionist approach to this problem, attempting to study specific viral, bacterial, or host factors that may contribute to the pathogenesis of infections. One possibility is that pneumococcus utilizes PAFR to establish infection through adherence to epithelial cells and further mediates the transition from the lung to blood compartments. Since PAFR can be activated by inflammatory stimuli (18), and might be made more accessible to lung-resident pneumococci by the sialidase action of the viral neuraminidase (17, 21), we have considered the hypothesis that PAFR is an important factor in the induction and enhancement of pneumococcal pneumonia following influenza (16). However, our preliminary studies testing this hypothesis were equivocal, requiring further work. The data reported here indicate that PAFR is not necessary for pneumococcal adherence to epithelial cells or induction of pneumococcal pneumonia following influenza and does not enhance secondary infections. In fact, our data suggest that the blockade of PAFR may be detrimental for the host in this setting.
PAFR is a ubiquitous G-protein coupled receptor that recognizes the phosphoryl choline moiety on the phospholipid mediator PAF. Several respiratory pathogens, including S. pneumoniae, display phosphoryl choline on their surfaces and can interact with the PAFR (18, 22, 23). This interaction has been shown to be important for invasion of endothelial cells by a β-arrestin dependent pathway (19). PAFR-mediated trafficking through endothelial cells appears to be critical for transitions of pneumococci between body compartments, such as from the lung to blood or across the blood-brain barrier (19, 24-26). Access to the brain and uptake into neurons through this mechanism has been implicated in the inflammatory response and poor outcomes associated with pneumococcal meningitis (27, 28). Indeed, our data support this role for PAFR as pneumococci present in the lung did not make the transition to the blood in mice lacking PAFR, as was observed in controls.
The importance to S. pneumoniae of PAFR expressed on respiratory epithelial surfaces is less certain, however, particularly in the setting of antecedent influenza virus infection. RSV, but not influenza virus, enhanced expression of PAFR on A549 cells and was associated with PAFR-dependent enhancement of adherence of pneumococcus (29). These results are in concordance with our data, which demonstrate that the enhanced adherence of pneumococcus observed following influenza virus is not PAFR dependent (Fig 1). In an earlier study using the same strain of mice lacking PAFR, a pneumococcal strain known to cause sepsis (given without prior influenza virus infection) engendered a more severe course of illness in WT controls, characterized by higher titers of bacteria in the blood and brain (25). An improved transition from lung to blood across endothelial surfaces consistent with the demonstrated role of PAFR in pneumococcal trafficking potentially accounts for this finding. In a post-influenza model similar to the one employed here, mice lacking PAFR had similar overall survival (1 of 11 survived) compared to wildtype controls (0 out of 11), despite lower rates of bacteremia in PAFR deficient mice (3 of 7) compared to wildtype (7 of 7) (30). In the current study, to control for the effect of PAFR on bacteremia and make the results more generalizable, we utilized 3 different strains of pneumococci, including one (A66.1) that does not cause bacteremia (20, 31), and consistent results were obtained with all 3 strains.
Although there was no statistically significant difference in survival in any of the comparisons between the mice lacking PAFR and controls, the trend in each case was for PAFR deficient mice to have worse outcomes (Fig 2). It is likely that this is due to the enhanced inflammatory response seen in the lung compartment when pneumococci were unable to access and utilize the PAFR (Fig. 4). This finding is consistent with histopathology data from our initial study (16) and cytokine data from Branger et al., who demonstrated higher levels of TNFα in mice deficient in PAFR compared to wildtype when challenged with nontypeable Haemophilus influenzae (32). Although there is a paucity of data regarding the normal interactions of pneumococcus with PAFR on epithelial surfaces, one could speculate that abrogation of these interactions and the confinement of the bacteria to the lung compartment allows increased activation of alternate pathways that lead to a pro-inflammatory response, such as those mediated by pneumolysin and cell wall (33, 34). This shift and subsequent enhancement of disease in the lung compartment may make strategies aimed at amelioration of sepsis through blockade of the PAFR less palatable (35).
Acknowledgments
This work was supported by ALSAC and U. S. Public Health Service Grants to JAM (AI-54802 and AI-66349).
Footnotes
The authors declare that no conflict of interest exists.
REFERENCES
- 1.Stohr K. Preventing and treating influenza. BMJ. 2003;326(7401):1223–4. doi: 10.1136/bmj.326.7401.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solorzano A, Swayne DE, et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310(5745):77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- 3.Yuen KY, Chan PK, Peiris M, Tsang DN, Que TL, Shortridge KF, et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet. 1998;351(9101):467–71. doi: 10.1016/s0140-6736(98)01182-9. [DOI] [PubMed] [Google Scholar]
- 4.Mote JR. Virus and rickettsial diseases. Harvard University Press; Cambridge, Massachusetts: 1940. Human and swine influenzas. pp. 429–516. [Google Scholar]
- 5.McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev. 2006;19(3):571–82. doi: 10.1128/CMR.00058-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simonsen L. The global impact of influenza on morbidity and mortality. Vaccine. 1999;17(Suppl 1):S3–10. doi: 10.1016/s0264-410x(99)00099-7. [DOI] [PubMed] [Google Scholar]
- 7.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA. 2003;289(2):179–86. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 8.Simonsen L, Reichert TA, Viboud C, Blackwelder WC, Taylor RJ, Miller MA. Impact of influenza vaccination on seasonal mortality in the US elderly population. Arch Intern Med. 2005;165(3):265–72. doi: 10.1001/archinte.165.3.265. [DOI] [PubMed] [Google Scholar]
- 9.Harford CG, Leidler V, Hara M. Effect of the lesion due to influenza virus on the resistance of mice to inhaled pneumococci. J Exp Med. 1949;89:53–67. doi: 10.1084/jem.89.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plotkowski MC, Puchelle E, Beck G, Jacquot J, Hannoun C. Adherence of type I Streptococcus pneumoniae to tracheal epithelium of mice infected with influenza A/PR8 virus. Am Rev Respir Dis. 1986;134(5):1040–4. doi: 10.1164/arrd.1986.134.5.1040. [DOI] [PubMed] [Google Scholar]
- 11.Stone WJ, Swift GW. Influenza and influenzal pneumonia at Fort Riley, Kansas. JAMA. 1919;72(7):487–93. [Google Scholar]
- 12.Wilson CB, Steer P. Bacteriological and pathological observations on influenza as seen in France during 1918. Br Med J. 1919:634–5. [Google Scholar]
- 13.Louria D, Blumenfeld H, Ellis J, Kilbourne ED, Rogers D. Studies on influenza in the pandemic of 1957−58. II Pulmonary complications of influenza. J Clin Invest. 1959;38:213–65. doi: 10.1172/JCI103791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin CM, Kunin CM, Gottlieb LS, Barnes MW, Liu C, Finland M. Asian influenza A in Boston, 1957−1958. I. Observations in thirty-two influenza-associated fatal cases. AMA Arch Intern Med. 1959;103(4):515–31. doi: 10.1001/archinte.1959.00270040001001. [DOI] [PubMed] [Google Scholar]
- 15.McCullers JA, Webster RG. A mouse model of dual infection with influenza virus and Streptococcus pneumoniae. In: Osterhaus ADME, Cox N, Hampson AW, editors. Options for the Control of Influenza IV. Elsevier Science B.V.; Amsterdam: 2001. pp. 601–7. [Google Scholar]
- 16.McCullers JA, Rehg JE. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis. 2002;186(3):341–50. doi: 10.1086/341462. [DOI] [PubMed] [Google Scholar]
- 17.McCullers JA, Bartmess KC. Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae. J Infect Dis. 2003;187(6):1000–9. doi: 10.1086/368163. [DOI] [PubMed] [Google Scholar]
- 18.Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature. 1995;377(5):435–8. doi: 10.1038/377435a0. [DOI] [PubMed] [Google Scholar]
- 19.Radin JN, Orihuela CJ, Murti G, Guglielmo C, Murray PJ, Tuomanen EI. beta-Arrestin 1 participates in platelet-activating factor receptor-mediated endocytosis of Streptococcus pneumoniae. Infect Immun. 2005;73(12):7827–35. doi: 10.1128/IAI.73.12.7827-7835.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith MW, Schmidt JE, Rehg JE, Orihuela C, McCullers JA. Induction of pro- and anti- inflammatory molecules in a mouse model of pneumococcal pneumonia following influenza. Comp Med. 2007;57(1):12–8. [PMC free article] [PubMed] [Google Scholar]
- 21.Peltola VT, Murti KG, McCullers JA. Influenza virus neuraminidase contributes to secondary bacterial pneumonia. J Infect Dis. 2005;192(2):249–57. doi: 10.1086/430954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swords WE, Buscher BA, Ver S,I, Preston A, Nichols WA, Weiser JN, et al. Nontypeable Haemophilus influenzae adhere to and invade human bronchial epithelial cells via an interaction of lipooligosaccharide with the PAF receptor. Mol Microbiol. 2000;37(1):13–27. doi: 10.1046/j.1365-2958.2000.01952.x. [DOI] [PubMed] [Google Scholar]
- 23.Soares AC, Pinho VS, Souza DG, Shimizu T, Ishii S, Nicoli JR, et al. Role of the platelet-activating factor (PAF) receptor during pulmonary infection with gram negative bacteria. Br J Pharmacol. 2002;137(5):621–8. doi: 10.1038/sj.bjp.0704918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ring A, Weiser JN, Tuomanen EI. Pneumococcal trafficking across the blood-brain barrier. Molecular analysis of a novel bidirectional pathway. J Clin Invest. 1998;102(2):347–60. doi: 10.1172/JCI2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rijneveld AW, Weijer S, Florquin S, Speelman P, Shimizu T, Ishii S, et al. Improved host defense against pneumococcal pneumonia in platelet-activating factor receptor-deficient mice. J Infect Dis. 2004;189(4):711–6. doi: 10.1086/381392. [DOI] [PubMed] [Google Scholar]
- 26.Miller ML, Gao G, Pestina T, Persons D, Tuomanen E. Hypersusceptibility to invasive pneumococcal infection in experimental sickle cell disease involves platelet-activating factor receptor. J Infect Dis. 2007;195(4):581–4. doi: 10.1086/510626. [DOI] [PubMed] [Google Scholar]
- 27.Fillon S, Soulis K, Rajasekaran S, edict-Hamilton H, Radin JN, Orihuela CJ, et al. Platelet-activating factor receptor and innate immunity: uptake of gram-positive bacterial cell wall into host cells and cell-specific pathophysiology. J Immunol. 2006;177(9):6182–91. doi: 10.4049/jimmunol.177.9.6182. [DOI] [PubMed] [Google Scholar]
- 28.Orihuela CJ, Fillon S, Smith-Sielicki SH, El Kasmi KC, Gao G, Soulis K, et al. Cell wall-mediated neuronal damage in early sepsis. Infect Immun. 2006;74(7):3783–9. doi: 10.1128/IAI.00022-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Avadhanula V, Rodriguez CA, DeVincenzo JP, Wang Y, Webby RJ, Ulett GC, et al. Respiratory viruses augment the adhesion of bacterial pathogens to respiratory epithelium in a viral species- and cell type-dependent manner. J Virol. 2006;80(4):1629–36. doi: 10.1128/JVI.80.4.1629-1636.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Sluijs KF, van Elden LJ, Nijhuis M, Schuurman R, Florquin S, Shimizu T, et al. Involvement of the platelet-activating factor receptor in host defense against Streptococcus pneumoniae during postinfluenza pneumonia. Am J Physiol Lung Cell Mol Physiol. 2006;290(1):L194–L199. doi: 10.1152/ajplung.00050.2005. [DOI] [PubMed] [Google Scholar]
- 31.Orihuela CJ, Gao G, McGee M, Yu J, Francis KP, Tuomanen E. Organ-specific models of Streptococcus pneumoniae disease. Scand J Infect Dis. 2003;35(9):647–52. doi: 10.1080/00365540310015854. [DOI] [PubMed] [Google Scholar]
- 32.Branger J, Wieland CW, Florquin S, Maris NA, Pater JM, Speelman P, et al. Platelet-activating factor receptor-deficient mice show an unaltered clearance of nontypeable Haemophilus influenzae from their respiratory tract. Shock. 2004;22(6):543–7. doi: 10.1097/01.shk.0000142818.91693.73. [DOI] [PubMed] [Google Scholar]
- 33.Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163(1):1–5. [PubMed] [Google Scholar]
- 34.Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, et al. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A. 2003;100(4):1966–71. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moreno SE, ves-Filho JC, Rios-Santos F, Silva JS, Ferreira SH, Cunha FQ, et al. Signaling via platelet-activating factor receptors accounts for the impairment of neutrophil migration in polymicrobial sepsis. J Immunol. 2006;177(2):1264–71. doi: 10.4049/jimmunol.177.2.1264. [DOI] [PubMed] [Google Scholar]