

Abstract
A large number of intracellular pathogens survive in vacuolar niches composed of host-derived membranes modified extensively by pathogen proteins and lipids. Although intracellular lifestyles offer protection from humoral immune responses, vacuole-bound pathogens nevertheless face powerful intracellular innate immune surveillance pathways that can trigger fusion with lysosomes, autophagy and host cell death. While many of the strategies used by vacuole-bound pathogens to invade and establish a replicative vacuole are well described, how the integrity and stability of these parasitic vacuoles are maintained is poorly understood. Here we identify potential mechanisms of pathogenic vacuole maintenance and the consequences of vacuole disruption by highlighting a select subset of bacterial and protozoan parasites.
Introduction
Adaptation to an intracellular lifestyle offers most pathogens the ability to escape recognition by humoral immune responses such as circulating antibodies and complement. However, within an infected cell a pathogen is further challenged by intracellular defense mechanisms. Prominent among these is the fusion of pathogen-containing vacuoles with lysosomal compartments (Ramachandra et al., 2009). The ability of infected cells to dispose of microbial invaders depends on the cell type and cytokine-dependent activation. Activated-macrophages and dendritic cells, for example, provide the least hospitable environment while non-immune cells are more permissive.
To avoid lysosomal fusion, a pathogen could potentially escape the membrane-bound vacuole. However, Nod-like receptors (NLRs) and Rig-like Receptors (RLRs), recognize pathogen associated molecular patterns (PAMPs) in the cytoplasm and induce the production of proinflammatory cytokines and chemokines. These molecules influence adaptive immune response and can trigger host cell death via activation of the inflammasome (Franchi, 2008; Yu, 2008). Additional antimicrobial responses include autophagosome formation on the surface of cytoplasm-exposed bacteria and their eventual fusion with lysosomes. Some pathogens like Listeria and Shigella have specifically adapted to life in the host cell cytoplasm by engaging in actin-based motility (Perrin, 2004) and by suppressing induction of autophagy (Ogawa et al., 2005).
Most intracellular pathogens studied to date replicate within membrane-bound compartments (Casadevall, 2008). It can be argued that perhaps in the context of an intact immune system and inflammatory responses, sequestration within membrane-bound vacuoles is a more desirable outcome for survival. Although the molecular mechanisms underlying the establishment of replicative niches by a number of membrane-bound intracellular pathogens are fairly well understood, how these pathogens maintain the stability and integrity of their vacuoles and the consequences of vacuole disruption on pathogenesis are not. Here we review literature for existing information on how pathogenic vacuoles are formed, their stability maintained, and describe pathways that specifically aid in the avoidance of innate immune responses.
1. Pathogenic vacuoles are customized host organelles
Intracellular pathogens invade their hosts via a cell entry process that culminates in the formation of a plasma membrane-derived pathogen containing vacuole. Shortly after entry, many intracellular pathogens hijack the endomembrane system of the host cell to prevent or delay their fusion with lysosomes. Initially, the pathogen-containing vacuole is composed of host-derived membranes and share molecular features of early endosomes. Soon thereafter, these compartments are modified by pathogen-derived proteins and lipids and their fusogenicity with vesicular carriers and other organelles is altered (Figure-1). As a result, pathogen-containing vacuoles become unique organelles with features of late endosomes/lysosomes (Salmonella), early endosomes (Mycobacteria) endoplasmic reticulum (Legionella), or appear devoid of any salient features (Chlamydia and Toxoplasma) (reviewed in (Meresse et al., 1999)).
Figure 1. Interaction of vacuolar niches of intracellular pathogens with host endomembrane system.

Bacterial pathogens invade their hosts via the endophagocytic pathway and establish vacuolar niches that share features with one or more host organelles. These include Mycobacteria pathogen vacuole (MPV), Salmonella containing vacuole (SCV), Brucella (BCV) and Legionella (LCV) containing vacuole. Others such as C. trachomatis inclusion (INC), and Toxoplasma (PV) appear devoid of any markers of the endocytic traffic. While avoiding fusion with degradative compartments these organelles retain the ability to selectively intercept host vesicular traffic for nutrient acquisition. EE- early endosomes, RE-recycling endosomes, LE-late endosomes, Ly-Lysosomes, Autophag- Autophagosomes,
The Salmonella containing vacuole (SCV)
Salmonella sps are facultative intracellular bacteria that cause gastroenteritis and enteric fever (Grassl and Finlay, 2008). Soon after entry the Salmonella-containing vacuole (SCV) displays markers of early endosomes such as Early Endosomal Antigen 1 and transferrin receptor. The SCV recruits Rab7 and a subset of lysosomal markers (Lamp1 and vATPase) by selective fusion with late endosomes (LE) and/or lysosomes. Some LE/Lysosomes markers such as the Mannose-6 Phosphate receptor (M6PR) are removed from the SCV via a Rab11-dependent recycling pathway (reviewed in (Bakowski, 2008)). The SCV then traffics along microtubules to the microtubule-organizing center (MTOC) in a process that is independent of SPI-2 secreted effectors (Ramsden et al., 2007). The SCV is then retained at this location via recruitment of the dynein complex by Rab7-Interacting Lysosomal Protein (RILP)-Dynein tethers. The bacterial effectors SseF and SseG are also required for SCV positioning at the MTOC (Abrahams, 2006; Deiwick, 2006). The SCV is a pleomorphic organelle characterized by filamentous projections termed Salmonella-induced filaments (Sifs) (Garcia del Portillo et al., 1993). Both Sif formation and juxtanuclear positioning of the SCV are dependent on interactions with dynein and kinesin and are required for efficient bacterial replication (Beuzon et al., 2002; Salcedo and Holden, 2003). The secreted Salmonella proteins SifA and SseJ modulate Sif formation, with mutations in sifA and sseJ leading to lower or higher Sif formation, respectively (Brummel, 2002a; Ruiz-Albert, 2002). Recent studies indicate that SseJ is a cholesterol acyltransferase which may regulate SCV membrane dynamics by removing free cholesterol (Lossi, 2008).
A small portion of intracellular Salmonella exits the SCV and resides in the host cytoplasm. These bacteria are targeted for ubiquitination (Perrin, 2004) and subsequently cleared by autophagy (Birmingham, 2006). How these bacteria escape the vacuole is unclear, although type-III secretion (T3S)-mediated damage of the SCV membrane has been proposed as one mechanism (Birmingham, 2006). Although the small proportion of cytosolic bacteria makes its significance questionable, sifA mutants readily spill into the cytoplasm (Beuzon et al., 2000) indicating a role for SifA in maintaining vacuole integrity. However, these mutants are not ubiquitinated and do not activate autophagy (Birmingham, 2006). While the basis for differential recognition of two forms of cytoplasmic Salmonella is unknown, it is possible that only bacteria associated with or enclosed in damaged SCV membranes are recognized. Indeed ubiquitinated forms of cytoplasmic wild-type bacteria, unlike sifA mutants, colocalize with SCV markers (Birmingham, 2006). In epithelial cells, cytoplasmic Salmonella populations replicate more efficiently than the SCV-enclosed ones (Beuzon et al., 2002). This is in contrast to murine macrophages and fibroblasts where sifA mutants grow poorly (Beuzon et al., 2002; Brumell et al., 2001). This likely reflects cell-type specific differences in the microbicidal capacity of cytoplasmic innate immune responses. Maintenance of SCV integrity is likely important for systemic disease since sifA mutants are attenuated in their ability to establish infection in mice (Stein et al., 1996).
The Mycobacterial phagosome
Mycobacterium tuberculosis, the causative agent of tuberculosis, replicates in macrophages (Cosma, 2003). The Mycobacteria pathogen vacuole (MPV) arrests at the early endosomal (EEA1, Rab5-positive) stage in a process partially mediated by bacterial phosphatidyl inositol mimics that inhibit PI3K activity (Philips, 2008). The MPV retains the ability to interact with early and recycling endosomes through the action of another mycobacterial lipid phosphatidylinositol mannoside (PIM) (de Chastellier, 2009) and Rabs 11 and 14, presumably to acquire nutrients delivered by endosomal recycling pathways (Kyei, 2006). In addition, proteins translocated by the ESX-1 type-VII secretion machinery are likely involved in mediating the arrest of MPV maturation (MacGurn and Cox, 2007).
Despite the arrest in MPV maturation, Mycobacteria can be delivered to phagolysosomes in macrophages after induction of autophagy with rapamycin treatment or in response to IFN-γ treatment (Gutierrez et al., 2004; Hope et al., 2004). As with the SCV, ubiquitination of bacterial products and autophagy are potent host defenses against establishment of the MPV (Alonso, 2007). Not surprisingly Mycobacteria have acquired mechanisms to minimize the induction of autophagy. One such mechanism proposed is via mycobacterial inhibition of PI3K, a central regulator of autophagy and phagosome maturation (Deretic, 2008).
Whether Mycobacteria reside exclusively within membrane compartments has been the subject of controversy. M. marinum, a close relative of M. tuberculosis, escapes from the phagosome by secreting ESAT-6, a pore forming substrate of the ESX-1 secretion system (Smith, 2008). Recent studies have revealed that during the lag phase between phagosome escape and initiation of actin-based motility, cytoplasmic M. marinum are targeted for ubiquitination by the host and subsequently engulfed in LAMP1 positive autophagosome-like compartments (Collins et al., 2009). Interestingly many bacteria escape this form of degradation by shedding cell wall material as ‘decoys’ (Collins et al., 2009). Although M. tuberculosis and M. leprae have also been reported to exit the MPV in dendritic cells and macrophages in an ESX-1 dependent manner (van der Wel, 2007) their fate in the cytoplasm has not been reported.
Brucella (BCV) and Legionella containing vacuoles (LCV)
For pathogens like Brucella abortus and Legionella pneumophila, avoidance of lysosomal compartments takes a circuitous route through the ER. The early Brucella containing vacuole (BCV) bears all the markers of early endosome (Rab5, EEA1 and transferrin receptor) (Gorvel, 2002). The BCV rapidly sheds these markers and embarks on a unique maturation pathway (Celli, 2003; Starr, 2008). After transient acidification, BCVs mature into organelles with features of both autophagosomes (e.g. LAMP-1 and monodansylcadaverine-positive) and ER (e.g. Calreticulin and Sec61b). These organelles likely represent ER-derived autophagosomes (Pizarro-cerda, 1998a). This branch of the autophagic pathway may be important for the pathogen since inhibition of PI3K increased Brucella killing while stimulation of autophagy by amino acid starvation enhanced replication (Pizarro-cerda, 1998b). Eventually the BCV becomes enriched in ER markers (Gorvel, 2002). The sustained acquisition of ER markers by the BCV requires Sar-1 and interaction with ER exit sites (Celli, 2005). In addition, acquisition of ER markers may involve the proliferation of ER-associated autophagosomes by Brucella-mediated activation of IRE1α a kinase, an inducer of the unfolded protein response (UPR) (Qin, 2008),
The Legionella containing vacuole (LCV) also interacts with ER-derived vesicles to mature into a vacuole primarily consisting of rough ER-derived membranes (reviewed extensively in (Isberg et al., 2009)). The LCV also frequently resembles autophagosomes with double membranes and associates with autophagy markers Atg7 and Atg8 (Amer and Swanson, 2005; Swanson and Isberg, 1995), although the role of autophagy in Legionella replication is unclear since bacterial growth is not impaired in autophagy deficient Dictyostelium (Otto et al., 2004).
Small GTPases Sar-1, Rab1 and Arf-1 are required for the LCV to acquire Sec22b containing ER-derived vesicles (Kagan and Roy, 2002). Rab1 recruitment and function at the LCV surface is modulated by Legionella effectors DrrA/SidM and LepB via their guanine nucleotide exchange (GEF) and activating (GAP) activities, respectively (Ingmundson et al., 2007; Machner and Isberg, 2007). The association of secreted Legionella effectors such as DrrA and SidC with the LCV surface is mediated by their affinity for PI4P, a lipid that is abundant on the LCV surface. (Brombacher et al., 2009; Ragaz et al., 2008).
Both Legionella and Brucella use a T4S apparatus to deliver effectors into host cells. It is likely that similar to T3S (Coombes and Finlay, 2005), T4S may also cause membrane damage that could compromise the integrity of the pathogenic vacuole. In macrophages and amoebae, L. pneumophila has been proposed to escape into the host cytoplasm late in infection (Molmeret et al., 2004). The significance of this is unclear.
The Chlamydia inclusion
Chlamydia sps are obligate intracellular bacterial pathogens that infect genital, ocular and pulmonary epithelial surfaces. In contrast to other bacterial vacuoles described above, the pathogenic vacuole (“inclusion”) is rapidly segregated from stereotypical endomembrane trafficking pathways. Like the SCV, the nascent inclusion travels on microtubules to the MTOC (Grieshaber, 2006) where it intimately interacts with the Golgi apparatus. At this stage, Chlamydia induces Golgi fragmentation by cleaving Golgin84, but the Golgi fragments remain in close association with the inclusion (Heuer et al., 2009), presumably to allow the efficient acquisition of sphingolipids and cholesterol.
Despite its segregation from classical endocytic traffic, a number of Rab GTPases (e.g Rab4, 11, 1, 6 and 10) associate with the inclusion (Rzomp, 2003), which suggest that the inclusion may selectively interact with ER and Golgi-derived vesicles. While no Rab7 or Rab9 are present on the inclusion, the vacuole can acquire markers of multivesicular body (MVB) endosomes (Beatty, 2006). The inclusion may also acquire lipids and nutrients by scavenging organelles from the host cytoplasm. For example, lipid droplets, neutral lipid storage organelles, are translocated across the inclusion membrane into the inclusion lumen (Cocchiaro, 2008).
The inclusion membrane is extensively modified by a set of poorly characterized integral membrane proteins (Incs) (Rockey et al., 2002). Inc proteins likely play central roles in inclusion biogenesis and maintenance by recruiting Rab and Rab effector proteins (Rzomp et al., 2006) and maintaining a fusogenic state (Hackstadt et al., 1999). IncA, for example may act as a SNARE mimic that permits homotypic fusion of inclusions (Delevoye, 2008).
Toxoplasma parasitopherous vacuole (PV)
Toxoplasma gondii is a widely disseminated protozoan parasite of human and animal cells. Attachment and invasion of host cells results in the formation of a plasma membrane-derived vacuole, the parasitophorous vacuole (PV)(Plattner, 2008) which is disconnected from classical host vesicular trafficking pathways (Plattner, 2008). Nevertheless, the PV acquires extracellular LDL-cholesterol by intercepting post-lysosomal cholesterol-loaded vesicles destined for the ER via a non canonical pathway independent of host fusion proteins (Sehgal, 2005). This likely occurs by direct translocation of cholesterol-loaded lysosomes into the PV lumen in a process mediated by Gra7, a parasite protein secreted from dense granules (Coppens, 2006b). The close apposition of PV membranes with mitochondria and ER, the latter mediated by parasite protein Rop2, (Sinai and Joiner, 2001; Sinai, 1997) may also allow lipid acquisition by a direct membrane transfer. Several dense granule proteins (Gra2, 4, 6, 9 and 12) localize to a membrane tubular network (MTN), connecting the PV membrane with the parasite, presumably to increase membrane surface area for nutrient acquisition. Interestingly, however, Gra2 deletion mutants that fail to form MTNs are attenuated for acute infection in mice but not in cultured fibroblasts (Mercier et al., 1998).
In HeLa cells, autophagy is required for parasite replication, and atg5−/− mouse embryo fibroblast cells are less permissive for growth (Wang, 2009). In contrast, activated macrophages use autophagosome formation and fusion with lysosomes to clear T. gondii infections (Andrade et al., 2006; Ling, 2006). In astrocytes parasite clearance, although dependent on atg5 is not inhibited by autophagy inhibitors and the PV is seen to fuse with the ER prior to its disruption (Halonen, 2009). Interestingly, mice with macrophage-specific atg5 deficiencies are more susceptible to T. gondii infection (Zhao et al., 2008) indicating that in the context of a systemic infection, the host autophagic pathways in immune effector cells are critical in pathogen control. However autophagy proteins such as Atg5 may mediate parasite clearance independent of their role in autophagy, via recruitment of interferon regulated GTPases (IGTP) (Zhao et al., 2008).
2. Molecular determinants of pathogenic vacuole integrity
While pathogens can escape from membrane bound compartments, it is apparent that residence in the cytoplasm is not necessarily an advantage, and can lead to the engagement of powerful antimicrobial responses in professional phagocytic cells and the onset of robust inflammatory responses. Many potential pathways may be critical for maintenance of pathogenic vacuoles.
Interactions with the cytoskeleton and cytoskeletal motors
The cytoskeleton and cytoskeletal motors play a critical role in organelle-positioning and membrane traffic. Not surprisingly, many pathogens co-opt cytoskeletal functions to maintain and stabilize their intracellular niches (Figure-2A). The mature SCV is surrounded by an F-actin network that requires the secreted bacterial effector SteC, a Raf-like kinase (Poh et al., 2008). SifA may also contribute to actin assembly at the SCV via its interaction with RhoA (Ohlson, 2008). Prolonged treatment with F-actin depolymerizing agents or inhibition of the actin motor myosin causes loss of SCV integrity and cytoplasmic exposure of bacteria (Meresse, 2001; Wasylnka, 2008).
Figure 2. Integrity of pathogenic vacuoles is essential for avoidance of host immune surveillance.
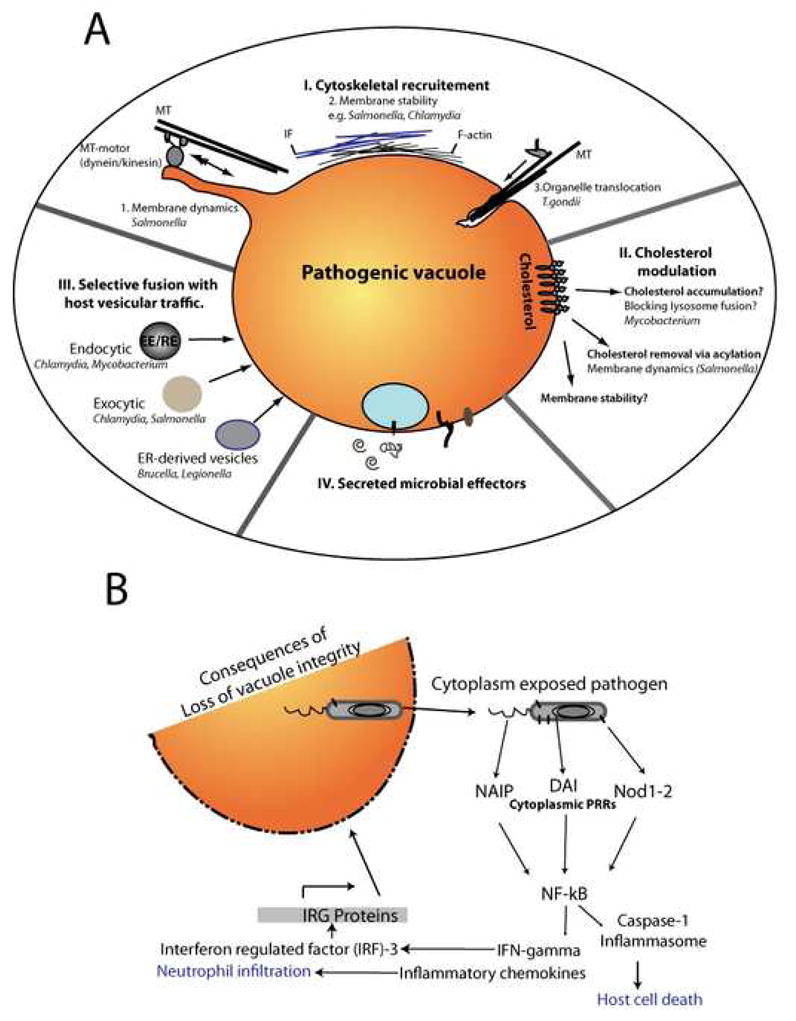
A. Pathogens such as Chlamydiae and Salmonella modulate interactions with host cytoskeleton and cytoskeletal motors (I), which may directly influence structural stability as well as membrane dynamics of their vacuoles. In T. gondii, interactions with host MTs mediate acquisition of host membranes via organelle scavenging. Interception of host membrane traffic is also mediated by differential interactions with host fusion machinery (II). Modulation of vacuolar membrane lipids especially cholesterol (III) may further influence vacuolar stability. Pathogens likely regulate the unique properties and interactions of their vacuoles via secretion of effector proteins (IV). B. Maintenance of vacuolar integrity is essential to avoid cytoplasmic immune defense pathways that include detection of microbial ligands by pathogen recognition receptors (PRRs). Such recognition is followed by activation of diverse pathways that can trigger not only host cell death and adaptive immune responses but also direct disruption of pathogenic vacuoles by IRG proteins.
The role of the actin cytoskeleton in the integrity of the MPV is less clear. In contrast to the SCV, assembly of F-actin is inhibited at the MPV (Anes, 2003). Because treatment of infected cells with lipids that stimulate actin assembly (ceramide and sphingosine) promote phagosomal maturation and bacterial killing, it has been proposed that Mycobacteria inhibits actin assembly at the MPV as a defense mechanism (Anes, 2003). Unlike the SCV and the MPV, the chlamydial inclusion is a large organelle that exhibits structural rigidity within intact infected cells. This is achieved by forming a structural scaffold consisting of F-actin and intermediate filaments (vimentin and cytokeratins) (Kumar, 2008). These structures are dynamic and require the GTPase RhoA and a bacterial protease (CPAF) to increase the flexibility of the intermediate filament network (Kumar, 2008). Disruption of this dynamic scaffold leads to a loss of vacuole integrity and the leakage of inclusion contents to the host cytoplasm. Other actin-binding proteins like α-adducin also localize to the inclusion and may play a role in F-actin ring assembly and maintenance (Chu, 2008).
The T. gondii PV recruits γ-tubulin and nucleates microtubule growth in vivo leading to a major reorganization of the microtubule network (Walker, 2008). Microtubule-dependent deformations of the PV membrane facilitates the internalization of host organelles such as lysosomes and recycling endosomes (Coppens, 2006a). In addition, like in Chlamydia vacuoles, vimentin is reorganized around the PV (Halonen, 1994). Although T. gondii replication is unaffected in vimentin deficient fibroblasts (Sehgal, 2005), whether by analogy to Chlamydia the intermediate filament reorganization influences PV stability, is unclear.
Host membrane trafficking pathways
MTs and actin filaments are required for the transport of vesicles between membrane-bound organelles. The specificity of membrane fusion events is controlled by SNAREs, Rab proteins and tethering factors (Pfeffer, 2007). Not surprisingly, many intracellular pathogens modulate Rab recruitment for the establishment of replicative vacuoles (Brumell and Scidmore, 2007). Expression of dominant negative forms of Rab7, or constitutively active forms of Rab5 disrupts integrity of the SCV and increases the frequency of cytoplasmic bacteria (Brummel, 2002b), indicating that membrane trafficking events mediated by these Rabs contributes to vacuolar integrity. The mycobacterial phagosome requires Rab14 to maintain its early endosome-like characteristics (Kyei, 2006) while inhibition of Rab5 reduces growth by limiting access to iron-rich early endosomes (Kelley and Schorey, 2003). Similarly, dominant negative Rab1 prevents delivery of ER markers to the LCV and impairs bacterial survival (Kagan et al., 2004). Whether interfering with Rab function disrupts MPV or LCV integrity has not been explored.
Cholesterol modulation at the pathogenic vacuole
Cholesterol is an important structural component of membranes and an essential organizer of membrane subdomains (Edidin, 2003). Many membrane-bound pathogens accumulate cholesterol on their PVs, and inhibition of cholesterol biosynthesis and transport pathways negatively impacts pathogen replication. Given its structural role, does modulation of cholesterol levels in PV membranes contribute to vacuole stability? Depletion of cholesterol in macrophages infected with M. avium triggers phagolysosomal fusion and bacterial degradation (de Chastellier and Thilo, 2006). In reconstituted liposomes, mycobacterial lipid Lipoarabinomannan (LAM) disrupts cholesterol rich membranes microdomains suggesting that this may be a mechanism by which it influences phagosome maturation (Hayakawa et al., 2007). The Salmonella effector SseJ, a glycerolipid-cholesterol acyltransferase, similarly depletes cholesterol from the SCV membrane via acylation of free cholesterol (Lossi, 2008). SseJ and SifA regulate membrane tubulation and Sif formation (Ohlson, 2008) indicating a potential role for cholesterol levels in SCV membrane dynamics. Cholesterol depletion from the BCV membrane by Cyclic β-glucans presumably shed from the Brucella periplasm facilitates lysosomal evasion and interactions with ER (Arellano-Reynoso et al., 2005). Cholesterol is also an abundant component of Chlamydia inclusion membranes and free cholesterol is incorporated into bacterial membranes (Carabeo et al., 2003). The role of cholesterol in inclusion stability is not known.
Cholesterol levels also regulate lysosomal function. Accumulation of cholesterol in late endosomes/phagosomes inhibits fusion with lysosomes (Huynh et al., 2008), while cholesterol-depletion (Deng et al., 2009) disrupts lysosome membrane permeability Therefore, cholesterol accumulation in pathogenic vacuoles could represent a strategy to limit lysosomal recognition. Alternatively association of Type-III secretion translocons of several pathogens has been shown to be cholesterol-dependent (Hayward et al., 2005) suggesting that secretion of effectors necessary for vacuole maintenance may require high levels of cholesterol. Although many studies have focused on the role of cholesterol-containing raft domains in pathogen entry, there is limited data on impact of cholesterol depletion on mature pathogenic vacuoles. Whether pathogenic strategies to modulate cholesterol directly or indirectly influences the stability of pathogenic vacuoles remains to be determined.
3. Consequences of disruption of pathogenic vacuoles: Perils and advantages of living in a vacuole
The evolution of complex strategies for intra-vacuolar survival hints at a significant selective advantage. Paradoxically, several lines of evidence indicate life in a vacuole may not be optimal for pathogen replication. In epithelial cells, cytoplasmic Salmonella has a shorter doubling time than membrane-enclosed bacteria (Beuzon et al., 2002). During the exponential growth phase in macrophages, M. tuberculosis is predicted to reside in the cytoplasm (van der Wel, 2007) while at late stages of infection Legionella can replicate in the macrophage cytoplasm (Molmeret et al., 2004). We speculate that the survival advantage gained by life in a membrane-bound organelle is derived from avoidance of cytosolic surveillance (Figure-2B) and the potent inflammatory signaling cascades that they activate.
A. Cytosolic surveillance pathways
The existence of a cytosolic immune surveillance pathway was first identified in studies of cytosolic pathogens Listeria, Francisella and Shigella. In these pathogens, mutants that cannot escape their vacuoles fail to activate NF-kB and Interferon regulated factor-3 (IRF3) dependent immune-related functions (Henry et al., 2007b; O’Riordan et al., 2002; Philpott et al., 2000). Cytoplasmic PRRs of the Nod-like receptor (NLR) family such as Nod1-2, NAIP and DNA-dependent activator of IFN-regulatory factors (DAI), have been implicated in recognition of bacterial ligands like peptidoglycan, DNA and flagellin in the cytoplasm (Martinon et al., 2009). Activation of this pathway leads to pro-inflammatory cytokine production, including type-I interferons, and activation of the inflammasome complex. Inflammasome-mediated cell death has emerged as a central immune defense mechanism against intracellular pathogens. Whether bacterial components of vacuole-bound pathogens can escape vacuoles and trigger similar signaling pathways is unclear although pathogens such as Chlamydia (Nagarajan et al., 2008) and Legionella (Opitz et al., 2006) activate Type-I interferon regulated pathways during infection. Additionally Type-I interferon regulated genes such as Nitric Oxide synthase and Immunity-related GTPases (IRGs) have been implicated in defense against a variety of vacuole-bound pathogens (Decker et al., 2005).
B. IRG P47 GTPases
In murine cells, a family of interferon-regulated GTPases (IRG) determine interferon-mediated resistance to a variety of membrane-bound intracellular pathogens (Taylor, 2007) including Toxoplasma (Taylor et al., 2000), Mycobacteria (MacMicking, 2005), Salmonella (Henry et al., 2007a) and Chlamydia (Bernstein-Hanley et al., 2006). In IFN-γ activated MEFs, IRG proteins accumulate at the surface of the PVs formed by avirulent T. gondii strains and may be involved in the subsequent rupture of PV membrane and parasite release into the host cytoplasm (Zhao, 2009). This is followed by host cell necrosis and parasite death. In contrast more virulent strains prevent the accumulation of IRG proteins to the PV surface. In macrophages, IRG proteins promote MPV maturation (MacMicking et al., 2003) and trigger autophagy-mediated destruction of the MP (Singh et al., 2006) suggesting that the IRG proteins may vary in their mechanism of action against vacuole-bound pathogens.
The microbicidal capacity of the mammalian cell cytoplasm varies significantly in different cell types. For instance while epithelial cells are more permissive for bacterial replication, macrophages are not, as exemplified in the differential ability of Salmonella to replicate in epithelial versus macrophage cytoplasm. Indeed several non-pathogenic bacteria will replicate efficiently in the cytoplasm of the host. For instance B. subtilis expressing Listeriolysin O (LLO) (Bielecki et al., 1990) and E. coli expressing Yersinia invasin and coated with LLO (Monack and Theriot, 2001), can escape intracellular vacuoles and replicate in the macrophage and epithelial cell cytoplasm respectively. The macrophage cytoplasm is rich in antimicrobial molecules such as ubiquicidin (Hiemstra et al., 1993; Hiemstra et al., 1999) requiring additional strategies by cytoplasmic pathogens to counter them. As a direct consequence of this, a pathogen’s choice of a vacuole-sequestered lifestyle may reflect their host cell tropism. Pathogens such as Mycobacterium, Salmonella, Brucella and Legionella target macrophages where vacuole sequestration may be essential for survival. Interestingly, ‘opportunistic’ vacuolar lifestyles have been observed in some cytoplasmic pathogens. In immunocompromised mice and less frequently in macrophages in culture, L. monocytogenes inhabits and replicates in LAMP-1 positive spacious Listeria-containing phagosomes (SLAPS) (Birmingham et al., 2008). In murine macrophages Francisella, after initial escape from phagosomes reenters and inhabits LAMP1 positive autophagosome-like vacuoles (Checroun et al., 2006). This suggests that a vacuolar lifestyle may be the preferred option in situations where host cytoplasmic environment is most potent in its microbicidal capacity with cytoplasmic replication being the exception rather than the rule. All in all vacuolar lifestyles may be an evolutionary response to antimicrobial defense strategies of the host cytoplasm.
Concluding remarks
For many pathogens the evolutionary choice of a sequestered lifestyle within specialized vacuoles over the nutrient rich cytoplasm appears to be based on compromising optimal growth in favor of avoidance of immune surveillance pathways. This may in turn govern both host cell tropism and ability of the pathogen to cause systemic and persistent infections. Although mechanisms of long-term maintenance of pathogenic vacuoles are poorly understood, the integrity and stability of their compartments may be central to pathogenicity. We predict that identification of critical determinants (e.g. microbial effectors and co-opted host pathways) of pathogenic vacuole stability will not only enhance our understanding of parasitic strategies but also offer novel therapeutic avenues.
Acknowledgments
We thank J.D. Dunn for helpful comments. This work was supported by funds from the Burroughs Wellcome Trust Fund. We apologize to scientists’ whose work could not be quoted due to space constraints.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahams GL, Muller P, Hensel M. Functional Dissection of SseF, a Type III Effector Protein Involved in Positioning the Salmonella-Containing Vacuole. Traffic. 2006;7:950–965. doi: 10.1111/j.1600-0854.2006.00454.x. [DOI] [PubMed] [Google Scholar]
- Alonso S, Pethe K, Rusell DG, Purdy GE. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc Natl Acad Sci USA. 2007;104:6031–6036. doi: 10.1073/pnas.0700036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amer AO, Swanson MS. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol. 2005;7:765–778. doi: 10.1111/j.1462-5822.2005.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest. 2006;116:2366–2377. doi: 10.1172/JCI28796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anes E, Kühnel MP, Bos E, Moniz-Pereira J, Habermann A, Griffiths G. Selected lipids activate phagosome actin assembly and maturation resulting in killing of pathogenic mycobacteria. Nat Cell Biol. 2003;5:793–802. doi: 10.1038/ncb1036. [DOI] [PubMed] [Google Scholar]
- Arellano-Reynoso B, Lapaque N, Salcedo S, Briones G, Ciocchini AE, Ugalde R, Moreno E, Moriyon I, Gorvel JP. Cyclic beta-1,2-glucan is a Brucella virulence factor required for intracellular survival. Nat Immunol. 2005;6:618–625. doi: 10.1038/ni1202. [DOI] [PubMed] [Google Scholar]
- Bakowski MA, Briaun V, Brumell JH. Salmonella-containing vacuoles: Directing Traffic and Nesting to Grow. Traffic. 2008;9:2022–2031. doi: 10.1111/j.1600-0854.2008.00827.x. [DOI] [PubMed] [Google Scholar]
- Beatty WL. Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. J Cell Sci. 2006;119:350–359. doi: 10.1242/jcs.02733. [DOI] [PubMed] [Google Scholar]
- Bernstein-Hanley I, Coers J, Balsara ZR, Taylor GA, Starnbach MN, Dietrich WF. The p47 GTPases Igtp and Irgb10 map to the Chlamydia trachomatis susceptibility locus Ctrq-3 and mediate cellular resistance in mice. Proc Natl Acad Sci U S A. 2006;103:14092–14097. doi: 10.1073/pnas.0603338103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuzon CR, Meresse S, Unsworth KE, Ruiz-Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, Holden DW. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 2000;19:3235–3249. doi: 10.1093/emboj/19.13.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuzon CR, Salcedo SP, Holden DW. Growth and killing of a Salmonella enterica serovar Typhimurium sifA mutant strain in the cytosol of different host cell lines. Microbiology. 2002;148:2705–2715. doi: 10.1099/00221287-148-9-2705. [DOI] [PubMed] [Google Scholar]
- Bielecki J, Youngman P, Connelly P, Portnoy DA. Bacillus subtilis expressing a haemolysin gene from Listeria monocytogenes can grow in mammalian cells. Nature. 1990;345:175–176. doi: 10.1038/345175a0. [DOI] [PubMed] [Google Scholar]
- Birmingham CL, Canadien V, Kaniuk NA, Steinberg BE, Higgins DE, Brumell JH. Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature. 2008;451:350–354. doi: 10.1038/nature06479. [DOI] [PubMed] [Google Scholar]
- Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006;281:11374–11383. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- Brombacher E, Urwyler S, Ragaz C, Weber SS, Kami K, Overduin M, Hilbi H. Rab1 Guanine Nucleotide Exchange Factor SidM Is a Major Phosphatidylinositol 4-Phosphate-binding Effector Protein of Legionella pneumophila. J Biol Chem. 2009;284:4846–4856. doi: 10.1074/jbc.M807505200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumell JH, Rosenberger CM, Gotto GT, Marcus SL, Finlay BB. SifA permits survival and replication of Salmonella typhimurium in murine macrophages. Cell Microbiol. 2001;3:75–84. doi: 10.1046/j.1462-5822.2001.00087.x. [DOI] [PubMed] [Google Scholar]
- Brumell JH, Scidmore MA. Manipulation of rab GTPase function by intracellular bacterial pathogens. Microbiol Mol Biol Rev. 2007;71:636–652. doi: 10.1128/MMBR.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummel JH, Goosney DL, Finlay BB. SifA, a type III secreted effector of Salmonella typhimurium, directs Salmonella-induced dilament (sif) formation along microtubules. Traffic. 2002a;3:407–415. doi: 10.1034/j.1600-0854.2002.30604.x. [DOI] [PubMed] [Google Scholar]
- Brummel JH, Tang P, Zaharik ML, Finlay BB. Disruption of Salmonella-containing vacuole leads to increased replication of Salmonella enterica serovar typhimurium in the cytosol of epithelial cells. Infection and Immunity. 2002b;70:3264–3270. doi: 10.1128/IAI.70.6.3264-3270.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabeo RA, Mead DJ, Hackstadt T. Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc Natl Acad Sci U S A. 2003;100:6771–6776. doi: 10.1073/pnas.1131289100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall A. Evolution of intracellular pathogens. Annu Rev Microbiol. 2008;62:19–33. doi: 10.1146/annurev.micro.61.080706.093305. [DOI] [PubMed] [Google Scholar]
- Celli J, de Chastellier C, Franchini D, Pizarro-cerda J, Moreno E, Gorvel J. Brucella Evades Macrophage Killing via VirB-dependent Sustained Interactions with the Endoplasmic Reticulum. J Exp Med. 2003;198:12. doi: 10.1084/jem.20030088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli J, Salcedo SP, Gorvel JP. Brucella coopts small GTPase Sar1 for intracellular replication. Proc Natl Acad Sci USA. 2005;102:1673–1678. doi: 10.1073/pnas.0406873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci U S A. 2006;103:14578–14583. doi: 10.1073/pnas.0601838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HG, Weeks SK, Gilligan DM, Rockey DD. Host alpha-adducin is redistributed and localized to the inclusion membrane in chlamydia- and chlamydophila-infected cells. Microbiology. 2008;154:3848–3855. doi: 10.1099/mic.0.2008/020941-0. [DOI] [PubMed] [Google Scholar]
- Cocchiaro JL, Kumar Y, FIscher E, Hackstadt T, Valdivia RH. Cytoplasmic lipid droplets are translocated into the lumen of the chlamydia trachomatis parasitophorous vacuole. Proc Natl Acad Sci USA. 2008;105:9379–9384. doi: 10.1073/pnas.0712241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, De Maziere A, van Dijk S, Carlsson F, Klumperman J, Brown EJ. Atg5-independent sequestration of ubiquitinated mycobacteria. PLoS Pathog. 2009;5:e1000430. doi: 10.1371/journal.ppat.1000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes BK, Finlay BB. Insertion of the bacterial type III translocon: not your average needle stick. Trends Microbiol. 2005;13:92–95. doi: 10.1016/j.tim.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Coppens I, Dunn JD, Romano JD, Pypaert M, Zhang H, Boothroyd JC, Joiner KA. Toxoplasma gondii Sequesters Lysosomes from Mammalian Hosts in the Vacuolar Space. Cell. 2006a;125:261–274. doi: 10.1016/j.cell.2006.01.056. [DOI] [PubMed] [Google Scholar]
- Coppens I, Sinai AP, Joiner KA. Toxoplasma gondii Exploits Host Low-Density Lipoprotein Receptor-mediated Endocytosis for Cholesterol Acquisition. J Cell Biol. 2006b;179:167–180. doi: 10.1083/jcb.149.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosma CL, Sherman DR, Ramakrishnan L. The secret lives of pathogenic mycobacteria. Annu Rev Microbiol. 2003;57:641–676. doi: 10.1146/annurev.micro.57.030502.091033. [DOI] [PubMed] [Google Scholar]
- de Chastellier C. The many niches and strategies used by pathogenic mycobacteria for survival within host macrophages. Immunobiology. 2009 doi: 10.1016/j.imbio.2008.12.005. epub. [DOI] [PubMed] [Google Scholar]
- de Chastellier C, Thilo L. Cholesterol depletion in Mycobacterium avium-infected macrophages overcomes the block in phagosome maturation and leads to the reversible sequestration of viable mycobacteria in phagolysosome-derived autophagic vacuoles. Cell Microbiol. 2006;8:242–256. doi: 10.1111/j.1462-5822.2005.00617.x. [DOI] [PubMed] [Google Scholar]
- Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675–687. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- Deiwick J, Salcedo SP, Boucrot E, Gilliland SM, Henry T, Petermann N, Waterman SR, Gorvel JP, Holden DW, Meresse S. The translocated Salmonella effector proteins SseF and SseG interact and are required to establish an intracellular replication niche. Infection and Immunity. 2006;74:6965–6972. doi: 10.1128/IAI.00648-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delevoye C, Nilges M, Dehoux P, Paumet F, Perrinet S, Dautry-Varsat A, Subtil A. SNARE protein mimicry by an intracellular bacterium. PLOS Pathog. 2008;14:e1000022. doi: 10.1371/journal.ppat.1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng D, Jiang N, Hao SJ, Sun H, Zhang GJ. Loss of membrane cholesterol influences lysosomal permeability to potassium ions and protons. Biochim Biophys Acta. 2009;1788:470–476. doi: 10.1016/j.bbamem.2008.11.018. [DOI] [PubMed] [Google Scholar]
- Deretic V. Autophagy, an immunological magic bullet: Mycobacterium tuberculosis phagosome maturation block and how to bypass it. Future Microbiol. 2008;3:517–524. doi: 10.2217/17460913.3.5.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edidin M. The state of lipid rafts: from model membranes to cells. Annu Rev Biophys Biomol Struct. 2003;32:257–283. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunological reviews. 2008;227:106–128. doi: 10.1111/j.1600-065X.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-del Portillo F, Zwick MB, Leung KY, Finlay BB. Salmonella induces the formation of filamentous structures containing lysosomal membrane glycoproteins in epithelial cells. Proc Natl Acad Sci U S A. 1993;90:10544–10548. doi: 10.1073/pnas.90.22.10544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorvel JP, Moreno E. Brucella intracellular life: from invasion to intracellular replication. Vet Microbiol. 2002;90:281–297. doi: 10.1016/s0378-1135(02)00214-6. [DOI] [PubMed] [Google Scholar]
- Grassl GA, Finlay BB. Pathogenesis of enteric Salmonella infections. Curr Opin Gastroenterol. 2008;24:22–26. doi: 10.1097/MOG.0b013e3282f21388. [DOI] [PubMed] [Google Scholar]
- Grieshaber SS, Grieshaber NA, Hackstadt T. Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J Cell Sci. 2006;7:3793–3802. doi: 10.1242/jcs.00695. [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Hackstadt T, Scidmore-Carlson MA, Shaw EI, Fischer ER. The Chlamydia trachomatis IncA protein is required for homotypic vesicle fusion. Cell Microbiol. 1999;1:119–130. doi: 10.1046/j.1462-5822.1999.00012.x. [DOI] [PubMed] [Google Scholar]
- Halonen SK. Role of autophagy in the host defense against Toxoplasma gondii in astrocytes. Autophagy. 2009;5:268–269. doi: 10.4161/auto.5.2.7637. [DOI] [PubMed] [Google Scholar]
- Halonen SK, Weidner E. Overcoating of Toxoplasma parasitopherous vacuoles with host cell vimentin type intermediate filaments. J Eukaryot Microbiol. 1994;41:65–71. doi: 10.1111/j.1550-7408.1994.tb05936.x. [DOI] [PubMed] [Google Scholar]
- Hayakawa E, Tokumasu F, Nardone GA, Jin AJ, Hackley VA, Dvorak JA. A Mycobacterium tuberculosis-derived lipid inhibits membrane fusion by modulating lipid membrane domains. Biophys J. 2007;93:4018–4030. doi: 10.1529/biophysj.107.104075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward RD, Cain RJ, McGhie EJ, Phillips N, Garner MJ, Koronakis V. Cholesterol binding by the bacterial type III translocon is essential for virulence effector delivery into mammalian cells. Mol Microbiol. 2005;56:590–603. doi: 10.1111/j.1365-2958.2005.04568.x. [DOI] [PubMed] [Google Scholar]
- Henry SC, Daniell X, Indaram M, Whitesides JF, Sempowski GD, Howell D, Oliver T, Taylor GA. Impaired macrophage function underscores susceptibility to Salmonella in mice lacking Irgm1 (LRG-47) J Immunol. 2007a;179:6963–6972. doi: 10.4049/jimmunol.179.10.6963. [DOI] [PubMed] [Google Scholar]
- Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J Exp Med. 2007b;204:987–994. doi: 10.1084/jem.20062665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer D, Lipinski AR, Machuy N, Karlas A, Wehrens A, Siedler F, Brinkmann V, Meyer TF. Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature. 2009;457:731–735. doi: 10.1038/nature07578. [DOI] [PubMed] [Google Scholar]
- Hiemstra PS, Eisenhauer PB, Harwig SS, van den Barselaar MT, van Furth R, Lehrer RI. Antimicrobial proteins of murine macrophages. Infect Immun. 1993;61:3038–3046. doi: 10.1128/iai.61.7.3038-3046.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiemstra PS, van den Barselaar MT, Roest M, Nibbering PH, van Furth R. Ubiquicidin, a novel murine microbicidal protein present in the cytosolic fraction of macrophages. J Leukoc Biol. 1999;66:423–428. doi: 10.1002/jlb.66.3.423. [DOI] [PubMed] [Google Scholar]
- Hope JC, Thom ML, McCormick PA, Howard CJ. Interaction of antigen presenting cells with mycobacteria. Vet Immunol Immunopathol. 2004;100:187–195. doi: 10.1016/j.vetimm.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Huynh KK, Gershenzon E, Grinstein S. Cholesterol accumulation by macrophages impairs phagosome maturation. J Biol Chem. 2008;283:35745–35755. doi: 10.1074/jbc.M806232200. [DOI] [PubMed] [Google Scholar]
- Ingmundson A, Delprato A, Lambright DG, Roy CR. Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature. 2007;450:365–369. doi: 10.1038/nature06336. [DOI] [PubMed] [Google Scholar]
- Isberg RR, O’Connor TJ, Heidtman M. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat Rev Microbiol. 2009;7:13–24. doi: 10.1038/nrmicro1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Roy CR. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat Cell Biol. 2002;4:945–954. doi: 10.1038/ncb883. [DOI] [PubMed] [Google Scholar]
- Kagan JC, Stein MP, Pypaert M, Roy CR. Legionella subvert the functions of Rab1 and Sec22b to create a replicative organelle. J Exp Med. 2004;199:1201–1211. doi: 10.1084/jem.20031706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley VA, Schorey JS. Mycobacterium’s arrest of phagosome maturation in macrophages requires Rab5 activity and accessibility to iron. Mol Biol Cell. 2003;14:3366–3377. doi: 10.1091/mbc.E02-12-0780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y, Valdivia RH. Actin and Intermediate filaments stabilize the Chlamydia trachomtis vacuole by forming dynamic structural scaffolds. Cell Host & Microbe. 2008;4:159–169. doi: 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyei GB, Vergne I, Chua J, Roberts E, Harris J, Junutula JR, Deretic V. Rab14 is critical for maintenance of Mycobacterium tuberculosis phagosome maturation arrest. EMBO J. 2006;25:5250–5259. doi: 10.1038/sj.emboj.7601407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJP, Yap GS. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med. 2006;203:2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossi NS, Rolhion N, Magee AI, Boyle C, Holden DW. The Salmonella SPI-2 effector SseJ exhibits eukaryotic activator-dependent phospholipase A and glycerophospholipid: cholesterol acyltransferase activity. Microbiology. 2008;154:2680–2688. doi: 10.1099/mic.0.2008/019075-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGurn JA, Cox JS. A genetic screen for Mycobacterium tuberculosis mutants defective for phagosome maturation arrest identifies components of the ESX-1 secretion system. Infect Immun. 2007;75:2668–2678. doi: 10.1128/IAI.01872-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machner MP, Isberg RR. A bifunctional bacterial protein links GDI displacement to Rab1 activation. Science. 2007;318:974–977. doi: 10.1126/science.1149121. [DOI] [PubMed] [Google Scholar]
- MacMicking JD. Immune control of phagosomal bacteria by p47 GTPases. Current Opinion in Microbiology. 2005;8:74–82. doi: 10.1016/j.mib.2004.12.012. [DOI] [PubMed] [Google Scholar]
- MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science. 2003;302:654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- Mercier C, Howe DK, Mordue D, Lingnau M, Sibley LD. Targeted disruption of the GRA2 locus in Toxoplasma gondii decreases acute virulence in mice. Infect Immun. 1998;66:4176–4182. doi: 10.1128/iai.66.9.4176-4182.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meresse S, Steele-Mortimer O, Moreno E, Desjardins M, Finlay B, Gorvel JP. Controlling the maturation of pathogen-containing vacuoles: a matter of life and death. Nat Cell Biol. 1999;1:E183–188. doi: 10.1038/15620. [DOI] [PubMed] [Google Scholar]
- Meresse S, Unsworth KE, Habermann A, Griffiths G, Fang F, Martinez-Lorenzo MJ, Watermann SR, Gorvel JP, Holden DW. Remodelling of the actin cytoskeleton is essential for replication of intravacuolar Salmonella. Cell Microbiol. 2001;3:567–577. doi: 10.1046/j.1462-5822.2001.00141.x. [DOI] [PubMed] [Google Scholar]
- Molmeret M, Bitar DM, Han L, Kwaik YA. Disruption of the phagosomal membrane and egress of Legionella pneumophila into the cytoplasm during the last stages of intracellular infection of macrophages and Acanthamoeba polyphaga. Infect Immun. 2004;72:4040–4051. doi: 10.1128/IAI.72.7.4040-4051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM, Theriot JA. Actin-based motility is sufficient for bacterial membrane protrusion formation and host cell uptake. Cell Microbiol. 2001;3:633–647. doi: 10.1046/j.1462-5822.2001.00143.x. [DOI] [PubMed] [Google Scholar]
- Nagarajan UM, Prantner D, Sikes JD, Andrews CW, Jr, Goodwin AM, Nagarajan S, Darville T. Type I interferon signaling exacerbates Chlamydia muridarum genital infection in a murine model. Infect Immun. 2008;76:4642–4648. doi: 10.1128/IAI.00629-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Riordan M, Yi CH, Gonzales R, Lee KD, Portnoy DA. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc Natl Acad Sci U S A. 2002;99:13861–13866. doi: 10.1073/pnas.202476699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- Ohlson MB, Huang Z, Alto NM, Blanc MP, Dixon JE, Chai J, Miller SI. Structure and function of salmonella SifA indicate that its interaction with SKIP, SseJ, and RhoA family GTPases induce endosomal tubulation. Cell Host & Microbe. 2008;4:434–446. doi: 10.1016/j.chom.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz B, Vinzing M, van Laak V, Schmeck B, Heine G, Gunther S, Preissner R, Slevogt H, N’Guessan PD, Eitel J, et al. Legionella pneumophila induces IFNbeta in lung epithelial cells via IPS-1 and IRF3, which also control bacterial replication. J Biol Chem. 2006;281:36173–36179. doi: 10.1074/jbc.M604638200. [DOI] [PubMed] [Google Scholar]
- Otto GP, Wu MY, Clarke M, Lu H, Anderson OR, Hilbi H, Shuman HA, Kessin RH. Macroautophagy is dispensable for intracellular replication of Legionella pneumophila in Dictyostelium discoideum. Mol Microbiol. 2004;51:63–72. doi: 10.1046/j.1365-2958.2003.03826.x. [DOI] [PubMed] [Google Scholar]
- Perrin AJ, Jiang X, Birmingham CL, So NSY, Brumell JH. Recognition of bacteria in the cytosol of mammalian cells by the ubiquitin system. Current Biology. 2004;14:806–811. doi: 10.1016/j.cub.2004.04.033. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR. Unsolved mysteries in membrane traffic. Annu Rev Biochem. 2007;76:629–645. doi: 10.1146/annurev.biochem.76.061705.130002. [DOI] [PubMed] [Google Scholar]
- Philips JA. Mycobacterial manipulation of vacuolar sorting. Cell Microbiol. 2008;10:2408–2415. doi: 10.1111/j.1462-5822.2008.01239.x. [DOI] [PubMed] [Google Scholar]
- Philpott DJ, Yamaoka S, Israel A, Sansonetti PJ. Invasive Shigella flexneri activates NF-kappa B through a lipopolysaccharide-dependent innate intracellular response and leads to IL-8 expression in epithelial cells. J Immunol. 2000;165:903–914. doi: 10.4049/jimmunol.165.2.903. [DOI] [PubMed] [Google Scholar]
- Pizarro-cerda J, Meresse S, Parton RG, van der goot G, Sola-landa A, Lopez-goni I, Moreno E, Gorvel J. Brucella abortus Transits through the Autophagic Pathway and Replicates in the Endoplasmic Reticulum of Nonprofessional Phagocytes. Infection and Immunity. 1998a;66:5711–5724. doi: 10.1128/iai.66.12.5711-5724.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-cerda J, Moreno E, Sanguedolce V, Mege J, Gorvel J. Virulent Brucella abortus prevents lysosome fusion and is distributed within autophagosome-like compartments. Infection and Immunity. 1998b;66:2387–2392. doi: 10.1128/iai.66.5.2387-2392.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner F, Soldati-Favre D. Hijacking of Host Cellular Functions by the Apicomplexa. Annu Rev Microbiol. 2008;62:471–487. doi: 10.1146/annurev.micro.62.081307.162802. [DOI] [PubMed] [Google Scholar]
- Poh J, Odendall C, Spanos A, Boyle C, Liu M, Freemont P, Holden DW. SteC is a Salmonella kinase required for SPI-2-dependent F-actin remodelling. Cell Microbiol. 2008;10:20–30. doi: 10.1111/j.1462-5822.2007.01010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Pei J, Ancona V, SHaw BD, Ficht TA, de Figueiredo P. RNAi screen of endoplasmic reticulum-associated host factors reveals a role for IRE1a in supporting Brucella replication. PLOS Pathog . 2008:4. doi: 10.1371/journal.ppat.1000110. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragaz C, Pietsch H, Urwyler S, Tiaden A, Weber SS, Hilbi H. The Legionella pneumophila phosphatidylinositol-4 phosphate-binding type IV substrate SidC recruits endoplasmic reticulum vesicles to a replication-permissive vacuole. Cell Microbiol. 2008;10:2416–2433. doi: 10.1111/j.1462-5822.2008.01219.x. [DOI] [PubMed] [Google Scholar]
- Ramachandra L, Simmons D, Harding CV. MHC molecules and microbial antigen processing in phagosomes. Curr Opin Immunol. 2009 doi: 10.1016/j.coi.2009.01.001. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden AE, Mota LJ, Munter S, Shorte SL, Holden DW. The SPI-2 type III secretion system restricts motility of Salmonella-containing vacuoles. Cell Microbiol. 2007;9:2517–2529. doi: 10.1111/j.1462-5822.2007.00977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockey DD, Scidmore MA, Bannantine JP, Brown WJ. Proteins in the chlamydial inclusion membrane. Microbes Infect. 2002;4:333–340. doi: 10.1016/s1286-4579(02)01546-0. [DOI] [PubMed] [Google Scholar]
- Ruiz-Albert J, Yu XJ, Beuzon CR, Blakey AN, Galyov EE, Holden DW. Complementary activities of SseJ and SifA regulate dynamics of the Salmonella typhimurium vacuolar membrane. Mol Microbiol. 2002;44:645–661. doi: 10.1046/j.1365-2958.2002.02912.x. [DOI] [PubMed] [Google Scholar]
- Rzomp KA, Moorhead AR, Scidmore MA. The GTPase Rab4 interacts with Chlamydia trachomatis inclusion membrane protein CT229. Infect Immun. 2006;74:5362–5373. doi: 10.1128/IAI.00539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzomp KA, Scholtes LD, Briggs BJ, Whittaker GR, Scidmore MA. Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infection and Immunity. 2003;71:5855–5870. doi: 10.1128/IAI.71.10.5855-5870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo SP, Holden DW. SseG, a virulence protein that targets Salmonella to the Golgi network. EMBO J. 2003;22:5003–5014. doi: 10.1093/emboj/cdg517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal A, Bettiol S, Pypaert M, Wenk MR, Kaasch A, Blader IJ, Joiner KA, Coppens I. Peculiarities of Host Cholesterol Transport to the Unique Intracellular Vacuole Containing Toxoplasma. Traffic. 2005;6:1125–1141. doi: 10.1111/j.1600-0854.2005.00348.x. [DOI] [PubMed] [Google Scholar]
- Sinai AP, Joiner KA. The Toxoplasma gondii protein ROP2 mediates host organelle association with the parasitophorous vacuole membrane. J Cell Biol. 2001;154:95–108. doi: 10.1083/jcb.200101073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinai AP, Webster P, Joiner KA. Association of host cell endoplasmic reticulum and mitochondria with the Toxoplasma gondii parasitophorous vacuole membrane: a high affinity interaction. J Cell Sci. 1997;100:2117–2128. doi: 10.1242/jcs.110.17.2117. [DOI] [PubMed] [Google Scholar]
- Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- Smith J, Manoranjan J, Pan M, Bohsali A, Xu J, Lu J, McDonald KL, Szyk A, LaRonde-LeBlanc N, Gao LY. Evidence for pore formation in host ell membranes by ESX-1 secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infection and Immunity. 2008;76:5478–5487. doi: 10.1128/IAI.00614-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T, Nd TW, Wehrly TD, Knodler LA, Celli J. Brucella Intracellular Replication Requires Trafficking Through the Late Endosomal/Lysosomal Compartment. Traffic. 2008;9:678–694. doi: 10.1111/j.1600-0854.2008.00718.x. [DOI] [PubMed] [Google Scholar]
- Stein MA, Leung KY, Zwick M, Garcia-del Portillo F, Finlay BB. Identification of a Salmonella virulence gene required for formation of filamentous structures containing lysosomal membrane glycoproteins within epithelial cells. Mol Microbiol. 1996;20:151–164. doi: 10.1111/j.1365-2958.1996.tb02497.x. [DOI] [PubMed] [Google Scholar]
- Swanson MS, Isberg RR. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun. 1995;63:3609–3620. doi: 10.1128/iai.63.9.3609-3620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor GA. IRG proteins: key mediators of interferon-regulated host resistance to intracellular pathogens. Cell Microbiol. 2007;9:1099–1107. doi: 10.1111/j.1462-5822.2007.00916.x. [DOI] [PubMed] [Google Scholar]
- Taylor GA, Collazo CM, Yap GS, Nguyen K, Gregorio TA, Taylor LS, Eagleson B, Secrest L, Southon EA, Reid SW, et al. Pathogen-specific loss of host resistance in mice lacking the IFN-gamma-inducible gene IGTP. Proc Natl Acad Sci U S A. 2000;97:751–755. doi: 10.1073/pnas.97.2.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ. M. tuberculosis and M. Leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. 2007;129:1287–1298. doi: 10.1016/j.cell.2007.05.059. [DOI] [PubMed] [Google Scholar]
- Walker ME, Hjort EE, Smith SS, Tripathi A, Hornick JE, Hinchcliffe EH, Archer W, Hager KM. Toxoplasma gondii actively remodels the microtubule network in host cells. Microbes Infect. 2008;10:1440–1449. doi: 10.1016/j.micinf.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Weiss LM, Orlofsky A. Host cell autophagy is induced by Toxoplasma gondii and contributes to parasite growth. J Biol Chem. 2009;284:1694–1701. doi: 10.1074/jbc.M807890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasylnka JA, Bakowski MA, Szeto J, Ohlson MB, Trimble WS, Miller SI, Brumell JH. A role for Myosin II in regulating positioning of Salmonella-containing vacuoles and intracellular replication. Infection and Immunity. 2008;76:2722–2735. doi: 10.1128/IAI.00152-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HB, Finlay BB. The Caspase-1 inflammasome: A pilot of innate immune responses. Cell Host & Microbe. 2008;4:198–208. doi: 10.1016/j.chom.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Zhao YO, Khaminets A, Hunn JP, Howard JC. Disruption of the Toxoplasma gondii parasitopherous vacuole by IFNgamma-inducible immunity-related GTPases (IRG proteins(triggers necrotic cell death. PLOS Pathog. 2009;5:e1000288. doi: 10.1371/journal.ppat.1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]