

Abstract
Trans-generational epigenetic phenomena, such as endocrine-disrupting chemicals (EDCs) that decrease fertility and the global methylation status of DNA in the offspring, are of great concern because they may affect the health of our children. However, of even greater concern is the possibility that trans-generational changes in the methylation status of the DNA might lead to permanent changes in the DNA sequence itself. By contaminating the environment with EDCs, mankind might be permanently affecting the health of future generations. In this chapter, we present evidence from our laboratory and others that trans-generational epigenetic changes in DNA might lead to mutations directed to genes encoding amino acid repeat-containing proteins (RCPs) that are important for adaptive evolution or cancer progression. Such epigenetic changes can be induced “naturally” by hormones or “unnaturally” by EDCs or environmental stress. To illustrate the phenomenon, we present new bioinformatic evidence that the only RCP ontological categories conserved from Drosophila to humans are “regulation of splicing,” “regulation of transcription,” and “regulation of synaptogenesis,” which are precisely the classes of genes that are important for evolutionary processes. Based on that and other evidence, we propose a model for evolution that we call the EDGE (Epigenetically Directed Genetic Errors) hypothesis for the mechanism by which mutations are targeted at epigenetically-modified “contingency genes” encoding RCPs. In the model, “epigenetic assimilation” of metastable epialleles of RCPs over many generations can lead to mutations directed to those genes, thereby permanently stabilizing the adaptive phenotype.
Keywords: Repeat Containing Proteins (RCPs), neuroendocrine signaling, evolution, cancer, epigenetics
1. Introduction
Nothing in biology makes sense except in the light of evolution.[1]
Theodosius Dobzhansky (1900–1975), who had a long, distinguished career as an evolutionary geneticist, wrote a short paper with the above title near the end of his life to emphasize the central role of evolution in understanding biological processes. The title is a paraphrase of a statement in his classic 1955 textbook on evolutionary biology [2]. In 1937 he had published a model of evolution called the “modern synthesis,” which links Darwinian evolutionary biology with Mendelian-Morganist genetics [3]. Also in 1937, he had published Genetics and the Origin of Species, a book in which he summarized many aspects of the “modern synthesis” model of evolution as "a change in the frequency of an allele within a gene pool" [4].
Dobzhansky’s training and initial research in Russia emphasized the role of the environment in influencing phenotypic traits. The role of the environment in shaping evolution is an area that his most illustrious student, Lewontin continued, as illustrated in Lewontin’s book, The Triple Helix [5]. However, most other practitioners of the modern synthesis minimized non-Mendelian ideas such as the role of the environment or the inheritance of acquired characteristics. The latter idea was first popularized in 1809 by Jean Baptiste Lamarck [6]. The classic Lamarckian example (although he did not actually use it himself) is that a giraffe has a long neck because successive generations have stretched their necks to eat the leaves on the tops of the trees.
We note that Darwin himself believed in Lamarckian-type inheritance [7]. That is succinctly stated in the last paragraph of Origin of the Species, where Darwin proposed that heritable variation stems from “use and disuse” [7]. In Darwin’s Lamarck-inspired “pangenesis” hypothesis, he proposed that any environmental phenomenon that affects a parent also affects “gemmules” that transfer through the blood (neuroendocrine hormones?) and accumulate in the male and female reproductive organs [7]. The modified “gemmules” then modify the phenotypes of the progeny in the same manner that the parents’ phenotype was modified by the environment [7].
Ironically, it was Darwin’s cousin, Francis Galton, who helped “disprove” (at least according to popular sentiment) the “pangenesis” hypothesis. In 1871, Galton transferred massive amounts of blood between two different colored rabbits and failed to find donor coat color inherited in the progeny [8]. For this and other reasons, Darwin’s “pangenesis” hypothesis went quickly out of favor. However, as noted above, one could loosely translated “gemmules” as blood-borne “neuroendocrine hormones,” which had not yet been discovered in Darwin’s time. Therefore, the “pangenesis” hypothesis can be construed as an 18th century version of a neuroendocrine-mediated evolutionary hypothesis lacking 21st century molecular knowledge.
Despite the ridicule and scorn that such non-genetic ideas still generate in many biologists, recent studies in epigenetic inheritance of traits that are induced by environmental agents has necessitated reconsideration of the controversial ideas of Lamarckian-type inheritance mechanisms. For example, as described in the next section, much has been learned about the role of epigenetics in cancer initiation and development. Based on the cancer studies, we wish to turn Dobzhansky’s quotation on its head and argue that nothing in evolution makes sense except in the light of cancer biology.
2. The role of epigenetics in cancer initiation
Feinberg and colleagues (2006) have proposed that cancer has an epigenetic origin [9]. Their hypothesis is summarized in the following quotation:
[T]umour cell heterogeneity is due in part to epigenetic variation in progenitor cells, and epigenetic plasticity together with genetic lesions drives tumour progression. This crucial early role for epigenetic alterations in cancer is in addition to epigenetic alterations that can substitute for genetic variation later in tumour progression. [9]
Feinberg et al. have recently proposed an “epigenetic progenitor origin” hypothesis for the development of cancer [9]. They propose that crucial events in cancer initiation are epigenetic and that further genetic or epigenetic alterations in the DNA or histones can lead to the progression of cancer to more aggressive states. The insight that cancer may be an initially epigenetic disease suggests that morphological evolution, or evolution in general, might also be an initially epigenetic process.
Cancer epigenetics is an active and well-accepted area of research, but the role of epigenetics in evolutionary processes remains controversial. In this chapter, we attempt to incorporate recent ideas on the epigenetic basis of cancer into a novel “neo-Larmackian” mechanism for rapid morphological evolution. Our model, which we term EDGE (Epigenetically Directed Genetic Error), proposes that heritable epigenetic changes can lead to directed mutations in DNA to stabilize an environmentally induced phenotype. That is, we propose that epigenetic modifications of the DNA or chromatin, such as methylation of the DNA or acetylation or methylation of histones, can lead to mutations in precisely the classes of genes that govern the phenotype during development and consequently increase survival of the organism. The idea is that survival-based selection of “epigenetic marks” on the DNA can lead to cancer progression within the lifespan of an individual or can accelerate changes in morphology or behavior over several generations via “epigenetic assimilation” of a novel phenotype.
In the following sections, we review literature evidence and present new evidence to support the EDGE hypothesis. In the process, we show how the EDGE hypothesis can explain aspects of cancer progression within an individual and rapid morphological evolution within a population.
3. The beginning of the EDGE
Barbara McClintock’s research on transposon mobilization in maize predated studies on DNA methylation or histone modifications. Nevertheless, she was an early pioneer in suggesting that the environment can affect genome structure. She said:
In the future, attention undoubtedly will be centered on the genome, with greater appreciation of its significance as a highly sensitive organ of the cell that monitors genomic activities and corrects common errors, senses unusual and unexpected events, and responds to them, often by restructuring the genome. [10]
McClintock made that remark when she received the 1983 Nobel Prize in Physiology or Medicine [10]. What she proposed in her Nobel lecture, which was controversial at the time, is that environmental stress causes the mobilization of transposons, in maize at least, and that the mobilization can lead to dramatic rearrangement of the plant’s genome in a last-ditch attempt to adapt to the new environment. In a book (which we will quote extensively in this chapter) that proposes an important role of epigenetic and environmental influences in evolution, Jablonka and Lamb cite McClintock’s Nobel Prize speech as one of the best and earliest examples of the renaissance in neo-Lamarckian thinking [11].
As noted by Jablonka and Lamb (2005), McClintock’s Nobel Prize speech brings to mind the controversial Lamarckian idea that environmental stress can directly influence the phenotype of the progeny of stressed individuals. In modern terms, McClintock proposed that the environment can “direct” mutations or rearrangements of the genome. For our purposes, we define a “directed mutation” as an environmentally-induced DNA change in a particular class of gene(s) that allows adaptation to a new environmental stress. However, there is a broad spectrum of different types of mutations: 1) completely random ones, which have no specificity; 2) semidirected ones, which is what McClintock had in mind because “controlling elements” (transposons) in maize show highly selective targeting; and 3) completely beneficial directed mutations. Many biologists consider a “directed mutation” as only the extreme case, such as the reversion of a nonsense point mutation to wild-type, but we will extend the term to include semidirected mutations in this chapter.
In a discussion of McClintock’s speech, Jablonka and Lamb comment as follows:
The new genetic variation that is produced in stressful conditions (e.g., after a sharp temperature change or prolonged starvation) is semidirected in the sense that it is a response to environmental signals, but it does not lead to a unique and necessarily adaptive response. It falls somewhere between totally blind variations, which are specific neither in their nature nor in the time and site in the genome where they occur, and totally directed variations, which are reproducible adaptive changes that occur at specific sites in response to specific stimuli. [11] , p. 89
We propose that the genomes of multicellular organisms, from Drosophila to humans, have “contingency genes” that encode repeat-containing proteins (RCPs), which are so called because they have (hypervariable) numbers of repeat sequences. However, unlike bacterial pathogens, which use “contingency genes” for evading host defense systems [12], animals use them for rapid morphological evolution, neuroendocrine-signaling, and other evolutionary processes. In the next few sections, we describe a possible mechanism by which random mutations can be directed to “contingency genes.”
4. Genetic and epigenetic assimilation experiments in Drosophila
In 1942, Conrad Waddington pioneered “genetic assimilation” experiments for new phenotypes induced by environmental stress [13]. However, recent studies in our laboratory have suggested that “epigenetic assimilation” of stress-induced phenotypes can also occur. The potential utility of “epigenetic assimilation” is illustrated in the following quotation from Jablonka and Lamb (2005):
The genetic system can transmit alternative states, but there are several reasons why epigenetic variants are often more appropriate. The first is that the rate at which they are produced is usually far greater than for mutations; if conditions change frequently, this can be a significant advantage. The second and related reason is that epigenetic variants are often readily reversible, whereas mutations usually are not. The third reason is that the production and reversion of epigenetic variants may be functionally linked to the changing circumstances; this is rarely true for mutations. [11] , p. 324
We became interested in the role of trans-generational epigenetic mechanisms in rapid morphological evolution accidentally. We were looking for genetic suppressors and enhancers of a small-eye phenotype in Drosophila that was caused by an allele of Krüppel (German for cripple), KrüppelIrregular facets-1 (KrIf-1). The mutation causes Krüppel, a zinc finger transcription factor, to be ectopically expressed in the eye imaginal disc during larval development. Krüppel is normally required for some thoracic and abdominal segmentation in the embryo, but ectopic expression in the eye imaginal disc triggers apoptotic cell death, and thereby reduces the size of the eye.
In a screen of random chromosomal regions that affect the KrIf-1 small-eye phenotype, we discovered that maternal loss of one copy of Hsp90 (called Hsp83 in Drosophila) or maternal loss of one copy of any one of a number of Trithorax Group (TrxG) genes, which encode chromatin-remodeling enzymes, causes ectopic large bristle outgrowths (ELBOs). ELBOs often look like proximal appendage tissues (Fig. 1b) [14]. What was remarkable about the ELBOs was that only maternal loss had an effect. Paternal loss of Hsp90 or TrxG genes did not generate ELBOs in KrIf-1 flies [14].
Figure 1. Genetic assimilation of crossveinless phenocopy and epigenetic assimilation of the ELBO phenotype.

a) Shown are a crossveinless wing and a wild-type wing (modified from [16]).
b) An eye containing an ectopic large bristle outgrowth (ELBO) in one eye and no outgrowth in the other eye in a KrIf-1/+ adult fly [14].
c) Genetic assimilation of the crossveinless phenotype (circles; data from [16]) and epigenetic assimilation of the ELBO phenotype (data from [14] for generations 2–13 and unpublished for generations 14–22).
Even more remarkably, the ELBOs generated by maternal loss of those factors still formed independently of whether the Hsp90 or TrxG mutations were inherited in the affected progeny [14]. That observation argues that depletion of Hsp90 or TrxG proteins in the egg predisposes the resulting embryos to abnormal development days later, after many rounds of cell division. Furthermore, breeding together otherwise wild-type flies bearing ELBOs increased the frequency of the ELBOs in subsequent generations, even when the Hsp90 or TrxG mutation was present only in the maternal ancestor that initially had the ELBO progeny (Fig. 1c, squares) [14]. In other words, the inheritance was strictly non-Mendelian.
Originally, we thought that we might be observing what Conrad Waddington called “genetic assimilation” [13] and Schmalhausen called “stabilizing selection” [15]. Waddington and colleagues showed that a crossveinless phenotype, in which the tiny crossveins in the wing are partially absent (Fig. 1a), can be induced by heat shocking genetically wild-type Drosophila larvae during a short window of development [16]. Eighteen generations of selection for the crossveinless phenotype led to a strain of “assimilated” flies of which over 95% had the phenotype [16] (Fig. 1c, circles). Furthermore, in the final rounds of selection, the crossveinless phenotype was present in over 95% of the flies even when the larvae were not heat shocked.
Waddington concluded, and future experiments confirmed, that existing genetic variation was selected to “canalize” (i.e., genetically and environmentally stabilize) the new crossveinless phenotype. According to Waddington, “Once the developmental path has been canalized, it is to be expected that many different agents, including a number of mutations available in the germplasm of the species, will be able to switch development into it. By such a series of steps, then, it is possible that an adaptive response can be fixed without waiting for the occurrence of a mutation” [13]. Waddington and others have represented “canalization” as an “energy well” with a ball (the phenotype) at the bottom of the well. Environmental stress or many genetic mutations pushes the ball one direction or the other, but the ball always returns to the lowest energy state, which is the “canalized” phenotype [11; 13].
Nearly a decade ago, Rutherford and Lindquist showed that Hsp90 is a “capacitor” [17] or an “adaptively inducible canalizer” [18] for morphological evolution. They showed that heat shock or pharmacological inhibition of Hsp90 causes morphological abnormalities in at least 7 different parts of adult flies, such as the eyes, wings, and legs [17]. As Waddington had done 45 years earlier, Rutherford and Linquist (1998) showed that multiple generations of selection of a particular abnormal phenotype allowed “genetic assimilation” of the phenotype, even in the presence of wild-type levels of Hsp90. They concluded that Hsp90 is the environmental sensor that probably explains Waddington’s observations [17; 19; 20].
A criticism of Lindquist’s work in the evolution research community is that it remains unclear whether the “capacitor” mechanism is adaptive or pathological; none of the phenotypes, for example leg and eye abnormalities, observed by Lindquist and colleagues, would seem to be adaptive. Meiklejohn and Hartl state in a critique of the Hsp90 capacitor model, “Over evolutionary time, the frequency with which a phenotypically revealed allele provides a selective advantage greater than the negative consequences of removing environmental canalization is likely to be extremely small” [18]. In his 1953 paper on assimilation of the crossveinless phenotype, Waddington persuasively responded to similar criticism [16], writing that, “There is, of course, no reason to believe that the phenocopy (the crossveinless phenotype) would in nature have any adaptive value, but the point at issue is whether it would be eventually genetically assimilated if it were favored by selection, as it can be under experimental conditions” [16].
The design and results of our ELBO experiments [14] resembled the assimilation experiments of Waddington [16] and Rutherford and Lindquist [17]. However, there were significant differences that suggested to us that what we were observing was “epigenetic assimilation” rather than “genetic assimilation” [20; 21]. First, as mentioned above, maternal loss of Hsp90 for one generation was sufficient to generate ELBOs for many generations, but multiple generations of heat shock [16] or genetic inactivation of Hsp90 [17] were required for the genetic assimilation experiments. Second, maternal loss of TrxG genes also induced ELBOs, and TrxG genes encode well-known chromatin-remodeling factors necessary for establishing chromatin conformations that can accommodate transcription. Third, and most convincingly, we eliminated most of the genetic variation in the KrIf-1 strain by backcrossing the mutation to an isogenic strain for 20 generations, but we still observed “epigenetic assimilation” of the ELBO phenotype induced by pharmacological inhibition of Hsp90 (Fig. 1c). In other words, since there was little or no genetic variation to select, we proposed that we were selecting for favorable “epigenetic variation” [14; 20].
The major difference between “genetic assimilation” and “epigenetic assimilation” is that the former is permanent and stable but the latter is transient and unstable. For example, if one were to mate to each other the rare flies with wild-type wings (~5%) in Waddington’s “genetic assimilation” experiment for the crossveinless phenotype, over 95% of the progeny would have the crossveinless phenotype [16]. The reason is that the genomes of the flies were “canalized” or stabilized for the crossveinless phenotype – i.e., they have a selected genetic repertoire that produces the crossveinless phenotype in ~95% of the flies. In contrast, in our “epigenetic assimilation” experiments, mating flies without the ELBO phenotype resulted in fewer than 5% of the progeny having the ELBO phenotype, presumably because epigenetic variation is unstable and requires continuous selection for the ELBO phenotype to be manifest [14]. We have continued the “epigenetic assimilation” experiment for over 5 additional years (over 100 additional generations), and the results remain essentially the same (D.M.R., unpublished).
Our interpretation is that, in the absence of almost all genetic variation, “canalization” of the ELBO phenotype cannot occur because there are no ELBO “stabilizing” genetic variants to select. Waddington’s or Shmalhausen’s “stabilizing selection” of genetic variants cannot occur if only epigenetic variants are being selected. Further evidence that the ELBO phenotype is generated by epigenetic rather than genetic variation is that histone deacetylase (HDAC) inhibitors partially suppress the ELBO phenotype [14]. Also, maternal loss of several suppressor-of-variegation (Suvar) mutations, which are mutations in genes that encode primarily histone-modifying enzymes, in the same isogenic background as the KrIf-1 mutation, partially suppress the ELBO phenotype (D.M.R., unpublished).
We believe that “genetic assimilation” and “epigenetic assimilation” are both important mechanisms in rapid morphological evolution. Again, to quote Jablonka and Lamb, who nicely summarized our “epigenetic assimilation” experiments and other related studies, “heritable epigenetic variants can do a holding job until genes catch up” [11] (p. 275).
Because it has no DNA methyltransferases I and III, Drosophila has little or no DNA methylation [22; 23] (but, interestingly, other insects have DNA methyl transferases I and III [24; 25; 26]). Therefore, “epigenetic assimilation” in Drosophila probably does not involve selection of appropriate genes with metastable DNA methylation patterns. Rather, Cavalli and Paro had earlier demonstrated that occupancy of Polycomb Response Elements/Trithorax Response Elements (PREs/TREs) by PcG (negative regulatory) or TrxG (positive regulatory) proteins is, in some situations, meiotically heritable [27; 28]. Therefore, we proposed that PRE/TRE occupancy at the KrIf-1 locus can be shifted from an active to a repressed chromatin state by reduction of Hsp90 activity, and that such a shift in occupancy is meiotically heritable [14]. However, whether DNA methylation or PRE/TRE occupancy explains “epigenetic assimilation” in Drosophila and other organisms remains an active area of investigation.
In our ELBO experiments, genes have unfortunately not yet “caught up” (i.e., there are no stabilizing mutations) even after over 100 generations of selection. Possible explanations are that population size was too small to find new mutations, the ELBO system cannot be genetically assimilated, or that epigenetically directed genetic errors (EDGE) cannot occur in Drosophila because, as noted above, there is little or no DNA methylation in that organism [22; 23]. Alternatively, even longer periods of selection might be required for the acquisition of genetic mutations to stabilize the epigenetically-induced ELBO phenotype.
Nevertheless, despite the fact that we have not yet seen an “EDGE effect” with the ELBO “epigenetic assimilation” system (i.e., genetic stabilizing mutations have not yet appeared) we nevertheless think that the epigenetically-directed genetic error (EDGE) mechanism for adaptive evolution is an attractive idea that warrants further investigation in other systems. As discussed in more detail in the next section, the EDGE model explains how “epigenetic assimilation” experiments can lead to new mutations directed at precisely the classes of genes that are likely to stabilize the selected phenotype.
5. Rapid morphological evolution in dogs
The dog is an excellent model for studying rapid morphological evolution because the species has undergone dramatic changes in morphology in just the past few thousand years. Dog evolution is a special case because it involves selection and interbreeding of specimens with particular phenotypes, such as the downward-pointing nose of the modern bull terrier or the flat face of the Pekinese.
Darwin believed that understanding dog evolution could help us understand evolution in general. In his book, Variation of Animals and Plants Under Domestication, Darwin wrote, “With respect to the precise causes and steps by which the several races of dogs have come to differ so greatly from each other, we are, as in most other cases, profoundly ignorant” [29]. Recently, Fondon and Garner (2004) and other dog geneticists have helped us to dispel the “profound ignorance” that Darwin noted [30].
Fondon and Garner (2004) showed that several “new” mutations that alter the dog phenotype are the result of repeat contractions and expansions within protein-coding sequences of genes [31]. For example, they showed that the downward sloping of the modern bull terrier’s skull, and consequently its downward sloping nose, is probably caused by a contraction of a repeat from 14 alanines in a row to 13 alanines in a row [31]. Similarly, they showed that the Great Pyrenees breed of dog has a 6th toe because of a contraction of 14 amino acids in a poly-proline-glutamine (poly-PQ) region of the Alx-4 gene [31]. Importantly, the extra-little-toe phenotype is identical to the phenotype of Alx-4 knockout mice [32], thus strongly supporting Alx-4 repeat contraction as the cause of the corresponding dog phenotype.
More recently, Sutter et al. have shown that the two-orders of magnitude difference in the weight of small-sized, as opposed to large-sized, dog breeds is associated with a single IGF1 haplotype containing several single-nucleotide polymorphisms (SNPs). Inactivation or reduction of the activity of that important growth factor is responsible for generating the small breeds of dog. A “haplotype,” according to Webster’s online dictionary, is a “group of alleles of different genes (as of the major histocompatibility complex) on a single chromosome that are closely enough linked to be inherited usually as a unit” (www.webster.com). It is not known if one of the SNPs or some other linked genetic event in the haplotype region is responsible for inactivation of the IGF1 gene, but it is interesting to note that the haplotype region in small dog breeds also contains a large contraction of a dinucleotide, (CA)n repeat in the IGF1 regulatory region [33]. It is, therefore, distinctly possible that contraction of the repeat is responsible for the evolution of small dogs.
Fondon and Garner argued that expansions and contractions of repeat sequences are likely responsible for a majority of changes in the phenotypes of the dog [31]. The main rationale for that hypothesis is that in prokaryotic models, the rate of expansions and contractions is up to five orders of magnitude higher than the rate of missense mutation [34; 35]. Also, the overall “purity” of the coding repeats (i.e., the fraction with the same codon in a repeat) is much higher in dogs than in humans [31]. Fondon and Garner argued that “purity” is proportional to the rate of evolution because higher rates expansions and contractions would lead to the corrections of sequence polymorphisms and thereby increase the purity [31]. They found that the purity of repeats in RCPs was much higher in dogs than in humans [31]. Since morphological evolution in dogs has proceeded at such an accelerated rate compared with humans, the hypothesis is attractive.
In a review inspired by Fondon and Garner’s paper, we proposed that repeat expansions and contractions might be epigenetically regulated by differential methylation of coding repeats at CpG sites in germline stem cells. That is, we proposed that coding repeats are a primary DNA location of “epigenetic marks”) [36]. Although we did not use the acronym “EDGE” in our earlier review, it constituted the first version of what we now refer to as the EDGE hypothesis.
In the revised EDGE hypothesis presented here (Fig. 2), we propose the following multi-step mechanism:
In non-stressful conditions, CpG-containing trinucleotide repeats are normally unmethylated in germline stem cells. (Originally, we proposed the reverse, that repeats are normally methylated, but see below.)
Environmental stress functionally inactivates Hsp90 because of the increased number of denatured proteins to which Hsp90 must bind.
Consequently, through the inactivation of the chromatin remodeling functions of Hsp90, such as its chaperoning of nuclear hormone receptors [36], the repeats become hyper-methylated (or “repressed chromatin” encompasses the RCGs).
Hyper-methylation of the repeats (or “repressed chromatin”) leads to an increase in the rate of mutation, and the increase in the rate of mutation enhances the rate of expansions and contractions of the repeats until a stress-relieving version of the gene that has a new adaptive phenotype is arrived at.
Finally, after stress has been reduced by the new adaptive phenotype, the repeat is de-methylated (or the chromatin is switched to an active conformation) and the repeat is again stabilized in the germline stem cell population (Fig. 2).
Figure 2. The Epigenetically directed Genetic Error (EDGE) hypothesis.

a) In germline stem cells, the trinucleotide repeat regions (short-thick arrows) are not methylated (blue circles), and the Hox-n gene has a high (normal) expression (long-thick arrow).
b) Under stressful conditions, the trinucleotide repeat becomes methylated (red circles) and the repeat region becomes unstable -- a mutation hot spot -- expressed at a lower level (long-thin arrow). The lower expression of Hox-n epigenetically reduces stress in the organism because the morphological change caused by the lower expression results in a morphological phenotype that can compensate for the stress. In the original version of the EDGE hypothesis [36], we proposed that stress leads to hypomethylation of repeats, but that idea has been revised because of new bioinformatic data (see text).
c) The trinucleotide repeat has undergone mutation and/or contraction. which stably (genetically) results in a lower steady-state level of Hox-n RNA (long thin arrow). Consequently, the epigenetic change (middle) has directed a mutation in the Hox-n gene to reduce the environmental stress more permanently.
In the EDGE model presented here, we postulate that methylated trinucleotide repeats are more likely than non-methylated repeats to mutate (Fig. 2). Our rationale for this part of the EDGE hypothesis is that 5-methyl-cytosine (5mC) is highly mutagenic. 5mC can spontaneously deaminate to T, whereas C deaminates to U, which is corrected by DNA repair enzymes. That deamination is a spontaneous analogue of the bisulfite treatment method, which is used extensively in studies of DNA methylation. Whitelaw and colleagues have found that methylated CpG dinucleotides mutate to TpG in retrotransposon sequences at up to 10-times the rate of unmethylated CpG dinucleotides [37]. Also, bioinformatic analyses in which CpG-containing repeats were compared with repeats that do not contain CpG dinucleotides (i.e., what we call “non-CpG repeats”) in humans and chimpanzees indicate that the CpG-containing repeats have a much higher mutation frequency (~ 10-fold) than do “non-CpG repeats” (Ruden et al., in preparation).
Figure 3 shows how the EDGE hypothesis might explain the rapid morphological evolution of the modern bull terrier skull. As of 1931, the modern bull terrier skull was quite similar to the ancestral wolf skull, and its nose had a gentle slope (θ1, Fig. 3a). As of 1950, the modern bull terrier nose was pointing downward more than was the earlier dog’s nose (θ2, Fig. 3a), and in 1976, even more so (θ3, Fig. 3a). Fondon and Garner sequenced the 1950 and 1976 specimens and showed that there was a stretch of 14 alanines in a row in Runx-2 in the 1950 dog, but 13 alanines in a row in the 1976 dog [31].
Figure 3. The EDGE hypothesis applied to dog evolution.

a) Modern bull terrier skull outlines from 1931, 1950, and 1976 prize-winning dogs and museum specimens (modified from [31; 36]). The slopes of the skull nasal sections are indicated (θ1–θ3). In the 1950 dog, there were 14 alanines in a row in the Runx-2 gene. In the 1976 dog, there was a contraction to 13 alanines in the Runx-2 gene. Information on the number of alanines in the 1931 dog is not available.
b) The alanine repeat (coded by GCC) is unmethylated in non-stressed germline stem cells (left). During epigenetic selection (epigenetic assimilation), the repeat is increasiningly methylated (red circles). The 1976 dog has a contraction to 13 alanines and no methylation.
c) Epigenetic selection for dogs with a more sloped nose leads to increased methylation of the GCC repeat (curves 1–3). As methylation increases, the nose phenotype increases in severity. Finally, EDGE-mediated contraction of the repeat from 14 to 13 alanines leads to a permanent (genetic) stabilization and enhancement of the severe nose phenotype (curve 4). That process can repeat as the nose continues to be selected for a downward bend.
DNA sequence analyses were not reported on the 1931 dog (it is not clear whether the skeleton is available for sequencing), but we propose the possibility that differential methylation of the DNA encoding the14 alanines (13 of which are encoded by GCC) altered expression of the Runx-2 gene such that the slope of the nose changed (θ2, Fig. 3a). Eventually, because the DNA encoding the alanine repeat was partially methylated, the DNA encoding the 14-alanine repeat was unstable and generated a 13-alanine repeat which is associated with a more dramatically bent nose (θ3, Fig. 3a).
For reasons known only to dog breeders, the downward-pointing nose is a favorable trait for pit bulls and selected for by the breeders. The modern bull terriers with more-downward-pointing noses won best-in-breed prizes in dog shows, and those prize dogs were then used to sire many other offspring. In a short period of time, under continuous human selection (a.k.a., “artificial selection”), the modern bull terrier nose continued its downward-pointing trek. Eventually, the Runx-2 gene encoding the 13-alanine repeat was selected as the 1976 best-of-breed modern bull terrier because it presented the most extreme skull phenotype (Fig. 3b).
We speculate on Runx-2 methylation to illustrate the EDGE model. In the EDGE hypothesis, we invoke a threshold effect to help explain the multi-generational aspects of the change in nose angle (Fig. 3c; modified from [21]). Phenotypic variations are shown in the figure as normal distributions. According to the hypothesis, the poly-alanine (GCC) repeat was initially unmethylated and all of the dogs had a phenotype characterized by the 1931 skull morphology (Fig. 3c, distribution 1). When the GCC repeat became partially methylated (or “repressed” chromatin encompassed the gene), expression of Runx-2 decreased, and the distribution moved further to the right, closer to the threshold (dashed line). A small percentage of dogs passed the threshold and had downward pointing noses (Fig. 3c, distribution 2). When the repeat became more heavily methylated (or the chromatin becomes more “repressed”), perhaps by “epigenetic assimilation” of the desirable trait, then expression of Runx-2 decreased further, and more of the dogs had downward pointing noses (Fig. 3c, distribution 3). Finally, because the heavily methylated (or “repressed” chromatin) GCC repeats were mutation hot spots, there was a contraction of 14 repeats to 13 repeats, which permanently (i.e., genetically rather than epigenetically) stabilized the most severe nose phenotype (Fig. 3c, distribution 4).
Although the EDGE hypothesis is highly speculative, it has the advantage that it generates testable hypotheses. In a later section, we will present some new bioinformatic analyses that we believe support the idea that the EDGE hypothesis is pertinent on evolutionary timescales. Finally, at the end of the review, we suggest further experiments that can test the hypothesis under laboratory conditions.
6. Rapid morphological evolution in foxes
Since evolution of dogs from wolves occurred over tens of thousands of years, it seems unlikely that we could understand the epigenetic and neuroendocrine basis of their behavioral evolution. However, classic studies by Shire and colleagues in the 1960’s and 1970’s demonstrated that mammals have measurable quantitative variation of their endocrine systems and that such variation can potentially be the building blocks for behavioral evolution [38]. Also, experiments performed by Belyaev and colleagues on silver foxes demonstrate that behavioral evolution studies are indeed feasible. Jablonka and Lamb (2005) summarize Belyaev’s remarkable “farm fox” experiments in the following quotation:
What is so interesting about the silver fox experiment is that a lot more than behavior was affected by selection for tameness. Within fewer than 20 generations, the reproductive season of the females had become longer, the time of molting had changed, and the levels of stress hormones and sex hormones had altered. There were physical changes too: the ears of some foxes drooped and the way some carried their tail was different; some had white spots on their fur; a few had shorter legs or tails, or a different skull shape [11], p. 259.
In 2005 we proposed a thought experiment, at the request of an anonymous reviewer, for testing the EDGE hypothesis by domesticating wolves. We wrote, “One could select for less wild progeny of these wolves over several generations. The ‘domestication genes’ could then be mapped and sequenced to determine whether the new alleles were caused through a genome-instability mechanism, such as trinucleotide repeat expansion or contraction” [36]. Little did we know that such a seemingly outlandish experiment, which presumably took thousands of years in nature, had already been accomplished by Dmitri Belyaev and colleagues over the past 50 years!
As described in the quotation at the top of this section, Siberian scientist Belyaev and his successor, Trut, obtained a strain of tame silver foxes in an ongoing, nearly 50-year-long selection experiment for tameness. In a sense, Belyaev wanted to repeat in a more controlled context the historical domestication of the dog, but instead of beginning with wolves, he began with silver foxes. He began his experiment in 1959 with 130 farm-bred silver foxes and used “tolerance to human contact” as the sole selection criterion for choosing the mating pairs. After only 8 generations, tolerance to human contact became a common behavioral phenotype of the selected silver fox lines [39].
Belyaev died in 1985, but his experimental program has been continued to this day by his successor, Trut [40; 41; 42]. In 1989, in an article in American Scientist that brought Balyaev’s work to the attention of scientists outside of Russia, Trut reported that, after 40 years of selective breeding, a group of 100 foxes that were as tame as a dog and had many other dog-like characteristics, such as floppy ears and spots had emerged out of over 45,000 animals in the overall study [39].
Belyaev and Trut believed that hormones and neurochemicals were critical to the rapid evolution of the silver foxes. According to Trut:
Because behavior is rooted in biology, selecting for tameness and against aggression means selecting for physiological changes in the systems that govern the body's hormones and neurochemicals. Those changes, in turn, could have had far-reaching effects on the development of the animals themselves, effects that might well explain why different animals would respond in similar ways when subjected to the same kinds of selective pressures. [39]
As Belyaev had predicted, other changes typical of domesticated animals appeared along with the tameness, even though they had not been explicitly selected for. The tame silver foxes had begun to show white patches on their fur, floppy ears, rolled tails, smaller skulls, and non-selected behavioral manifestations such as fawning behavior toward their master [39].
Belyaev and Trut argued that what they were observing in the “farm fox” experiments was not simply “genetic assimilation” of alleles at quantitative trait genes that enhance tameness. First, the experimental design was such that, “Through outbreeding with foxes from commercial fox farms and other standard methods, we have kept the inbreeding coefficients for our fox population between 0.02 and 0.07” [39]. Most genetic assimilation experiments, such as those of Waddington discussed above [16; 43], used brother-sister matings (with an inbreeding coefficient of 0.50) or breeding strategies with much higher inbreeding coefficients. It is doubtful whether inbreeding coefficients between 0.02 and 0.07 would be effective in a strict genetic assimilation experiment unless something else, such as “epigenetic assimilation,” was also occurring. Second, and more convincingly, many of the traits were dominant or partially dominant [39], suggesting that they were caused by new mutations (possibly via the EDGE mechanism).
If not quantitative traits, what did Belyaev believe he was selecting for? Trut says, “According to Belyaev, the answer is not that domestication selects for a quantitative trait but that it selects for a behavioral one” [39]. However, since behavior is influenced by quantitative traits, as has been demonstrated for numerous honey bee behaviors [44; 45], it is not clear to us why Trut makes the distinction between quantitative traits and behavioral ones. Indeed, Trut uses a traditional “genetic assimilation” mechanism to predict which genes are being selected in the farm fox experiment.
Which genes are they? Although numerous genes interact to stabilize an organism's development, the lead role belongs to the genes that control the functioning of the neural and endocrine systems. Yet those same genes also govern the systems that control an animal's behavior, including its friendliness or hostility toward human beings. So, in principle, selecting animals for behavioral traits can fundamentally alter the development of an organism. [39]
We agree with Trut that the neural and endocrine systems are being affected in the assimilation experiments. However, instead of “genetic assimilation” as she proposes, we propose “epigenetic assimilation” followed by genetic mutation to stabilize the new phenotypes. That is, we invoke the EDGE hypothesis. We propose that tame foxes are likely to have heritable epigenetic modifications and EDGE-induced genetic dominant mutations in gene networks regulated by neuroendocrine signaling molecules. In other words, not only do quantitative traits affect behavior, as has been shown in honey bees [44; 45], but behavior can also induce novel quantitative traits by changing the DNA via the proposed “EDGE effect.”
How might neuroendocrine-mediated behavioral changes induce heritable epigenetic changes in the genome? In an earlier review on transgenerational epigenetics, we proposed a model for the generation of epigenetic alterations by nuclear hormone receptors, such as the estrogen receptor, in germline stem cells [46]. In that model, ligand binding induces nuclear hormone receptors to recruit chromatin-remodeling enzymes. That process leads to activation of transcription by making the chromatin a hospitable environment, e.g., by histone 3 trimethylation on lysine 4 and acetylation on lysine 9 [46].
However, when the level of the nuclear hormone ligand is reduced, as is observed for glucocorticoids in the farm fox experiment, the chromatin is altered by histone-modifying enzymes that lead to a repressed state of chromatin, e.g., deacetylation followed by methylation of histone 3 lysine 9 [46]. The repressed chromatin can in turn recruit chromodomain proteins such as HP1 (histone 3, lysine 9, trimethyl-binding protein) and de novo DNA CpG-dependent DNA methyl transferase Dnmt3a, leading to methylation of the nuclear hormone target genes. We proposed that DNA or histone methylation, or other repressive chromatin marks, were they to occur in germline stem cells, could be transmitted to the next generation at a frequency dependent on the “stubbornness” of the marks [36].
It is possible that altered hormone and neuroendocrine signaling pathways can be transmitted to the next generation by altered DNA or histone methylation of the target genes or of the signaling molecules themselves [46]. Once a target gene’s DNA or chromatin is methylated, for instance, it can remain in the “removed ligand” OFF state throughout the animal’s lifespan and, if the methylation (or other chromatin) mark is sufficiently “stubborn,” into the next generation [36].
Szyf’s laboratory has shown in mice that early stress can alter life-long DNA methylation of the glucocorticoid receptor gene, with long-term behavioral consequences [47; 48]. Of course, for such “stubborn” marks to be transmitted to the next generation, the process has to occur in the germline stem cells that produce the eggs and the sperm. As discussed in a later section, that is a testable hypothesis.
Trut and Belyaev had the insight that stress can be a strong mediator of rapid morphological evolution [39]. The EDGE hypothesis might help explain how the new mutations they observed might have arisen so rapidly. An example is the Star mutation, a metastable epiallele that causes piebald spotting in silver foxes [49]. It would be interested to determine whether the new Star mutation is a result of aberrant DNA methylation or repeat expansion or contraction, as predicted by the EDGE hypothesis. The EDGE hypothesis predicts that aberrant DNA methylation in a repeated region in the Star gene would be a first step in its alteration, and that this would be followed by a contraction or an expansion of the repeat. However, the molecular identity of the Star locus has not yet been determined, so whether or not the Star mutation is a result of the “EDGE effect” is speculative.
We end this section with a colorful summation by Trut [39]:
By intense selective breeding, we have compressed into a few decades an ancient process that originally unfolded over thousands of years. Before our eyes, "the Beast" has turned into "Beauty," as the aggressive behavior of our herd's wild progenitors entirely disappeared. We have watched new morphological traits emerge, a process previously known only from archaeological evidence. Now we know that these changes can burst into a population early in domestication, triggered by the stresses or captivity, and that many of them result from changes in the timing of developmental processes. [39]
7. Evolutionary conservation of repeat-containing proteins (RCPs)
As mentioned above, Fondon and Garner (2004) predicted that developmental proteins such as transcription factors would turn out to be the principal repeat-containing proteins (RCPs) conserved across the phylogenies. In bioinformatic analyses that we present for the first time here, Fig. 4 verifies their prediction and expands on it.
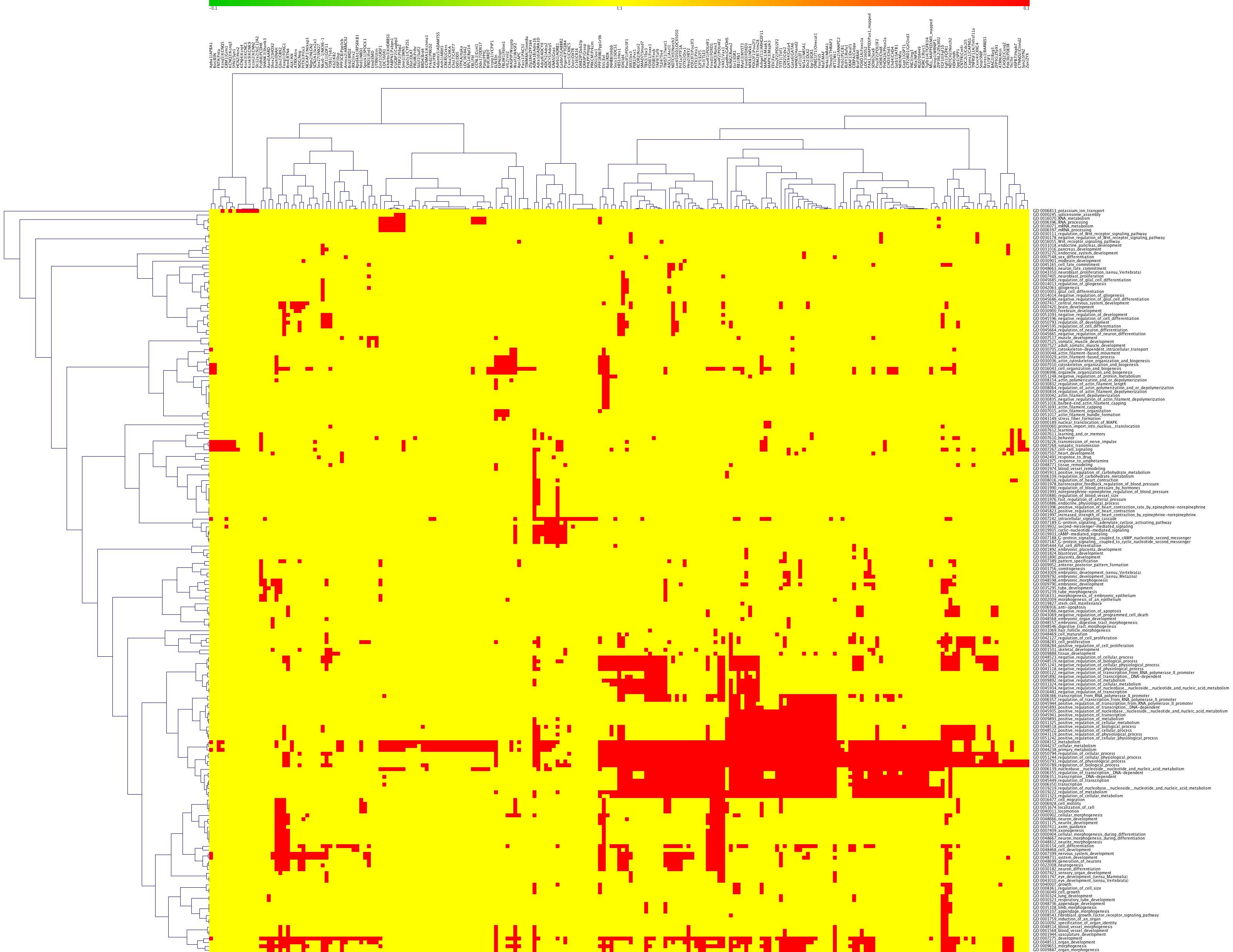
Figure 4. Cluster analysis of repeatcontaining proteins (RCPs) in Drosophila, mouse, rat, and humans.



a) Thumbprint of cluster analysis of RCPs with at least 6 amino acids indicates that the “splicing”, “positive regulation of transcription”, “negative regulation of transcription”, and “neurogenesis” gene ontology (GO) categories are conserved in RCP containing proteins.
b) Enlargement of the top two clusters in 4a, “splicing” and “positive regulation of transcription”, showing the individual GO categories of those types. In this case, the axes are labeled with the types of repeats (top) and the GO categories (side). The types of repeats are indicated on top as follows: organism (rat, Drosophila (dm), mouse (mus), or human (hs)_CpG or no CpG_ size of repeat (n6 means at least 6 amino acids)_simple (s) or complex (c).
c) A small region of a two-dimensional CIM (2D-CIM) showing a cluster of Drosophila genes (top) encoding positive regulators of transcription (side). This analysis is from the 2D-CIM named “dm_nocpg_n6_s” defined in 4b. Full-sized CIMs for 4b and 4c (for all 4 types of repeats in all four organisms) are in the Supplementary Material.
In a series of whole-genome bioinformatic analyses, we compared RCPs, which we define as having six or more amino acids (n6), across R. norvegicus, M. musculus, H. sapiens, and D. melanogaster. Similar analyses were performed by Faux and colleagues but they used different approaches that led to different conclusions (see “Standard GO Analyses” section in Methods) [50]. We classified the RCPs as follows (see Fig. 4b):
“simple” (s): containing a single amino acid repeat (e.g., poly-alanine (An), where n ≥ 6)
“complex” (c): containing two or more repeated amino acids (e.g., poly-alanine, arginine (AR)n, where n ≥ 3)
CpG-containing (cpg)
Non-CpG containing (nocpg)
We used the program GoMiner to analyze the conservation of RCPs from Drosophila to humans. GoMiner is a tool for biological interpretation of 'omic' data, including data from gene expression microarrays [51]. It leverages the Gene Ontology (GO) to identify “biological processes,” “molecular functions,” and “cellular components” represented in a list of genes. High-Throughput GoMiner (HTGM) [52], which was used for the analyses reported here, is an enhancement of GoMiner that efficiently performs the computationally-challenging task of automated batch processing of an arbitrary number of such gene lists.
Newly-developed multiple-species cluster analyses of GO categories determined by HTGM shows that transcription factors, splicing factors, and neurodevelopment genes are the main conserved classes of genes with repeats (Fig. 4a). The GO categories that are conserved in the clustering across all four species, from Drosophila to humans, are “splicing,” “positive regulation of transcription,” “negative regulation of transcription,” and “synaptogenesis” (Fig. 4a). Figure 4a shows what we call a “thumbnail” CIM (clustered image map) because the full CIM, containing labels for the individual genes and specific GO categories, is too big to fit on a printed page.
Zooming in on the clusters in a “detailed CIM” shows many of the GO categories that are in the “splicing” cluster (“RNA splicing,” “RNA processing,” “RNA metabolism,” etc.), the “transcription” clusters (“transcription,” “transcription DNA dependent,” “positive regulation of transcription,” “negative regulation of transcription,” etc.), and the “synaptogenesis” cluster (“synaptogenesis,” “synapse formation and biogenesis,” etc.) (Fig. 4b). Finally, we developed what we call a “two-dimensional CIM” (2D-CIM) that shows all of the individual RCPs in the four organisms and the GO categories to which they map (Fig. 4c). The full-sized 2D-CIMs for R. norvegicus, M. musculus, H. sapiens, and D. melanogaster are too large and detailed to represent here, but they are available in the Supplementary Material (see Table 1 for a list of Supplementary Figures).
Table 1.
Parameter Settings for High-Throughput GoMiner.
| organism | datasource | lookup settings |
|---|---|---|
| H. sapiens | UniProt | Enhanced Names |
| M. musculis | MGI | null |
| R. norvegicus | RGD | null |
| D. melanogaster | FB | null |
For all organisms, auto-generate was used to produce the total-genes file, the evidence level was set to 1, the FDR threshold was 0.10, and the number of randomizations was 100.
If DNA methylation is a “stubborn” transgenerational epigenetic mark that can facilitate evolutionary processes, then the EDGE hypothesis predicts that CpG-containing repeats might be over-represented in the conserved RCP-containing GO clusters. The clusters (i.e., “transcriptional regulators” and “synaptogenesis”) are enriched in genes containing both CpG and noCpG repeats (Fig. 4b). That observation suggests that repeat expansion and contraction of those genes are not epigenetically regulated by CpG methylation. However, the GO categories related to “splicing” are enriched in genes containing CpG repeats and deficient in genes containing noCpG repeats. That enrichment occurs for Drosophila and mouse, partially for human, but not at all for rat. Since Drosophila has little or no DNA methylation, the enrichment of RCPs in splicing categories (top section of Figure 4b) is not likely to be related to DNA methylation.
Instead of inducing direct DNA methylation of the repeats, heritable chromatin states or heritable PRE/TRE occupancy might be “stubborn marks” that epigenetically regulate repeat expansion and contraction, especially of the noCpG repeats that cannot be methylated.
Alternatively, repeats might expand or contract in a non-epigenetically regulated manner (as Fondon and Garner proposed [31]). Since expansions and deletions of repeats can be detected in the laboratory (e.g, in Drosophila [53]), experiments can be devised to determine whether or not expansions and contractions of CpG and noCpG repeats are epigenetically regulated.
8. Repeat-containing genes (RCGs) and human neurological diseases
In this section, we discuss how the EDGE hypothesis might apply to human neurological diseases that are characterized by dramatic expansion of repeats.
A large number of human neurological diseases are caused by expansions of trinucleotide repeats in either the coding or non-coding regions of genes (reviewed in [54]). For example, carriers at risk for Fragile X-associated tremor and ataxia syndrome have “pre-mutation” FMR1 alleles with 55–200 CGG repeats in the 5' UTR. Through an unknown mechanism, maternally-transmitted “pre-mutation” alleles are at risk of expansion of the repeat tract into the "full mutation" range (>200 repeats) [54].
When there are >200 repeats (full mutation), the CGG trinucleotide repeat region and adjacent promoter CpG island become hypermethylated, rendering FMR1 transcriptionally inactive [55]. It is not known when the CGG trinucleotide repeat becomes methylated in the FMR1 gene, but the EDGE hypothesis predicts that the repeats are methylated in the germline stem cells of the mother and that the methylation might potentiate the rapid expansion of the repeat. That prediction could, in principle, be tested by epigenetic analysis of ova from ovarectomy specimens. Several laboratories have developed mouse models of FMR1 that recapitulate many of these effects [56; 57]. The EDGE hypothesis also predicts that maternal stress, such as an endocrine disruption by hormones or man-made chemicals can contribute to the methylation of the repeat.
Trinucleotide repeats that do not contain CpG dinucleotides (e.g., the CTG repeat in myotonic dystrophy 1) also have pre-mutation and full mutations forms. In the involved genes, DNA methylation cannot directly mediate the rapid expansion of the repeat, but nearby CpG islands might be involved. However, it is possible that other PRE/TRE or chromatin-based epigenetic alterations, induced by stress or endocrine disruption, might also affect the process [54].
9. Life on the EDGE – endocrine disruptors and their effects on evolution
In this section, we use the EDGE hypothesis to predict the long-term effects of endocrine-disrupting chemicals on evolutionary processes. That concept is emphasized in the following quotation from Jablonka and Lamb (2005):
Animal studies suggest that stress and hormone treatments can also have effects lasting several generations. Epidemiological research programs and medical practice will have to accommodate information like this, so that we know how to avoid passing on the effects of our sins or misfortunes to future generations. [11] p. 365
The farm fox experiment of Trut and Belyaev, discussed in a previous section, showed that physiological levels of neuroendocrine molecules can influence rapid behavioral and morphological evolution of the silver fox [39; 58]. Likewise, man-made chemicals with endocrine-disrupting properties can perhaps have pathological effects on evolution.
For over 30 years, Retha Newbold and colleagues have been studying the effects of the highly estrogenic chemical diethylstilbesterol (DES) on human sexual development and carcinogenesis [59]. They have shown that mothers who take DES during critical periods of their pregnancies have children, and even grandchildren, with reproductive system abnormalities. Their daughters and granddaughters also have higher incidences of uterine cancer, possibly because the DES causes epigenetic changes in their DNA that are trans-generationally inherited [59]. Newbold and colleagues have reproduced many of the transgenerational effects of DES in rodent models [59], but the precise effects of DES on future generations remains to be seen. As discussed above, we have proposed a mechanism by which nuclear hormone receptors can mediate transgenerational epigenetic phenomena [46].
Recently, Skinner and colleagues studied gestating female rats during gonadal sex determination. Exposure to the endocrine-disrupting pesticides vinclozolin (an antiandrogenic compound) or methoxychlor (an estrogenic compound) increased incidence of male infertility and hypomethylation of sperm DNA in the next generation. Analogously to what was observed with DES in females, those effects were transferred through the male germ line to nearly all males through the F4 generation [60]. Likewise, Crews and colleagues have shown that effects of vinclozolin can affect the behavior of the female progeny for three generations [61].
Those trans-generational effects of DES, vinclozolin, and methoxychlor are frightening in their own right. However, if our EDGE hypothesis is correct, then it is possible that such endocrine-disrupting chemicals can have permanent effects on the DNA in humans and other mammals by setting the stage for genetic mutations. Clearly, more research is needed to understand the roles of hormones and neuroendocrine molecules during normal and pathological evolution.
10. Possible mechanisms of transgenerational epigenetic inheritance
Jablonka and Lamb stress the importance of understanding “epigenetic marks” in the following quotation:
The evolution of all types of heritable chromatin marks must have been closely associated with variations in the DNA sequences that carry them. Since only a subset of marks is heritable, we have to ask what type of DNA sequences are capable of carrying the “stubborn marks” that are transmitted to subsequent generations. [11] , p. 332
A question raised in the above quotation is how “epigenetic marks” are passed down through the germline. Most DNA methylation is erased in the genital ridge during embryogenesis, but a few methylated cytosines, such as some of those in repetitive sequences, are more resistant to erasure [62]. However, Whitelaw and colleagues have argued that it is not the inheritance of methylated cytosines that is important in transgenerational epigenetic phenomena, but rather the inheritance of the Polycomb Group (PcG) or Trithorax Group (TrxG) complexes on the chromatin [63]. Whitelaw and colleagues found that all cytosine methylation is erased at the Avy locus during embryogenesis, so they argued that DNA methylation cannot be the repressive “epigenetic mark” for that variegated gene [63]. Rather, they showed that mice mutant for a PcG protein have increased transgenerational epigenetic inheritance in the male [63]. Consistent with the idea that PcG and TrxG complexes are involved in the formation of “epigenetic marks,” as mentioned above, Cavalli and Paro have shown that PcG and TrxG complexes at Polycomb Response Elements (PREs) are stable through at least one meiosis [27; 28].
In the EDGE hypothesis, we propose that the “stubborn marks” are in the regions of genes that encode amino acid repeats in RCPs (Fig. 2). We propose that repeats containing CpG dinucleotides are methylated in germline stem cells in a stress- or hormone-dependent manner. The proposed DNA methylation could increase the mutation rate of the repeat and thereby increase the rate of expansions and contractions. However, in repeats that do not have CpG dinucleotides, or in organisms such as Drosophila that have little or no DNA methylation, there might be other epigenetic mechanisms that regulate expansion and contraction of repeats. In light of the finding that PRE/TRE occupancy might be a heritable “epigenetic mark,” it is possible that PcG and TrxG proteins are also involved in epigenetic regulation of the expansion and contraction of repeats. Those are fertile issues for future research.
11. Beyond the EDGE –implications for evolution and cancer
We consider EDGE to be a working hypothesis that can be refined and updated as experiments and bioinformatic analyses test its various aspects. Ongoing bioinformatic studies in our laboratory comparing the human and chimpanzee RCPs circumstantially support the version presented here, but there is no body of direct experimental evidence to support it. One would have to analyze the global methylation status of RCPs in stressed and unstressed germline stem cells to verify or refute salient aspects of the EDGE hypothesis. For example, methylation-sensitive restriction enzymes (or possibly bisulfite-based methods) could be applied to the germline cells – presumably most easily in animal models of stress or disease but possibly in ovarectomy specimens. This type of analysis is now feasible because single-cell and pauci-cell methods of analysis are available (e.g, [64; 65]).
If some predictions of the EDGE hypothesis are valid, there are implications for research on evolution and cancer. One implication, mentioned in the previous section, is that endocrine-disrupting chemicals might induce both epigenetic and genetic effects in animals. One implication for cancer research goes beyond Feinberg and colleagues’ “epigenetic progenitor origin” theory for the progression of cancer [9]. The EDGE model suggests that epigenetic selection can directly induce mutations in classes of genes that lead to progression of cancer. If that is true, treating the epigenetic alterations in early stages of cancer might, in principle, prevent later DNA mutations involved in carcinogens, invasiveness, or metastasis.
In this chapter, we have also provided conceptual tests to determine whether the EDGE hypothesis is a valid model for evolution. For example, bioinformatic analyses suggest that polymorphisms in developmental RCPs such as splicing factors, transcription factors, and synaptogenesis proteins are the key mutations that differentiate closely related species The importance of the RCP polymorphisms in distinguishing two species or two variants within a species can be tested by swapping the critical RCPs and determining whether that exchange leads to partial phenotypic conversion. The importance of RCP polymorphisms in cancer progression and their possible epigenetic regulatory mechanisms should also be more thoroughly investigated, for example by analyzing the sequencing data being generated in The Cancer Genome Atlas project (www.cancergenome.nih.gov) [66].
12. Methods
High-Throughput GoMiner (HTGM)
Gene Ontology (GO) [67] categorization was determined using HTGM [52], which is freely available to academic and private sector users on the internet (http://discover.nci.nih.gov/gominer/GoCommandWebInterface.jsp). The “changed genes” files, which are lists of genes with amino acid repeats containing six or more amino acids, were computed by C. Jamison and are available upon request. The HTGM parameter settings are given in Table 1.
Clustered image maps (CIMs)
Clustered image maps (i.e., clustered heatmaps) [68] have been used very widely in the “postgenomic era” for display and first-order analysis of data from microarrays and other high-throughput experimental platforms. In conjunction with HTGM, several specialized forms were developed to represent relationships between genes and Gene Ontology categories. Several of the HTGM output files are suitable for submission to a program such as Genesis [69] for making the heatmaps. The HTGM output files are described in detail at the HTGM web site (http://discover.nci.nih.gov/gominer/htgmoutput.jsp). Before submission to the Genesis program, CIM files were filtered to remove large, generic categories containing ≥ 300 genes. In a few rare cases, CIM files with very large numbers of categories that met the FDR = 0.01 threshold were pruned by removing categories with FDRs > 0.00. (As described in [52], we compute FDRs by resampling randomized sets of genes. An FDR of 0.00 indicates that there were no instances in which a resampled category achieved a p-value that was at least as good as that for the real category). The individual CIMs display categories versus genes, and the cumulative CIMs display categories versus microarray experiments (or in our case, versus the four repeat classes in each animal species analyzed). A new type of heatmap, which we term a "combination CIM," was generated by performing a "logical join" of categories across two or more individual or cumulative CIMs. That technique was particularly useful for analysis and interpretation of cross-species results.
Standard GO analyses
As mentioned in section 8, Faux and colleagues argued that they failed (but we argue succeeded) to find support for Fondon and Garner’s hypothesis.
Fondon and Garner (2004) hypothesized that repeat expansion and contraction can provide a mechanism for rapid morphological evolutionary changes and that this process may be particularly important in those genes that are involved in development (such as transcription factors). Comparison of the rank order of the biological process and the functional classifications between groups A and B did not reveal any such bias. [70]
A key prediction of Fondon and Garner’s model for rapid morphological evolution is that transcription factors and other developmental genes must have trinucleotide repeats in the coding regions for them to be highly evolvable [31]. A recent paper by Faux and colleagues, quoted above, presented evidence that they conclude does not support Fondon and Garner’s hypothesis [70]. However, we argue that reinterpretation of their data and more extensive clustering analyses presented below do, in fact, support the hypothesis.
Faux and colleagues analyzed repeat-containing proteins (RCPs) in 13 species, including human, rat, mouse, Drosophila, and Arabidopsis. They defined an RCP as containing 7 or more identical repeated amino acids (note that we define an RCP in this chapter as containing 6 or more identical amino acids. They classified 3 types of RCPs, conserved across H. sapiens, R. norvegicus, and M. musculus, as follows:
Group A: proteins that contain only conserved homopeptides (i.e., all homopeptides are present across all three species in the same positions; 314 proteins); Group B: proteins that contain only nonconserved homopeptides (1129 proteins); and Group C: proteins that contain a mixture of conserved and nonconserved homopeptides (86 proteins). [70]
Faux et al. determined that the primary GO categories of RCPs in Group A were in: 1) “physiological processes” (35%), 2) “regulation of biological process” (26%), 3) “development” (15%), 4) “cellular process” (14%), 5) “biological process unknown” (5%), 6) “response to stimulus” (2%), and 7) “growth” (2%) [70]. The primary GO categories of RCPs in Group B were in:1) “physiological processes” (34%), 2) “regulation of biological process” (22%), 3) “cellular process” (15%), 4) “biological process unknown” (14%), 5) “development” (9%), and 6) “response to stimulus” (3%) [70]. All of the other GO categories were represented in less than 1% of the RCPs.
Faux et al. argued that one way to test Fondon and Garner’s hypothesis that expansions and contractions are particularly important in developmental genes is to determine whether there is any bias in the rank order of GO categories in Group A (conserved RCPs) compared with Group B (non-conserved RCPs) in humans, mice, and rats [70]. As quoted in the lead to this section, their analyses “did not reveal any such bias” [70]. However, Faux et al. did not explicitly state why bias of rank order is an important consideration for testing Fondon and Garner’s hypothesis, and what result would support that model.
Our interpretation of Fondon and Garner’s hypothesis is that conserved RCPs (Group A) would have a higher bias for developmental genes than non-conserved RCPs (Group B). In fact, this is what Faux et al. observed: 15% of the conserved RCPs, and 9% of the non-conserved RCPs, respectively, mapped to the GO category "development." Since we do not have access to the original data, we cannot assess whether 15% and 9% are significantly different. If those values are, in fact, significantly different, then we believe that that difference supports Fondon and Garner’s hypothesis.
The reason we argue that Fondon and Garner’s hypothesis predicts that Group A would show a higher bias for the GO category “development” than Group B is that expansions and contractions of a “conserved” repeat (i.e., the homopeptide is in the same location across all three species) would drive morphological evolution. In contrast, Group B repeats would require an additional mechanism of insertion of repeated amino acids into novel locations in developmental genes to drive morphological evolution, and that would be a mechanism different from the one proposed by Fondon and Garner. Therefore, since the difference between Groups A and B is not great, we reinterpret the analyses of Faux et al. as weakly supporting Fondon and Garner’s hypothesis. Furthermore, the fact that “development” is a primary GO category in both Groups A and B argues that insertion of novel repeats into developmental genes (i.e., Group B) might be an additional mechanism for rapid morphological evolution.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 2.
List of Supplementary Figures Available Online.
| Figure | Description |
|---|---|
| S01_dm_CpG_c.CIM.png | Drosophila n6 CpG complex repeats |
| S02_dm_CpG_s.CIM.png | Drosophila n6 CpG simple repeats |
| S03_dm_noCpG_c.CIM.png | Drosophila n6 noCpG complex repeats |
| S04_dm_noCpG_s.CIM.png | Drosophila n6 noCpG simple repeats |
| S05_hs_CpG_c.CIM.png | Human n6 CpG complex repeats |
| S06_hs_CpG_s.CIM.png | Human n6 CpG simple repeats |
| S07_hs_noCpG_c.CIM.png | Human n6 noCpG complex repeats |
| S08_hs_noCpG_s.CIM.png | Human n6 noCpG simple repeats |
| S09_mus_cpg_n6_c.CIM.png | Mouse n6 CpG complex repeats |
| S10_mus_cpg_n6_s.CIM.png | Mouse n6 CpG simple repeats |
| S11_mus_noCpG_n6_c.CIM.png | Mouse n6 noCpG complex repeats |
| S12_mus_noCpG_n6_s.CIM.png | Mouse n6 noCpG simple repeats |
| S13_rat_CpG_n6_c.CIM.png | Rat n6 CpG complex repeats |
| S14_rat_CpG_n6_s.CIM.png | Rat n6 CpG simple repeats |
| S15_rat_noCpG_n6_c.CIM.png | Rat n6 noCpG complex repeats |
| S16_rat_noCpG_n6_s.CIM.png | Rat n6 noCpG simple repeats |
| S17_rat.CIM_mus.CIM_subset.dm.CI M_subset.hs.CIM.png | Rat, Mouse, Drosophila, and Human combined CIM |
Acknowledgements
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research to B.N.Z. and J.N.W., NIH R01 grants (ES012933 and CA105349) to D.M.R., , and a Center for Nutrient Gene Interaction in Cancer Prevention(CNGI) grant to M. D.G. Finally, we thank David Crews and Anna Jang for critically reading the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dobzhansky T. Nothing in biology makes sense except in the light of evolution. The American Biology Teacher. 1973;35:125–129. [Google Scholar]
- 2.Dobzhansky TG. Evolution, genetics, and man. New York: Wiley; 1955. [Google Scholar]
- 3.Dobzhansky TG. Genetics and the origin of species. New York: Columbia Univ. Press; 1937. [Google Scholar]
- 4.Dobzhansky TG. Genetics and the origin of species. New York: Columbia University Press; 1982. [Google Scholar]
- 5.Lewontin RC, Lewontin RC. The triple helix : gene, organism, and environment. Cambridge, Mass: Harvard University Press; 2000. [Google Scholar]
- 6.Lamarck JBP. Zoological Philosophy. Chicago: Chicago Press; 1809. [Google Scholar]
- 7.Darwin C. The Origin of the Species. Random House; 1859. [Google Scholar]
- 8.Galton F. Experiments in pangenesis, by breeding from rabbits of a pure variety, into whose circulation blood taken from other varieties had previously been transfused. Proc R Soc Lond B. 1871;19:393–410. [Google Scholar]
- 9.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 10.McClintock B. The significance of responses of the genome to challenge. Science. 1984;226:792–801. doi: 10.1126/science.15739260. [DOI] [PubMed] [Google Scholar]
- 11.Jablonka E, Lamb MJ. Evolution in four dimensions : genetic, epigenetic, behavioral, and symbolic variation in the history of life. Cambridge, Mass: MIT Press; 2005. [Google Scholar]
- 12.Rando OJ, Verstrepen KJ. Timescales of genetic and epigenetic inheritance. Cell. 2007;128:655–668. doi: 10.1016/j.cell.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 13.Waddington CH. Canalization of development and the inheritance of acquired characters. Nature. 1942;150:563–565. doi: 10.1038/1831654a0. [DOI] [PubMed] [Google Scholar]
- 14.Sollars V, Lu X, Xiao L, Wang X, Garfinkel MD, Ruden DM. Evidence for an epigenetic mechanism by which Hsp90 acts as a capacitor for morphological evolution. Nat Genet. 2003;33:70–74. doi: 10.1038/ng1067. [DOI] [PubMed] [Google Scholar]
- 15.Shmalhausen II. Factors of evolution; the theory of stabilizing selection. Philadelphia: Blakiston Co.; 1949. [Google Scholar]
- 16.Waddington CH. Genetic Assimilation of an acquired character. Evolution. 1953;7:118–126. [Google Scholar]
- 17.Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396:336–342. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- 18.Meiklejohn CD, Hartl DL. A single mode of canalization. Trends in Ecology and Evolution. 2002;17:468–473. [Google Scholar]
- 19.McLaren A. Too late for the midwife toad: stress, variability and Hsp90. Trends Genet. 1999;15:169–171. doi: 10.1016/s0168-9525(99)01732-1. [DOI] [PubMed] [Google Scholar]
- 20.Ruden DM, Garfinkel MD, Sollars VE, Lu X. Waddington's widget: Hsp90 and the inheritance of acquired characters. Semin Cell Dev Biol. 2003;14:301–310. doi: 10.1016/j.semcdb.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 21.Rutherford SL, Henikoff S. Quantitative epigenetics. Nat Genet. 2003;33:6–8. doi: 10.1038/ng0103-6. [DOI] [PubMed] [Google Scholar]
- 22.Lyko F. DNA methylation learns to fly. Trends in Genetics. 2001;17:169–172. doi: 10.1016/s0168-9525(01)02234-x. [DOI] [PubMed] [Google Scholar]
- 23.Lyko F, Ramsahoye BH, Jaenisch R. DNA methylation in Drosophila melanogaster. Nature. 2000;408:538–540. doi: 10.1038/35046205. [DOI] [PubMed] [Google Scholar]
- 24.Mampumbu AR, Mello ML. DNA methylation in stingless bees with low and high heterochromatin contents as assessed by restriction enzyme digestion and image analysis. Cytometry A. 2006;69:986–991. doi: 10.1002/cyto.a.20312. [DOI] [PubMed] [Google Scholar]
- 25.Schaefer M, Lyko F. DNA methylation with a sting: an active DNA methylation system in the honeybee. Bioessays. 2007;29:208–211. doi: 10.1002/bies.20548. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Jorda M, Jones PL, Maleszka R, Ling X, Robertson HM, Mizzen CA, Peinado MA, Robinson GE. Functional CpG methylation system in a social insect. Science. 2006;314:645–647. doi: 10.1126/science.1135213. [DOI] [PubMed] [Google Scholar]
- 27.Cavalli G, Paro R. The Drosophila Fab-7 chromosomal element conveys epigenetic inheritance during mitosis and meiosis. Cell. 1998;93:505–518. doi: 10.1016/s0092-8674(00)81181-2. [DOI] [PubMed] [Google Scholar]
- 28.Cavalli G, Paro R. Epigenetic inheritance of active chromatin after removal of the main transactivator. Science. 1999;286:955–958. doi: 10.1126/science.286.5441.955. [DOI] [PubMed] [Google Scholar]
- 29.Darwin C. Variation in animals and plants under domestication. New York: Appleton and Co; 1883. [Google Scholar]
- 30.Sutter NB, Ostrander EA. Dog star rising: the canine genetic system. Nature Reviews Genetics. 2004;5:900–910. doi: 10.1038/nrg1492. [DOI] [PubMed] [Google Scholar]
- 31.Fondon JW, Garner HR. Molecular origins of rapid and continuous morphological evolution. Proc Natl Acad Sci U S A. 2004;101:18058–18063. doi: 10.1073/pnas.0408118101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qu S, Niswender KD, Ji Q, van der Meer R, Keeney D, Magnuson MA, Wisdom R. Polydactyly and ectopic ZPA formation in Alx-4 mutant mice. Development. 1997;124:3999–4008. doi: 10.1242/dev.124.20.3999. [DOI] [PubMed] [Google Scholar]
- 33.Sutter NB, Bustamante CD, Chase K, Gray MM, Zhao K, Zhu L, Padhukasahasram B, Karlins E, Davis S, Jones PG, Quignon P, Johnson GS, Parker HG, Fretwell N, Mosher DS, Lawler DF, Satyaraj E, Nordborg M, Lark KG, Wayne RK, Ostrander EA. A single IGF1 allele is a major determinant of small size in dogs. Science. 2007;316:112–115. doi: 10.1126/science.1137045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellegren H. Microsatellite mutations in the germline: implications for evolutionary inference. Trends in Genetics. 2000;16:551–558. doi: 10.1016/s0168-9525(00)02139-9. [DOI] [PubMed] [Google Scholar]
- 35.Ellegren H. Microsatellites: simple sequences with complex evolution. Nature Reviews Genetics. 2004;5:435–445. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- 36.Ruden DM, Garfinkel MD, Xiao L, Lu X. Epigenetic Regulation of Trinucleotide Repeat Expansions and Contractions and the "Biased Embryos" Hypothesis for Rapid Morphological Evolution. Curr Genomics. 2005;6:145–155. [Google Scholar]
- 37.Garrick D, Fiering S, Martin DI, Whitelaw E. Repeat-induced gene silencing in mammals. Nat Genet. 1998;18:56–59. doi: 10.1038/ng0198-56. [DOI] [PubMed] [Google Scholar]
- 38.Shire JG. The forms, uses and significance of genetic variation in endocrine systems. Biol Rev Camb Philos Soc. 1976;51:105–141. doi: 10.1111/j.1469-185x.1976.tb01121.x. [DOI] [PubMed] [Google Scholar]
- 39.Trut LN. Early canid domestication: the farm fox experiment. Am Scientist. 1989;87:160–165. [Google Scholar]
- 40.Kukekova AV, Trut LN, Oskina IN, Johnson JL, Temnykh SV, Kharlamova AV, Shepeleva DV, Gulievich RG, Shikhevich SG, Graphodatsky AS, Aguirre GD, Acland GM. A meiotic linkage map of the silver fox, aligned and compared to the canine genome. Genome Res. 2007;17:387–399. doi: 10.1101/gr.5893307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hare B, Plyusnina I, Ignacio N, Schepina O, Stepika A, Wrangham R, Trut L. Social cognitive evolution in captive foxes is a correlated by-product of experimental domestication. Curr Biol. 2005;15:226–230. doi: 10.1016/j.cub.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 42.Trut LN, Pliusnina IZ, Os'kina IN. [An experiment on fox domestication and debatable issues of evolution of the dog] Genetika. 2004;40:794–807. [PubMed] [Google Scholar]
- 43.Waddington CH. Genetic Assimilation of the Bithorax Complex. Evolution. 1956;10:1–13. [Google Scholar]
- 44.Hunt GJ. Flight and fight: a comparative view of the neurophysiology and genetics of honey bee defensive behavior. J Insect Physiol. 2007;53:399–410. doi: 10.1016/j.jinsphys.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hunt GJ, Amdam GV, Schlipalius D, Emore C, Sardesai N, Williams CE, Rueppell O, Guzman-Novoa E, Arechavaleta-Velasco M, Chandra S, Fondrk MK, Beye M, Page RE., Jr Behavioral genomics of honeybee foraging and nest defense. Naturwissenschaften. 2007;94:247–267. doi: 10.1007/s00114-006-0183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruden DM, Xiao L, Garfinkel MD, Lu X. Hsp90 and environmental impacts on epigenetic states: a model for the trans-generational effects of diethylstilbesterol (DES) on uterine development and cancer. Hum Mol Genet. 2005;14:R147–R155. doi: 10.1093/hmg/ddi103. [DOI] [PubMed] [Google Scholar]
- 47.Meaney MJ, Szyf M, Seckl JR. Epigenetic mechanisms of perinatal programming of hypothalamic-pituitary-adrenal function and health. Trends Mol Med. 2007;13:269–277. doi: 10.1016/j.molmed.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 48.Szyf M, Weaver I, Meaney M. Maternal care, the epigenome and phenotypic differences in behavior. Reprod Toxicol. 2007;24:9–19. doi: 10.1016/j.reprotox.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 49.Belyaev DK, Ruvinsky AO, Trut LN. Inherited activation-inactivation of the star gene in foxes: its bearing on the problem of domestication. J Hered. 1981;72:267–274. doi: 10.1093/oxfordjournals.jhered.a109494. [DOI] [PubMed] [Google Scholar]
- 50.Faux NG, Bottomley SP, Lesk AM, Irving JA, Morrison JR, de la Banda MG, Whisstock JC. Functional insights from the distribution and role of homopeptide repeat-containing proteins. Genome Res. 2005;15:537–551. doi: 10.1101/gr.3096505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, Bussey KJ, Riss J, Barrett JC, Weinstein JN. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zeeberg BR, Qin H, Narasimhan S, Sunshine M, Cao H, Kane DW, Reimers M, Stephens RM, Bryant D, Burt SK, Elnekave E, Hari DM, Wynn TA, Cunningham-Rundles C, Stewart DM, Nelson D, Weinstein JN. High-Throughput GoMiner, an 'industrial-strength' integrative gene ontology tool for interpretation of multiple-microarray experiments, with application to studies of Common Variable Immune Deficiency (CVID) BMC Bioinformatics. 2005;6:168. doi: 10.1186/1471-2105-6-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315:1857–1859. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
- 54.Wang YH. Chromatin structure of repeating CTG/CAG and CGG/CCG sequences in human disease. Front Biosci. 2007;12:4731–4741. doi: 10.2741/2422. [DOI] [PubMed] [Google Scholar]
- 55.Dahl C, Gronskov K, Larsen LA, Guldberg P, Brondum-Nielsen K. A homogeneous assay for analysis of FMR1 promoter methylation in patients with fragile X syndrome. Clin Chem. 2007;53:790–793. doi: 10.1373/clinchem.2006.080762. [DOI] [PubMed] [Google Scholar]
- 56.Lauterborn JC, Rex CS, Kramar E, Chen LY, Pandyarajan V, Lynch G, Gall CM. Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J Neurosci. 2007;27:10685–10694. doi: 10.1523/JNEUROSCI.2624-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2007;104:15537–15542. doi: 10.1073/pnas.0707484104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trut LN, Naumenko EV, Belyaev DK. Change in the pituitary-adrenal function of silver foxes during selection according to behavior. Sov Genet. 1974;8:585–591. [PubMed] [Google Scholar]
- 59.Newbold RR, Padilla-Banks E, Jefferson WN. Adverse effects of the model environmental estrogen diethylstilbestrol are transmitted to subsequent generations. Endocrinology. 2006;147:S11–S17. doi: 10.1210/en.2005-1164. [DOI] [PubMed] [Google Scholar]
- 60.Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466. doi: 10.1126/science.1108190. [see comment] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crews D, Gore AC, Hsu TS, Dangleben NL, Spinetta M, Schallert T, Anway MD, Skinner MK. Transgenerational epigenetic imprints on mate preference. Proc Natl Acad Sci U S A. 2007;104:5942–5946. doi: 10.1073/pnas.0610410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lane N, Dean W, Erhardt S, Hajkova P, Surani A, Walter J, Reik W. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis. 2003;35:88–93. doi: 10.1002/gene.10168. see page 49: Weinstein JN, Myers TG, O'Connor PM, Friend SH, Fornace AJ, Kohn KW, Fojo T, Bates SE, Rubinstein LV, Anderson NL, Buolamwini JK, van Osdol WW, Monks AP, Scudiero DA, Sausville EA, Zaharevitz DW, Bunow B, Viswanadhan VN, Johnson GS, Wittes RE, Paull KD. An information-intensive approach to the molecular pharmacology of cancer. Science. 1997;275:343–349. doi: 10.1126/science.275.5298.343.
- 63.Blewitt ME, Vickaryous NK, Paldi A, Koseki H, Whitelaw E. Dynamic reprogramming of DNA methylation at an epigenetically sensitive allele in mice. PLoS Genet. 2006;2:e49. doi: 10.1371/journal.pgen.0020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O'Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tateishi K, Ashihara E, Takehara N, Nomura T, Honsho S, Nakagami T, Morikawa S, Takahashi T, Ueyama T, Matsubara H, Oh H. Clonally amplified cardiac stem cells are regulated by Sca-1 signaling for efficient cardiovascular regeneration. J Cell Sci. 2007;120:1791–1800. doi: 10.1242/jcs.006122. [DOI] [PubMed] [Google Scholar]
- 66.Check E. Cancer atlas maps out sample worries. Nature. 2007;447:1036–1037. doi: 10.1038/4471036a. [DOI] [PubMed] [Google Scholar]
- 67.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weinstein JN, Myers TG, O'Connor PM, Friend SH, Fornace AJ, Jr, Kohn KW, Fojo T, Bates SE, Rubinstein LV, Anderson NL, Buolamwini JK, van Osdol WW, Monks AP, Scudiero DA, Sausville EA, Zaharevitz DW, Bunow B, Viswanadhan VN, Johnson GS, Wittes RE, Paull KD. An information-intensive approach to the molecular pharmacology of cancer. Science. 1997;275:343–349. doi: 10.1126/science.275.5298.343. [DOI] [PubMed] [Google Scholar]
- 69.Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18:207–208. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- 70.Faux NG, Huttley GA, Mahmood K, Webb GI, de la Banda MG, Whisstock JC. RCPdb: An evolutionary classification and codon usage database for repeat-containing proteins. Genome Res. 2007;17:1118–1127. doi: 10.1101/gr.6255407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.