

Abstract
The validity of the popular point dipole approximation for interpretation of the zero-field splitting (ZFS) parameter (D-value) in EPR spectroscopy is studied. This approximation is of central importance for the determination of distances by analysis of EPR data. In this work, a detailed experimental (EPR spectroscopy and X-ray crystallography) and theoretical study for a model system (2,2’,5,5’-tetra(t-butyl)-4,4’-bis(ethoxy-carbonyl)-3,3’-bipyrrolyl-1,1’-dioxyl) was performed to understand the scope and limitations of the point dipole model in EPR spectroscopy. For this diradical, the radical-radical distance derived with the point-dipole approximation deviates significantly (by ~40%) from the results derived from the X-ray analysis. Explicit quantum chemical calculation of the D-value on the basis of B3LYP density functional calculations leads to excellent quantitative agreement with the measured D-value. The quantitative accuracy of the employed methodology was confirmed for two additional systems that have previously been experimentally characterized. We therefore analyzed the contributions to the D-value of the target system in detail. This analysis leads to insight into the reasons for the failure of the point-dipole approximation. The analysis was then extended to an in silico study of five classes of model systems. Linkers of varying length and bond saturation were introduced between the radical-carrying groups. This allows for the analysis of the distance dependence of the D-parameter as well as the through-bond and through-space spin-spin interaction. From these results we established the limitations of the point-dipole approximation. The results of this analysis demonstrate that even very modest amounts of spin delocalization can cause significant deviations from pure point-dipole behavior and consequently cause the EPR derived distances to deviate from the N-O midpoint distance by up to several Ångström. If unsaturated linkers are used, the distance dependence of D does not follow the inverse cubic behavior predicted by the point dipole model. However, for commonly-used non-aromatic nitroxide rings connected by a saturated linker, the point dipole approximation works well. Among the various point dipole variants tested in this work for delocalized spins, the most successful one is based on distributed point dipoles with spin populations derived from quantum chemical calculations. The distance dependence of the isotropic Heisenberg exchange parameter J has also been studied theoretically. The decay was found to be monoexponential with a decay constant of ~1 Å−1. Thus at linker lengths between 6–8 carbon atoms between a nitroxide radical pair a switch from the strong to the weak exchange limit is predicted.
Keywords: Point dipole approximation, EPR spectroscopy, spin-spin interaction, density functional theory
Introduction
Determination of structure and conformation in macromolecular systems, either biomolecular or synthetic, requires determination of distances between specified points in the system. An important, and rapidly developing application of electron paramagnetic resonance (EPR) is pulsed electron-electron double resonance (PELDOR) and double quantum coherence (DQC) for measurement of interspin distances, or distributions of distances, up to 60 or 70 Å in proteins and polymers.1–3 The probes frequently are nitroxyl radicals, and data analysis typically is based on a point dipole approximation in which it is assumed that the unpaired electron is localized on the nitrogen-oxygen moiety. Distances determined by PELDOR for dinitroxyls with well defined structures have been shown to be in good agreement with expected interspin distances in the range of 20 4 to 28 Å. 5 However, it is also known that at shorter distances or in delocalized systems the use of the point dipole approximation is inadequate to relate the zero field splitting observed in EPR spectra to intercenter distances. 6
In order to evaluate the limitations of the point dipole approximation when applied to dinitroxyls, it is necessary to perform detailed quantum chemical calculations on systems of significant size. Fortunately, concomitant with the progress in EPR spectroscopy, there has been considerable progress in the quantum chemistry of spin-Hamiltonian parameters.7 Reasonably accurate values can now be calculated for organic radicals, based on density functional theory (DFT), for hyperfine couplings,8–12 g-tensors,8 quadrupole couplings,8, 13 and more recently also for the two parts of the zero-field splitting.14–19 It is particularly this latter interaction that is of importance for distance measurements. The biquadratic ZFS D-tensor contains a first order contribution from the direct magnetic dipole magnetic dipole spin-spin (SS) interaction and a second-order contribution from the spin-orbit coupling (SOC). While the general equations have been known for a long time20, 21 their realization in a DFT framework has been less than obvious and it is only recently that general, efficient programs became available that allow the full treatment of the ZFS in organic15–17 as well as inorganic18, 19 systems.
Diradical [6] (see Scheme 1) was selected for this study because it has been characterized by single-crystal X-ray crystallography and by EPR spectroscopy in a rigid glass matrix. It is shown that analysis of the zero-field splitting by the point-dipole approximation gives an intercenter distance that is not consistent with the X-ray structure. After demonstrating that explicit quantum chemical calculations provide a ZFS that agrees with experiment, we present an analysis of factors that contribute to the observed ZFS and identify the parts that are missing from the point dipole treatment. This analysis provides insight into the scope and limitations of this important approximation that is widely used in determination of intercenter distances by EPR. We then discuss the distance dependence of the zero-field splitting in nitroxide diradicals and study the ‘through-bond’ and ‘through-space’ interactions. The predictions are compared with experimental results for diradicals with longer intercenter distances. The types of linkages that typically are used to attach a spin label to a macromolecule tend to disrupt delocalization, which decreases the through-bond' interactions, and explains why the point dipole approximation works reasonably well in these systems.
Scheme 1.
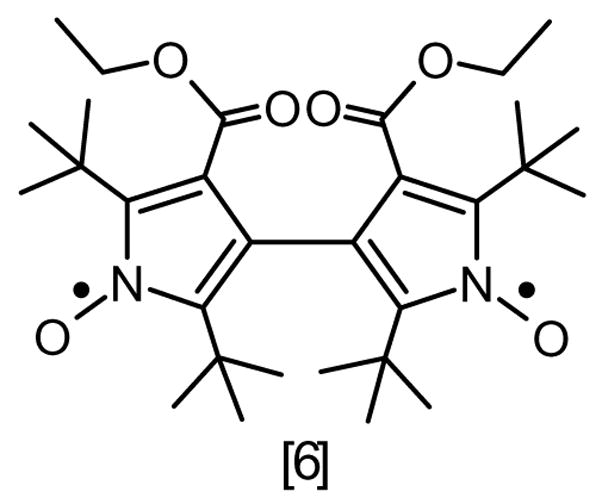
Structural diagram of diradical [6].
1. Experimental Details
General Methods
All chemicals were of reagent grade unless indicated otherwise. For column chromatography, silica gel was used (230–400 mesh; EMD Chemicals, distributed by VWR International, Bridgeport, NJ). IR spectra were recorded on a FT-IR spectrometer (Perkin-Elmer, Norwalk, CT) in CHCl3. 1H NMR spectra were obtained on a GE QE-300/Tecmag NMR spectrometer. Melting points were measured on a Thomas Hoover capillary melting point apparatus and were corrected. Synthesis of the dinitroxyl [6] is outlined in Scheme 2 and detailed below.
Scheme 2.

Route of synthesis for diradical [6].
Ethyl 1,2-dipivaloylpropionate [3]
To an unstirred solution of sodium ethoxide, prepared by adding sodium metal (1.15 g, 50 mmol) to absolute ethanol (30 mL), was added ethyl pivaloylacetate [1] (8.6 g, 50 mmol, TCI America) in absolute ethanol (20 mL) over 1 h. Thereafter, the reaction mixture was cooled in an ice bath, and 1-bromopinacolone [2] (8.95 g, 6.7 mL, 50 mmol, Aldrich Chemical Co.) was added over 3 h with stirring. After addition was complete, the reaction mixture was stirred at room temperature for 12 h. Water (400 mL) was added and the mixture was extracted with ether (5 × 100 mL). The combined ether extracts were dried over anhydrous MgSO4, filtered, and evaporated in vacuo to yield an oil which, upon double distillation through a long Vigreux column, afforded ethyl 2,3-dipivaloylpropionate [3] as a colorless liquid, bp, 66–69 °C at 0.05 mm Hg, 6 g (45%). IR (CHCl3): 1728 (C=O), 1704 (C=O) cm−1. 1H NMR (CDCl3): 1.16, 1.21 (21H, 2 t-butyl and -OCH2CH3), 3.02 (2H, m, -OCH2CH3), 4.14 (2H, m, -COCH2CH-), 4.40 (1H, m, -COCHCO-). Anal. (C15H26NO4): calcd: C = 66.64%, H = 9.69%; found: C = 66.72%, H = 9.76%.
1-Hydroxy-2,5-di(tert-butyl)-3-ethoxycarbonylpyrrole [4]
To a solution of 2,3-dipivaloylpropionate [3] (1 g, 3.7 mmol) in acetic acid (20 mL) was added an aqueous solution (10 mL) of hydroxylamine hydrochloride (0.38 g, 5.5 mmol) and sodium acetate (0.8 g, 9.8 mmol). The reaction mixture was heated for a week at 60–65°C in an oil bath. The reaction mixture was carefully made alkaline with saturated aqueous Na2CO3, and extracted with ether (5 × 100 mL). The ether layer was dried over anhydrous Na2SO4, filtered, and evaporated in vacuo to yield an oil. The oil was purified by column chromatography: The starting material was eluted with petroleum ether/ether (95:5); subsequent elution with petroleum ether/ether (80:20) afforded 1-hydroxy-2,5-di(tert-butyl)-3-ethoxycarbonylpyrrole [4] (0.44 g, 45%), which recrystallized from petroleum ether/benzene as a white solid, mp = 159–161 °C.22 IR (CHCl3): 3275 (N-OH), 1683 (C=O) cm−1. 1H NMR (CDCl3): 1.34 (3H, t, J = 7 Hz, -OCH2CH3), 1.35 (9H, s, 5-t-butyl), 1.51(9H, s, 2-t-butyl-), 4.23 (2H, q, J = 7 Hz, -OCH2CH3), 5.82, 6.02 (2H, s, 3-H), 7.36 (1H, s, 4-H).
2,5-Di(tert-butyl)-3-ethoxycarbonyl-4-hydroxy-1-pyrroloxyl [5a], 2,5-di(tert-butyl)-3-ethoxycar-bonylpyrrol-4-one-1-oxide [5b], and 2,2’,5,5’-tetra(t-butyl)-4,4’-bis(ethoxycarbonyl)-3,3’-bipyrrol-yl-1,1’-dioxyl] [6]
Nickel peroxide (250 mg, 2.75 mmol, Aldrich Chemical Co) was added to a solution of 1-hydroxy-2,5-di(tert-butyl)-3-ethoxycarbonylpyrrole [4] (50 mg, 0.18 mmol) in 15 mL benzene.23 After being stirred for 30 min in the presence of air at room temperature, the reaction mixture was filtered. The residue obtained after evaporating the filtrate in vacuo was purified by column chromatography (petroleum ether/dichloromethane, 10:3). A faster-eluting, dark purple minor product, which formed nearly black crystals, was shown by X-ray crystallography to be bipyrroloxyl [6]. The second, major product [5a], initially isolated as a yellow oil, was paramagnetic and had an EPR spectrum characteristic of a pyrroloxyl.21 A benzene solution of [5a], on standing in air for several days, yielded yellow prismatic crystals, which X-ray crystallography revealed to be 2,5-di(tert-butyl)-3-ethoxycarbonylpyrrol-4-one-1-oxide [5b]. Performing the oxidation under N2 again yielded compounds [5a] and [6], but with the latter as the major product.
EPR spectroscopy
X-band EPR spectroscopy was performed on a Bruker E580 spectrometer. Simulations were performed with the Bruker XSophe software and least-squares minimization.24 The parameters used to simulate the CW EPR spectrum of [6] are given in table 1. Since the axes of the ZFS are not coincident with the magnetic axes of the dinitroxyl, the g values for the diradical are effective values. The hyperfine coupling constants (A) for [6] were within the line widths and hence are not provided in the table.
Table 1.
Parameters used for simulation of spectra of dinitroxyl [6] in decalin at 89 K.
| gxx | gyy | gzz | |D| [G] | |E/D| | Azz [G] | |
|---|---|---|---|---|---|---|
| Conformation1a | 2.0064b | 2.0052b | 2.0097b | 211 | 0.061 | |
| Conformation2a | 2.0064b | 2.0052b | 2.0097b | 244 | 0.026 | |
| Monoradical | 2.0110 | 2.0059 | 2.0028 | 15.7c |
The populations of Conformation1 : Conformation 2 are 70 : 30.
The g values for the diradical [6] are effective values along the axes of the spin-spin interaction tensor.
For the monoradical the nitrogen hyperfine splitting along the x and y axes is less than linewidth.
X-ray Crystallography
X-ray crystallography was performed at the X-ray Crystallographic Center in the Department of Chemistry and Biochemistry, at the University of Maryland, College Park. X-ray intensity data were collected at 220 ± 2 K on a 3-circle diffractometer (Smart 1000, Bruker AXS Ltd., Madison, WI) equipped with a CCD area detector and a MoKα fine-focus sealed tube source (λ = 0.71073 Å, operated at 50 kV and 30 mA) in conjunction with a graphite monochromator. Data frames were acquired with a scan width of 0.5° in ω and 23 sec exposure time per frame. The frames were integrated with SAINT software (Bruker 2001) using a narrow-frame integration algorithm. Data were corrected for absorption effects with the semi-empirical from equivalents method using SADABS software.25 SHELXS-97 and SHELXL-97 software26, 27 was used to solve and refine structures.
2,2’,5,5’-tetra(t-butyl)-4,4’-bis(ethoxycarbonyl)-3,3’-bipyrrolyl-1,1’-dioxyl [6]
A crystalline prism of compound [6], measuring approximately 0.19 × 0.19 × 0.24 mm3, was used for X-ray crystallographic analysis. Over 9.2 h, 1,098 data frames were collected. Integration of the data using a tetragonal unit cell yielded a total of 29,588 reflections (maximum θ of 20.01°), of which 2,966 were independent (99.8% completeness, Rint = 7.16%, Rsig = 4.17%) and 2,135 were greater than 2σ(I). The final cell dimensions of a = b = 18.5020(12) Å, c = 36.985(5) Å, α = β = γ = 90°, and V = 12661(2) Å3, are based on refinement of the XYZ-centroids of 2,966 reflections with 2.2° < θ < 15.6° using SAINT software.28 Analysis of the data showed negligible decay during data collection. The minimum and maximum transmission coefficients were 0.970 and 0.985. The structure was solved and refined in the space group I41/a, with Z = 16 for the formula unit C30H46N2O6. The final anisotropic full-matrix least-squares refinement on F2 with 397 variables converged at R1 = 5.42 % for the observed data and wR2 = 12.21 % for all data. The goodness-of-fit was 1.000. The largest peak on the final difference map was 0.227 e−/Å3 and the largest hole was −0.157 e−/Å3. On the basis of the final model, the calculated density was 1.114 g·cm−3 and F(000), 4,608 e−. Crystallographic details for [6] including atomic coordinates, anistropic displacements are included in the Supplementary Material.
2,5-di(tert-butyl)-3-ethoxycarbonylpyrrol-4-one-1-oxide
A yellow prism of compound [5b], with approximate dimensions 0.22 × 0.40 × 0.50 mm3, was used for X-ray crystallographic analysis. Over 15.28 h, 1,824 data frames were collected. Integration of the data using a triclinic unit cell yielded a total of 8,309 reflections (maximum θ of 25.00°,), of which 2,862 were independent (99.7% completeness, Rint = 2.16%, Rsig = 1.91%) and 2,507 were greater than 2σ(I). The final cell dimensions of a = 9.1990(6) Å, b = 9.8972(6) Å, c = 10.4887(6) Å, α = 112.724(1)°, β = 92.009(1)°, γ = 109.404(1)°, V = 816.00(9) Å3, are based on refinement of the XYZ-centroids of 5,317 reflections with 2.1° < θ < 28.5° using SAINT software. Analysis of the data showed no decay during data collection. The minimum and maximum transmission coefficients were 0.917 and 0.982. The structure was solved and refined in the space group P1, with Z = 2 for the formula unit C15H23NO4. The final anisotropic full-matrix least-squares refinement on F2 with 212 variables converged at R1 = 3.82 % for the observed data and wR2 = 7.92% for all data. The goodness-of-fit was 1.000. The largest peak on the final difference map was 0.174 e−/Å3 and the largest hole was −0.156 e−/Å3. On the basis of the final model, the calculated density was 1.145 g·cm−3 and F(000), 304 e−.
2. Experimental Results and Analysis
The dinitroxyl [6] was unexpectedly obtained as a product of the oxidation of 1-hydroxy-2,5-di(tert-butyl)-3-ethoxycarbonylpyrrole [4] with nickel peroxide. The structure of dinitroxyl [6], determined by X-ray crystallography, is shown in Fig. 1. Key features of the structure are summarized below. Owing to steric constraints, the two pyrrole rings are far from coplanar; indeed, the dihedral angle between the mean planes defined by the two sets of ring atoms (N1-C1-C2-C3-C4 and N2-C5-C6-C7-C8) is 80.77°. The C3-C7 bond linking the two rings has a length of 1.476 ± 0.005 Å. The nitrogen-oxygen bond lengths are: N1-O1, 1.279 ± 0.004 Å; N2-O2, 1.281 ± 0.004 Å. The distance between the two nitrogen atoms is 5.830 Å; the distance between the midpoints of the two N-O bonds is 7.006 Å. There is moderate disorder in the structure: One of the two ethoxycarbonyl groups (attached to C6, Fig. 1) is found in two orientations, with corresponding site occupancy factors of 0.592 ± 0.017 and 0.408±0.017.
Figure 1.

Structure of dinitroxyl [6]. Anisotropic atomic displacement ellipsoids for non-hydrogen atoms are shown at the 30% probability level. Hydrogens are displayed with arbitrarily small radii.
The X-band EPR spectrum of dinitroxyl [6] in glassy decalin at 89 K is shown in Figure 2. The insert in the upper right of Figure 2 shows the feature at about 3400 G which is assigned as a small amount of monoradical, presumably [5], based on the g- and A-values and ease of power saturation. To match all other features of the spectrum it was necessary to assume two conformations. For the dominant conformation the zero-field splitting parameter (D) = 211 G (0.020 cm−1) and E = 13 G and for the minor component D = 244 G (0.023 cm−1), and E = 6 G. If the point dipole approximation is used, these values of D correspond to interspin distances of 5.1 and 4.8 Å, respectively, which are significantly shorter than the 7.0 Å distance between the centroids of the N-O bonds found by X-ray crystallography. The molecular structure suggests that extensive delocalization of the unpaired electron might contribute to this discrepancy, so this system was selected for detailed computational studies.
Figure 2.

X-band EPR spectrum of [6] in glassy decalin at 89 K. An expanded scan of the monoradical [5] signal is shown in the inset. The dashed lines are simulations obtained using the Bruker XSophe software.
3. Theoretical Considerations
3.1. Quantum chemical treatment
The spin-spin (SS) contribution to the zero-field splitting (ZFS) parameter D(SS ) dominates the ZFS of organic triplets and diradicals. As discussed recently,15 within a DFT framework, the tensor components of D(SS ) may be calculated from the equation of McWeeny and Mizuno:29
| (1) |
where ge is the free electron g-value (2.002319…), α is the fine structure constant (~1/137 in atomic units), the indices μ,v,k,τ refer to basis functions, is an element of the spin-density matrix and is the operator for the electron-electron magnetic dipole-dipole interaction.
In analogy to Hartree Fock theory we can distinguish between two different contributions to . The part of equation (1) is a ‘Coulomb’ contribution, while the term is an ‘exchange’ contribution that arises from the antisymmetry requirement of the N-electron molecular wavefunction. Thus, even the direct dipolar spin-spin interaction contains an exchange contribution that is of fundamentally different origin than the isotropic Heisenberg type exchange interaction frequently used in the modeling of interacting spins.30, 31 The Heisenberg isotropic exchange interaction is responsible for the splitting of the energy levels into singlet- and triplet states of the interacting radical pair. The exchange contribution to the spin-spin interaction in equation (1) on the other hand does not preserve singlet- and triplet spin symmetry and represents a quantum mechanical correction to the magnetic dipole-dipole interaction.
3.2. Coulomb versus Exchange Contributions
In the following we will concentrate on a system with two unpaired electrons in the singly occupied molecular orbitals (SOMOs) i and j. If we expand equation (1) in the MO basis we get:
| (2) |
Four terms arise that represent a self-interaction of the unpaired electrons and that cancel out identically. Thus, one is left with only two terms that represent the Coulombic and exchange contributions to the SS interaction. Unfortunately, the division of the total spin-spin interaction into these two contributions is not unique. This is readily seen upon performing a unitary transformation of the SOMOs (| iq〉 =|i〉cos q +|j〉sin q ,|j q〉 = −|i〉sin q +|j〉cos q , arbitrary rotation angle q ). Such a transformation leaves the sum of Coulomb and exchange terms invariant but changes their relative magnitudes. Thus, since the SOMOs of a spin system with S > 1/2 are only specified up to an arbitrary unitary transformation, one has to specify the representation that one wishes to work with. For interpretation purposes, we argue that transforming the SOMOs to a local representation (iL , jL) is most revealing. In this case, the two interactions smoothly go into ‘classical’ limits upon separating the spin-carrying fragments.
If the localized orbitals are ‘compressed’ to δ-functions (say, at the center of gravity of iL and jL given by RiL = 〈iL | r | iL〉 and RJL = 〈jL | r | jL〉), the Coulomb part of the SS-interaction simply becomes:
| (3) |
This clearly is a point dipole type equation with the effective intercenter distance vector RLjL = RiL − RjL.
Under the plausible assumption that the orbitals (iL , jL ) decay exponentially at large distances from their respective centroids, the exchange contribution should also decay very rapidly. A Mulliken type approximation would suggest that the leading term goes as (with constant κ) - that is, much faster than the Coulomb interaction.
3.3. Multicenter Contributions
A different way to partition the SS interaction is to recognize that the basis functions are tied to parent atomic centers. Hence, one obtains one- through 4-center contributions to the SS interaction as in equation (4) and Table 2:
| (4) |
Table 2.
Definition of the 1- to 4-center- contributions to D(SS ) in equation (4).
| ZFS-Contribution | Definition |
|---|---|
| 1-center | A=B=C=D |
| 2-center-Coulomb | A=B, C=D, A≠C |
| 2-center-Exchange | A=C, B=D, A≠B |
| 2-center-Hybrid | A=B=C≠D or A=B=D≠C or A=C=D≠B or B=C=D≠A |
| 3-center-Coulomb | A=B, A≠C≠D or C=D, A≠B≠C |
| 3-center-Exchange | A=C, A≠B≠D or B=D, B≠A≠C or A=D, A≠B≠C or B=C, B≠A≠D |
| 4-center | A≠B≠C≠D |
The various interaction types have a transparent physical interpretation: The (AA|AA) one-center contributions represent the interactions of local magnetic dipoles, that arise through a certain amount of triplet character at a given atom A. The contributions (AA| BB) represent the quasi-classical point dipole interactions while (AB|AB) exchange and (AA|AB) hybrid type two-center integrals provide quantum mechanical corrections that depend primarily on the overlap of spin-carrying orbitals on centers ‘A’ and ‘B’ respectively. The hybrid type (AA|AB) integral may also be interpreted as the dipole-dipole interaction of unpaired electrons in the bond A-B with unpaired electrons localized on center A. Likewise, the three-center contribution (AA|BC) represents the interaction of unpaired electrons in the bond B-C with unpaired electrons on A. Finally, the four center contributions (AB|CD) are interpreted as ‘distant’- and the three-center exchange integrals (AB|AC) as ‘adjacent’ bond-bond interactions.
3.4. Distributed Point Dipole Model
The popular distributed point dipole model32 is obtained upon contracting the individual basis functions (instead of the entire localized SOMOs) to δ-functions. Assuming the spin-density matrix to be diagonal in this basis and neglecting the exchange interaction, one then obtains:
| (5) |
where A and B sum over nuclei and is the ‘gross’ spin population on atom A. Equation (5) describes the interaction of collections of point dipoles centered at atomic positions where each atom pair is weighted by the product of the spin populations that reside on these atoms. Models of this type have been frequently used in EPR investigations 6 where the spin populations have either been estimated from measured hyperfine couplings or from quantum chemical calculations. Note however, that our justification of these approaches does not involve the net spin populations as output by quantum chemical program packages within various schemes (Mulliken, Löwdin, natural population analysis (NPA33) ) but the gross atomic spin populations. The differences between the two sets of numbers are not negligible. As an illustrative example we give in table 3 (a) the net Mulliken spin population, (b) the gross atomic Mulliken spin population and (c) the spin population obtained with a NPA for our truncated model system (see 4.1).
Table 3.
Net and gross atomic Mulliken spin population obtained with a NPA for the truncated dinitroxyl model system (orthogonal configuration, numbering as in Scheme 3 and 4).
| atom | Net Mulliken spin population | Gross atomic Mulliken spin population | NPA spin population |
|---|---|---|---|
| O1 | 0.50 | 0.46 | 0.48 |
| N2 | 0.19 | 0.21 | 0.21 |
| C3 | 0.03 | 0.03 | 0.05 |
| C4 | 0.12 | 0.14 | 0.10 |
| C5 | 0.13 | 0.10 | 0.10 |
| C6 | 0.03 | 0.06 | 0.05 |
The calculated spin distribution suggests that the dominant resonant structures are the ones shown in Scheme 3.
Scheme 3.
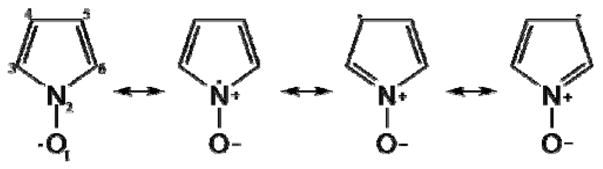
This description emphasizes the delocalization of the spin onto the ring-system of the nitroxide – an important factor in the analysis that follows below.
3.5 Effective Point Dipole Model
If the distance between two spin-carrying fragments is large enough, it may be possible to reduce equation (5) to a single term where RAB refers to an ‘effective’ distance. Quite frequently, the origins of the spin-distributions are simply fixed in an ad hoc manner based on chemical intuition (for nitroxides commonly the center of the N-O bond is chosen).34 A perhaps somewhat more rational approach is the following: First calculate the centers of gravity (COG) of the spin populations on fragments ‘F1’ and ‘F2’ as:
| (6) |
| (7) |
and then obtain the ZFS D-value as:
| (8) |
where R12 = R(F 2) − R(F1) is the distance vector between the centers of gravity of fragment F1 and F2. We will explore all the approximations and their limitations below.
4. Computational Details
All computations in this work were carried out with a development version of the ORCA program package.35 Geometry optimizations were performed with the BP8636–38 density functional employing the TZVP39 basis set.
4.1. Model Systems for the Analysis of Coulomb, Exchange and Multicenter Contributions
Ten different sets of calculations were carried out. Two sets of calculation (a–b) for the full system as shown in Figure 1 and eight sets of calculation (c–j) for the truncated system as shown in Scheme 4: (a) a structure in which only the positions of the hydrogen atoms were relaxed, (b) a structure that was fully optimized, (c) a truncated system (substituents replaced by hydrogen atoms, see Scheme 4) where only the hydrogen positions were optimized, (d) a fully optimized truncated system (see Scheme 4) and (e) a relaxed surface scan of the truncated system with the dihedral angle defined by the atoms 12, 11, 5 and 6 fixed at a chosen and stepwise modified value in order to scan a full rotation of the two rings. The model system was constructed in an effort to separate electronic and steric effects that may arise from the bulky substituents of [6].
Scheme 4.
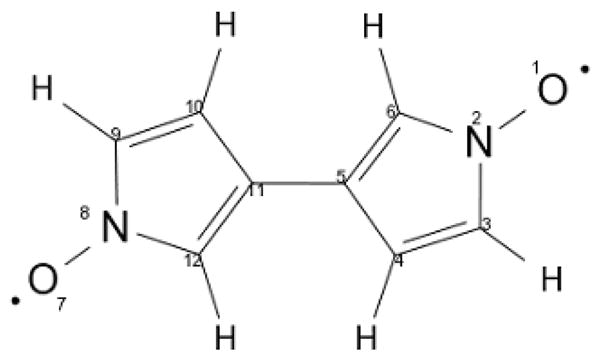
The truncated dinitroxyl model system studied in this work together with its atom numbering scheme.
4.2. Model Systems for the Analysis of ‘Through-Bond’ and ‘Through-Space’ Contributions
To analyze the distance dependence of the ZFS with respect to ‘through-bond’ and ‘through-space’ contributions, we constructed additional model systems in which we inserted a linker between the two interacting nitroxyl radical fragments. This part of the study is designed to investigate the effective distance dependence of the ZFS interaction to find out under which electronic and geometric circumstances the widely used traditional point-dipole approach to the ZFS provides accurate distances.
The linkers were inserted between the two ring fragments (atoms 5 and 11 in Scheme 4). We constructed (see also scheme 5): (f) Fully optimized structures of the ring fragments with an unsaturated allyl linker consisting of consecutive ethenyl-groups (C2nH2n, n=1-10), (g) partially optimized structures of the ring fragments with a saturated alkyl linker consisting of consecutive ethyl-groups (C2nH4n, n=1–10) and (h) partially optimized structures of the ring fragments with a partially saturated linker (C4n+2H4n+4, n=0–4). Furthermore we constructed model systems with non-aromatic nitroxyl rings: (i) Fully optimized structures of the ring fragments with an unsaturated allyl linker and (j) partially optimized structures of the ring fragments with a saturated linker. The structures of (g), (h) and (j) were optimized while constraining the carbon skeleton of the linker to be planar to have approximately the same distance in space in all sets of systems.
Scheme 5.

Model systems with selected linkers
4.3 Zero-Field Splitting Calculations
The spin-spin contribution to the ZFS parameter D(SS ) was obtained from additional single-point calculations. For this purpose, the BP86 density functional was applied in combination with the EPR-II basis set40. As discussed previously, the spin-spin term was calculated on the basis of the UNO determinant.15 The SOMOs were localized according to the Pipek-Mezey criterion41 as implemented in ORCA. Calculations with the much larger EPR-III basis set42 led to changes in the results of 2–7%, which was deemed insignificant. Hence for the present purposes the EPR-II basis set was considered to be appropriate and was employed in all calculations reported below. Results obtained with this method will be designated as DI in the following.
The spin population needed for the calculation of the COG approximation and for the distributed point dipole approximation were obtained from a natural population analysis (NPA) of the localized SOMOs of the UNO determinant using the “gennbo” code developed by Weinhold and coworkers,33 that is interfaced to ORCA. In these calculations, atoms 1 through 6 plus the adjacent hydrogen atoms (see Scheme 4) were attributed to one spin center and atoms 7 through 12 plus the adjacent hydrogen atoms were attributed to the other spin center. If a linker was present it was cut into two halves and the atoms of each half were attributed to the neighboring ring fragment.
For the calculation of the ZFS within the commonly used point dipole approximation the two spin centers were located in the middle of the NO-group. Results obtained via the COG approximation (DCOG ), distributed point dipole approximation (DDPD ) or point dipole approximation (DPD ) will be designated collectively as DII in the following.
4.3 Heisenberg isotropic exchange coupling constant
The Heisenberg isotropic exchange coupling constant J was obtained from additional single-point calculations. The BP86 density functional used for the calculation of D(SS ) would not have been appropriate for the calculation of the exchange coupling constant and thus the hybrid density functional B3LYP43, 44 was chosen for the prediction of J.45 For this purpose, a broken symmetry calculation with the B3LYP density functional was applied in combination with the TZVP basis set.39 The Yamaguchi approach46, 47 was used to estimate the Heisenberg exchange coupling constant J. Our J-values are based on the Spin-Hamiltonian HDvV = 2JŜA ŜB.
4.4 Analysis of distance dependence
For the model systems with linkers we analyzed the distance dependence of the D-values obtained with the different approximations by least-squares fitting the D-values to the function
| (8) |
with x being the length of the linkers and being the prefactor for the dipole-dipole-interaction already described in equation (1); a and b are fit-parameters.
5. Quantum Chemical Results
5.1. Geometry optimizations
Full and constrained optimizations were performed for the complete systems (starting from the X-ray structure) and for the truncated model system. For the complete system the relaxation of the coordinates of all atoms leads to very limited changes compared to the optimization where only the hydrogen atom positions were relaxed (see Fig. 3). In particular, the relative orientation of the central spin-carrying ring systems changed by only about 10° and even the dangling carboxylate groups rotate by only about 30° relative to the original structure.
Figure 3.
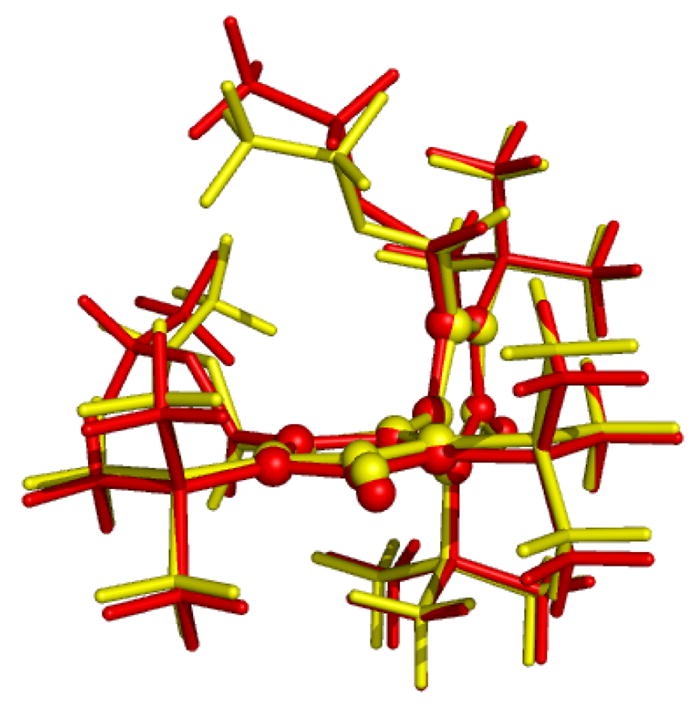
Overlay of the fully optimized structure (BP86/TZVP) in red and of the constrained optimization (with fixed heteroatoms) structure in yellow. The atoms of the two aromatic nitroxyl rings, that are used in our model system, are shown in the ball and stick representation, the substituents are shown as sticks only.
By contrast, the relaxation of all degrees of freedom in the truncated model systems led to large changes in the geometry. Without the steric constraints provides by the bulky substituents, the two rings are predicted to be coplanar compared to a dihedral angle of 104.6° observed experimentally and 95.3° obtained in the optimization of the complete system.
Given that the dihedral angle between the two ring systems is an important degree of freedom for the truncated systems, we investigated the properties of the system as a function of this angle. This provides essential insight into the electronic communication between the two subsystems. A relaxed surface scan was therefore performed of the truncated system (Fig. 4). Compared to the orthogonal configuration, the coplanar structures are stabilized by 4.0 and 5.7 kcal/mol, respectively.
Figure 4.
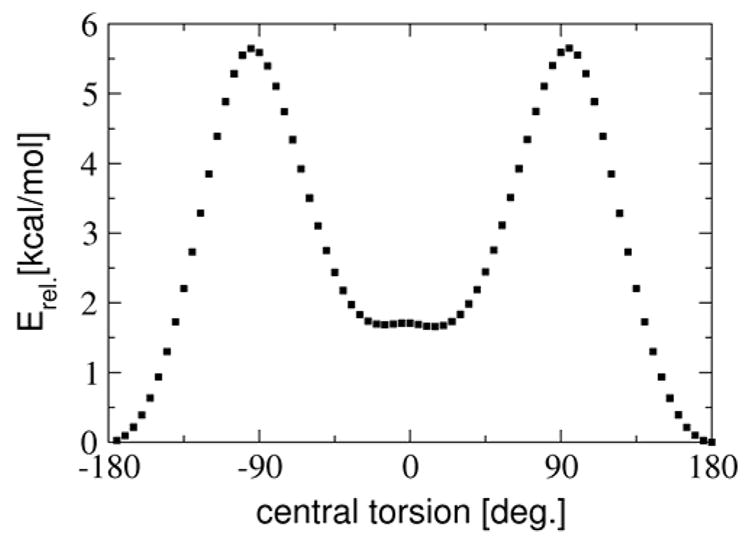
Calculated energy for the truncated model system as a function of the constrained central torsion employing the BP86 density functional with the TZVP basis set.
5.2. Electronic Structure
The localized SOMOs are shown in Fig. 5 for three configurations: the two coplanar configurations at 0° and 180° and a perpendicular configuration at −90°. Not surprisingly, in all configurations the SOMOs are π-orbitals, that are delocalized over the N-O group and the parent ring system.48–50 However, in the coplanar configurations even the localized SOMOs have significant contributions on the neighboring ring (up to 9% delocalization). In the perpendicular configuration the SOMOs show only small orthogonalization tails into the σ-system of the adjacent ring system (~2%).
Figure 5.

Localized SOMOs at three different configurations: On the left side at a central torsion of 180°, in the middle a central torsion of 0° and on the right side an orthogonal configuration with a central torsion of − 90°. The population of the SOMOs on the two fragments is indicated as percentages.
5.3. Zero-Field Splitting: Comparison to Experiment
The spin-spin contribution to the ZFS was calculated for both the fully relaxed and the partially optimized untruncated system. Both optimized structures gave the same DI of − 0.020 cm−1 which is in excellent agreement with the experimentally determined value of 0.020 cm−1. Thus, we conclude that the electronic structure of [6] is well represented by the truncated model system. The distance between the N-O midpoints in the experimental structure is 7.006 Å. Assuming the point dipole approximation to be valid leads to a predicted | DII | of 0.008 cm−1 which represents only about 40% of the experimental (or the quantum chemically calculated) value. As pointed out above, if we employ the point dipole approximation to interpret the measured D-value and estimate an intercenter distance, the result is 5.1 Å. Thus, an error as large as 2 Å would result from the use of the point dipole approximation! Since the full calculation reproduces the experimental value accurately, the reason for this dramatic failure must be of quantum mechanical origin. Hence, it is important to investigate in detail at which stage deviations from the point dipole behavior occur and under which circumstances one will have to exercise more care.
Interestingly, for the fully relaxed structure of the truncated system with completely coplanar rings the magnitude of DI increases by as much as 65% to 0.033 cm−1 although the intercenter distance does not change at all. This finding together with the various approximations to the D-value will be analyzed in detail below.
5.4. Zero field splitting analysis
To obtain insight into the nature of the failure of the point dipole approximation a series of calculations was carried out. We found it instructive to compare the behavior of the individual contributions to DI along the twisting coordinate. This will clearly show which parts of the D-value arise quantum mechanically and which are of purely classical origin.
5.4.1 Coulomb versus Exchange Contributions
DI was calculated for the structures obtained with the relaxed surface scan. Its dependence on the orientation of the two rings is shown in Fig. 6. The magnitude of DI is minimal for the perpendicular configuration and increases to a maximum value for the coplanar configuration.
Figure 6.
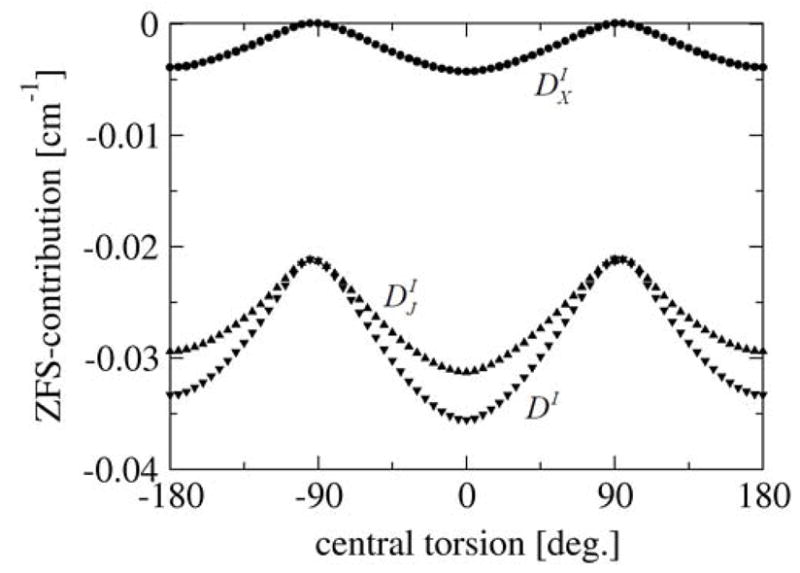
Calculated spin-spin (SS) contribution to the ZFS parameter DI for the dinitroxyl as a function of the constrained central torsion, employing the EPR-II basis set. Results are given for DI (black filled lower triangles), Coulomb-like contribution ( , black filled upper triangles) and exchange-like contribution ( , black filled circles).
To analyze this phenomenon we split DI into an exchange-like contribution ( ) and a Coulomb-like contribution ( ) (Fig. 6). Three interesting observations are made: (1) is zero for the perpendicular configuration; (2) is of the same sign as and (3) its magnitude in the coplanar configuration is about 20% of DI. This ~20% contribution is a quantum mechanical correction to the magnetic dipole interaction and it can obviously not be neglected in an analysis that aims to be quantitative.
The magnitude of increases upon approaching the coplanar conformation. A much weaker geometry dependence would be expected if the spins behaved as classical magnetic dipoles situated at the midpoint of the N-O bond. As will be elaborated below, we attribute this finding to electron delocalization effects that maximize in the coplanar orientation.
5.4.2 Multicenter contributions
If a point-dipole model is assumed with spins localized on individual centers, it is exclusively the (AA|BB) class of integrals that would contribute to the final D-value. The extent to which this is true can be investigated by studying the one- through four-center contributions to the DI -value evaluated quantum mechanically (Fig. 7) and defined in Table 2. According to the results of these calculations, there are two main contributions to DI. The largest contribution originates – as expected – from the 2-center-Coulomb integrals (AA|BB) that make up . This contribution follows the same trend as the final DI -value, but significantly (9–23%) overestimates its magnitude. Perhaps unexpectedly, the 3-center-Coulomb integrals (AA|BC), , account for the second largest contribution. also shows the same trend as DI, but with opposite sign. The opposite sign results from the antibonding character of the SOMOs that reduces the spin density in the N-O bonding region. Hence, this important bond correction compensates for the overestimate of the ‘pure’ 2-center-Coulomb integral contribution.
Figure 7.
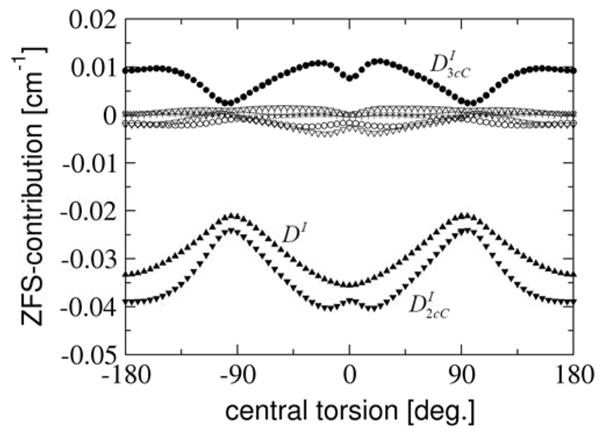
Calculated spin-spin (SS) contribution to the ZFS parameter DI from the 1–4 center contributions defined in Table 2 for the truncated dinitroxyl as a function of the constrained central torsion, employing the EPR-II basis set. Results are given for DI (black filled upper triangles), 1-center-Coulomb- (+), 2-center-Coulomb- ( , black filled lower triangles), 2-center-Exchange- (grey filled lower triangles), 2-center-Hybrid- (open lower triangles), 3-center-Coulomb- ( , black filled circles), 3-center-Exchange- (open circles) and 4-center-integral contribution (x).
5.4.3 Comparison of point dipole models
In Fig. 8 DI, the Coulomb part of the 2-center-Coulomb contribution and the DII -values obtained at different levels of approximation are plotted. With the approximations made to obtain the ‘distributed point dipole’ like equations, DDPD should reproduce the Coulomb part of the 2-center-Coulomb contribution to DI. For the orthogonal configuration, where the two SOMOs show only very small delocalization tails onto the neighboring spin center, this is indeed the case. The distributed dipole model approximates the Coulomb contribution to DI to within 2% here, but still has an error of 15% compared to the full DI -value since it does not include the multicenter and exchange interactions properly. However, compared to an error of 40% observed for the ‘naïve’ point-dipole model, the distributed dipole approximation is much more successful and reduces the error by more than a factor of two.
Figure 8.
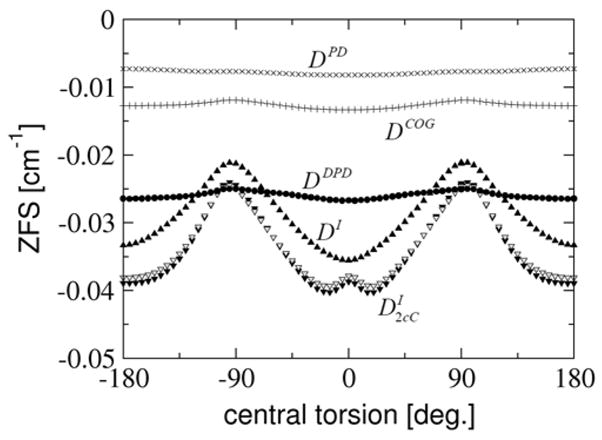
Comparison of DI and its important contributions calculated with the EPR-II basis set, and DII obtained from spin populations calculated with the TZVP basis set for the truncated dinitroxyl as a function of the constrained central torsion. Results are given for DI (black filled upper triangles), distributed dipole approximation (DDPD, black filled circles), center of gravity approximation (DCOG, +), point dipole approximation (DPD, x), 2-center-Coulomb ( , black filled lower triangles) and the Coulomb part of the 2-center-Coulomb contribution (grey filled lower triangles).
On top of the 15% error relative to the full DI -value, there are additional failures of the distributed point dipole model: foremost, the DDPD has too weak an orientation dependence and thus strongly underestimates the -value for the coplanar structure. For these configurations both SOMOs are significantly delocalized over the neighboring fragment (see Fig. 5). Thus, the magnetic dipole-dipole interaction occurs (on average) at much smaller distances than can be modeled within the ‘distributed point dipole’ type approximation where the spin centers are necessarily separated. The observed small orientation-dependence of DDPD is due to the fact that the spin density within the rings increases at the expense of the spin density on the oxygens with increasing planarity of the rings.
The center of gravity approximation yields DCOG, which shows only about half of the magnitude of DDPD, but the same orientation dependence. For a highly delocalized system this approximation highly underestimates the contribution of the dipole-dipole interaction of atom pairs at small distances, since it just takes an average distance of the unpaired electrons.
Within the simplest approximation, the point dipole approximation, DPD is even smaller in magnitude than DCOG. Again this arises from the fact that the spin density is delocalized over the ring system. The distance between the two nitroxyl groups is essentially independent of the actual conformation and thus there is an even smaller dependence on the orientation of the two rings than with the two other approximations.
Taken together, the deviations of the naïve point dipole model (DPD ) from the precisely calculated values may be summarized as follows: The point dipole model, assuming the unpaired electrons in the middle of the N-O-bond, strongly underestimates the D-value. Depending on the amount of exchange contribution and of electron delocalization this simplest possible model accounts for only 22–36% of DI. The center of gravity approximation, which takes the delocalization of the SOMOs on the ring atoms to some extent into account, results in 38–56% of DI. The more elaborate introduction of the electron delocalization by the distributed dipole approximation yields a D-value that is 75–118% of DI. This oscillation around the exact value is due to different errors occurring at different configurations. Comparing the DDPD more rigorously with the Coulomb part of the 2-center-Coulomb contribution we reach 70–100% of , depending on the configuration of the two rings and thus on electron delocalization onto the neighboring ring.
5.5 ‘Through-Bond’ versus ‘Through-Space’ Interactions
The analysis presented above suggests that, at the heart of the failure of the point dipole approximation for dinitroxyl [6] is the close proximity of the two electronic systems and the significant electron delocalization effects, which leads to large quantum mechanical corrections to the point dipole approximation.
We therefore asked at which distance and separation of the two spin-systems does the (naïve) point dipole approximation become accurate? In order to investigate this question we have carried out an in silico study in which we inserted a linker of increasing length between the two nitroxyl spin-centers. Two types of linkers were investigated: (a) an unsaturated linker that would allow for through bond interactions, and (b) a saturated linker that greatly attenuates through-bond interactions.
The (partially optimized) structures and the localized SOMOs for these systems are shown in Fig. 9 for the inserted unsaturated and saturated C10-linkers. All structures with the allyl linker are planar. The structures with the alkyl linker were optimized such that both rings and the heteroatomic part of the linker are planar. The SOMOs of both systems are of similar shape as in the case without linkers. However, the contour plots show that the unsaturated linker with its conjugated π-electron system functions as a kind of conductor and thus even the localized SOMOs extend over the whole linker into the remote ring system. By contrast, the saturated linker cuts the electronic connection between both spin centers. Here we find only a slight delocalization into the σ-system of the first few carbon atoms.
Figure 9.

Localized SOMOs for the unsaturated linker - C10H10 – on the left side and the saturated linker - C10H20 – on the right side. The population of the SOMOs on the fragments is indicated in percentages. It is given separately for the ring fragments and for the linker halves.
The calculated D-values for the unsaturated linker are shown in Fig. 10 as a function of intervening linker length. Not surprisingly, the magnitude of all DII -values decreases with increasing separation of the spin centers. However, most importantly, the absolute D-value decreases much faster with interfragment distance for all point-dipole based schemes than for DI, which is used as the standard for evaluating other methods.
Figure 10.
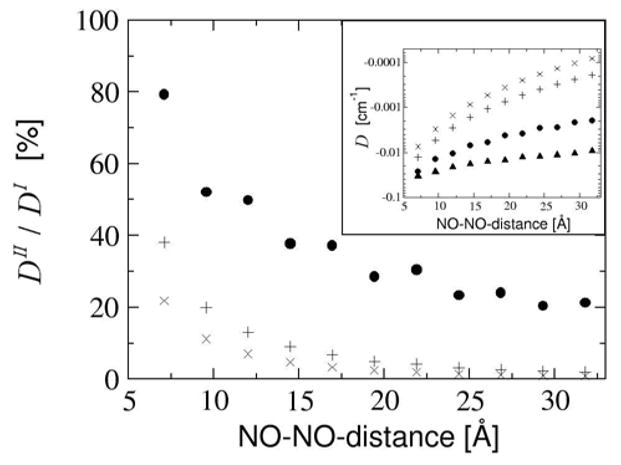
Unsaturated linker. Comparison of DI and DII as a function of the NO-NO-midpoint-distance with the unsaturated linker. The D-value is given as percentage of DI and as absolute values in the inlay diagram. Results are given for DI (black filled upper triangles), distributed dipole approximation (DDPD, black filled circles), center of gravity approximation (DCOG, +) and point dipole approximation (DPD, x).
Apparently, the least erroneous approximation is the distributed dipole model. Without additional linker atoms (which corresponds, at the same time, to a NO-NO-bond midpoint distance of about 5.7 Å), it accounts for 80% of DI. However, this fraction decreases to 50% of DI with a C4-linker. The decline of the quality of the distributed dipole approximation is due to the fact that the delocalization of both SOMOs over the neighboring fragment is supported by the unsaturated linker, which cannot be modeled by this approximation, where the spin centers are localized.
The center of gravity approximation accounts for only 40% of DI without any additional linker atoms. This fraction decreases even faster than that of DDPD. With increasing linker length of the unsaturated linkage and thus increasing delocalization over the linker atoms the contribution to DI from the dipole-dipole interaction of the spin density on the ring fragments decreases very fast. Nevertheless, the majority of the spin population can still be found on the ring atoms. The interaction at small distances is thus strongly underestimated and DCOG is only 10% of DI with a C6-linker.
The point dipole approximation does not account for the conducting bridge at all. Without additional linker atoms DPD yields 20% of DI. This fraction declines to less than 10% with a C4-linker.
Thus, no matter what the chain length is, any point dipole model produces incorrect results if the two spin carrying fragments are connected via an unsaturated bridge. Effective distances determined for such systems via the point dipole model will therefore have serious errors.
The calculated D-values for the saturated linker (shown in Fig. 11) are significantly different. Comparing DI for the saturated linker with those of the unsaturated linker we see, that the magnitude of DI decreases much faster for the saturated bridge (compare Fig. 10). Thus, the different point dipole approximations become more and more accurate with increasing spin center distance.
Figure 11.
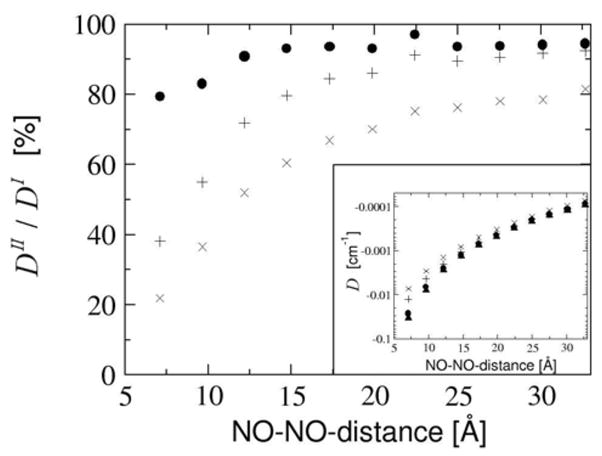
Saturated linker. Comparison of DI and DII as a function of the NO-NO-midpoint-distance for the saturated linker. The D-values are given as percentage of DI and as absolute values in the inset. Results are given for DI (black filled upper triangles), distributed dipole approximation (DDPD, black filled circles), center of gravity approximation (DCOG, +) and point dipole approximation (DPD, x).
Within the ‘distributed point dipole’ like approximation the calculated DII -value DDPD accounts for more than 90% of DI with a linker of 4 C atoms and reaching a limit of around 94% with a C8-linker. For a C4-linker the fraction of the exchange contribution falls below 1% of the total DI (see also Fig. 12). Thus for linkers with more than 4 C atoms, the remaining errors of the distributed dipole approximation are those introduced by the spin population analysis together with the slight errors arising from the ‘compression’ of the spin density onto the atomic centers.
Figure 12.

Comparison of the exchange- and the Coulomb-contribution to DI as a function of the NO-NO-midpoint-distance with the unsaturated linker (left figure) and with the saturated linker (right figure). Results are given for the exchange- ( , black filled circles) and for the Coulomb-contribution ( , black filled squares).
The center of gravity approximation can account for more than 80% of DI for a C6-linker, and for more than 90% for a C12-linker. Since the SOMOs in the systems with saturated linkers are more or less independent of the length of the linker, the centers of gravity of the SOMOs are constantly around 4.4 Å more distant than the length of the linker. With increasing linker length the underestimation of the dipole-dipole-interaction of atom pairs at small distances becomes less important due to the 1/R3 behavior.
Finally, the simplest approximation, the point dipole approximation, only very slowly approaches DI. It reaches 80% of DI only for a linker with 20 C atoms! The point dipoles representing the SOMOs in this approximation are always about 5.7 Å further apart than the length of the linker and thus the magnitude of DPD is always below that of DCOG.
To study whether there is a difference between the interaction through vacuum and through the saturated linker, we replaced the saturated linkers of different lengths by a hydrogen atom on each ring, relaxed the hydrogen positions, calculated DI and compared the results to DI with the saturated, non-conducting bridge. We found a significant difference of up to 25% of the D-value only at small linker lengths. This deviation decreased quickly to only 5% at a NO-NO-distance of 20 Å (i.e. with C10-linker, if present). This increase in the D-value is not due to a different type of interaction, but can be traced back to the slight delocalization of the SOMOs into the σ-system of the first few carbon atoms (see also Fig. 9). Thus, the saturated linker itself behaves essentially identical to vacuum. However, since the small tails of the spin-density (caused by hyperconjugation) are likely to be present in many of the real systems, the point dipole approximation still introduces a recognizable error whenever there is any chemical contact between the two spin carrying fragments.
5.6 Power laws
To compare the distance dependence of the D-values for the different applied models and the different linkers we fitted the data to a power function. The results can be seen in table 5. Here the parameter ‘a’ measures the decline of the D-values with increasing linker length. Assuming the SOMOs to be contracted to one point, the parameter ‘b’ can be interpreted as an extrapolation of the location of the real “contracted” point dipoles.
Table 5.
Distance Dependence of D for the different approximations. Least squares fit to .
| Saturated linker | Unsaturated linker | |||
|---|---|---|---|---|
| ZFS calculation model | a | b [Å] | a | b [Å] |
| DI | 3.1 | −2.9 | 1.5 | 11.2 |
| Distributed point dipole | 3.1 | −2.6 | 2.1 | 2.0 |
| Center of gravity | 3.0 | −1.1 | 2.8 | −0.2 |
| Point dipole | 3.0 | 0.1 | 3.0 | 0.1 |
For the saturated linker we find about the same expected inverse third power law for all four methods of calculation, since the delocalization of the SOMOs does not change. The only difference lies in the localization of the “contracted” point dipoles (parameter b). As already pointed out, the simple point dipole and the center of gravity approximation overestimate the distance of the point dipoles.
For the unsaturated linker the power laws predicted by different approximations to the D-value vary significantly. The center of gravity and the distributed point dipole approximations both lead to a significantly faster decay than DI . The origin of this effect is that both approximations take insufficient account of the spin-delocalization onto the bridge. Hence, for DI , where the delocalization is exactly treated, the decay is found to be slowest. Strikingly, the D-value through an unsaturated bridge only decays as R−1.5 rather than the expected R−3.
Furthermore the “contracted” point dipoles for DI or the distributed dipole approximation based D-value are not even found within the ring systems. Rather, the positive sign of b indicates that they are found outside of the molecule (5.6 Å away from the NO-midpoint for DI ). This reflects the fact that the electronically correct effective intercenter distance is much smaller than the one that is read off the geometric structure.
To disrupt conjugation in the unsaturated tether we inserted linkers consisting of allyl-groups separated in the middle by an alkyl-group. The partially optimized structure and the localized SOMOs for the partially saturated C10-linker are shown in Fig. 13. In comparison to the completely unsaturated linker, the SOMOs are delocalized up to the saturated alkyl-group, which ‘insulates” between the two conjugated π-electron systems.
Figure 13.
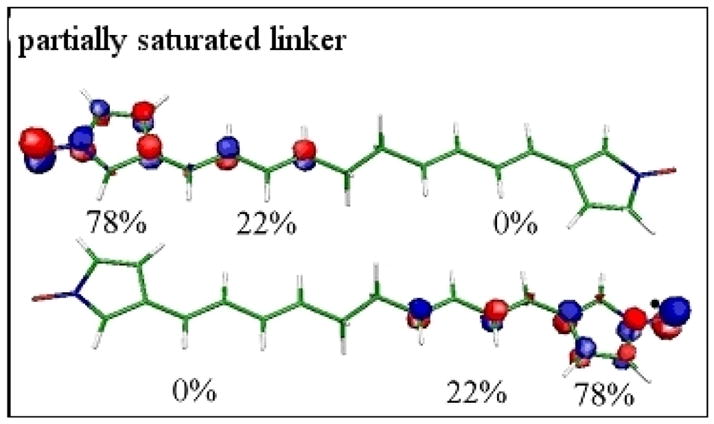
Localized SOMOs for the partially saturated linker - C10H12. The population of the SOMOs on the fragments is indicated as percentages. It is given separately for the ring fragments and for the linker halves.
The calculated D-values for the partially saturated linker are shown in Fig. 14 as a function of intervening linker length. Compared to the unsaturated linker the distributed dipole model follows DI rather closely. Only DCOG and DPD decrease much faster.
Figure 14.
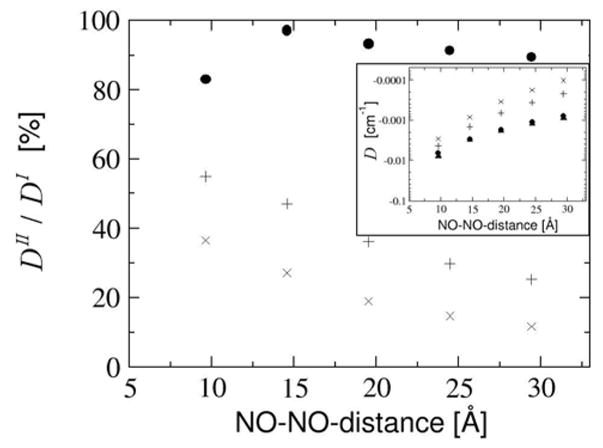
Partially saturated linker. Comparison of DI and DII as a function of the NO-NO-midpoint-distance with the partially saturated linker. The D-value is given as a percentage of DI and as absolute values in the inset diagram. Results are given for DI (black filled upper triangles), distributed dipole approximation (black filled circles), center of gravity approximation (+) and point dipole approximation (x).
As expected, the distributed dipole model performs well for this system, since the SOMOs are delocalized only up to the saturated center of the system. Nevertheless a slight decline of the quality of the distributed dipole approximation (evidenced by the ratio D DPD / DI ) is observed beyond a linker length of 10 Å. This behavior can be traced back to the growing impact of the errors in the spin population analysis. The center of gravity approximation and the point dipole approximation perform slightly better than for the unsaturated linker, but they still greatly underestimate the target DI -value.
5.7 Errors in Point Dipole Based ‘Experimental Distances’
In many experimental cases measured D-values are used to determine distances.1–5 Thus, it is of critical importance to determine the errors in distance that are introduced in the experimental analysis by assuming the point dipole approximation to be valid. To this end we compared (Fig. 15) the NO-NO-midpoint distances in the molecular structures with the NO-NO-midpoint distances that we calculate using the point dipole approximation and the values of DI .
Figure 15.

Comparison of the NO-NO-midpoint-distances in the molecular structures (molec.) and the NO-NO-midpoint-distances calculated using the point dipole approximation and the DI -value for the aromatic nitroxyl ring (left figure) and for the non-aromatic nitroxyl ring (right figure). Results are given for the saturated linker (lower triangles), the unsaturated linker (circles) and the partially saturated linker (open squares).
For the saturated linker the derived distances are 1–2 Å smaller than the distances in the molecular structures. The spin delocalization from the N-O group into the aromatic ring is much less significant for non-aromatic nitroxyl spin labels, since in this case the saturated ring effectively prevents delocalization of the spin onto the bridge. As expected, the error is larger at small linker lengths, since in this regime the neglect of the exchange-contribution to the D-value introduces a non-negligible error in the derived distance. The distances derived for the unsaturated linker increase only slightly with increasing linker length. Here the approximation that the spin is localized on the nitroxyl group is not valid and introduces substantial error for shorter linkers. The errors in distances derived for the partially saturated linker are in between the extremes for saturated and unsaturated linkages.
For the non-aromatic nitroxyl rings that are used in most spin labeling experiments51 the SOMOs are much more localized on the nitroxyl group. In this situation the point dipole approximation becomes significantly better. If the rings are connected by a saturated linker, the error in distance calculated by the point dipole approximation ranges from 0.5 Å at the smallest distance to 1 Å at a NO-NO-distance of 32 Å. Fitting the data to the power law we find a perfect inverse third power law with the parameter b of −0.5. With a N-O-bond length of 1.3 Å the “contracted” point dipoles can thus be found predominantly on the nitrogen nucleus. For the unsaturated linker the results predicted become worse again. Here the errors of the predicted distance accumulate up to 8.5 Å for an experimental distance of 32 Å. Although the bonds next to the nitroxyl group are σ-bonds, both SOMOs delocalize over them and a small – but critical for the distance measurements - spin population (about 1%) is on the unsaturated linker. Due to this effect the power function for the unsaturated linker decays slightly more slowly (as R−2.9 with b ~0.3). In this case, the “contracted” point dipoles are located essentially in the middle of the N-O-bond.
The bottom line of this analysis is that, whenever there are significant spin-delocalization effects, the point dipole approximation is not a reliable way to interpret dipolar splittings measured by EPR.
5.8 Further Validation through Comparison of Calculated and Experimental Zero-Field Splittings
We further validate our study by examining two biradicals (scheme 6) for which interspin distances were derived from double electron-electron resonance (DEER) measurements by using the point dipole approximation.4, 5 Initially we compared the above interspin distance and the experimental D-value with our calculated values from DFT optimized models. For these DFT models we first built model structures for the biradicals, then optimized the molecule structures and finally calculated DI without any further approximation as explained in the Computational Details section. We then derived an interspin distance from the calculated DI -value. If the point dipole approximation is valid, this calculation should give the same distance as is measured from the optimized molecular structures. For both biradicals the interspin distances measured from the optimized structure and calculated from the DFT spin densities are in reasonable agreement (see table 6). Biradical 7a gives a N-N-distance which is 0.7 Å shorter than the point dipole interspin distance. For biradical 8a the N-N-distance was measured from X-ray diffraction and is in the range of experimental uncertainty of the point dipole derived interspin distance. The DFT results show a deviation of about 0.3 Å between the N-N-distance and the point dipole derived interspin distance. These results demonstrate that the point dipole approximation is valid in these systems, but that uncertainties in the derived distance of a few tenth of an Ångström must be tolerated.
Scheme 6.
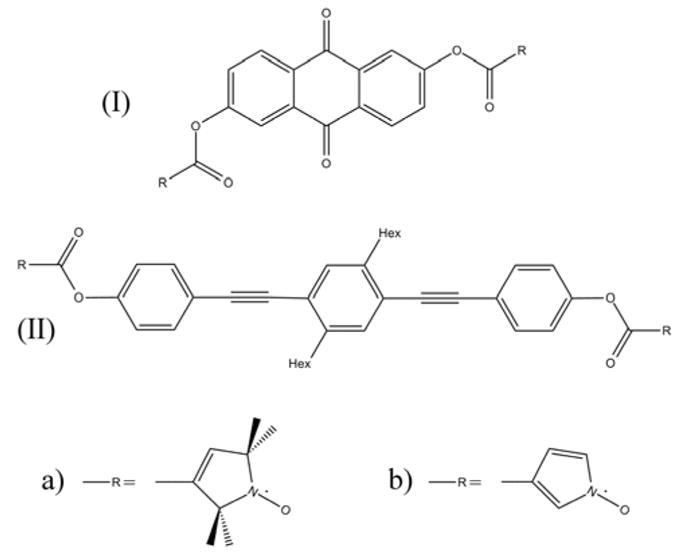
Structural diagram of biradical 7 (I) and biradical 8 (II). These biradicals were studied with non-aromatic oxypyrrolin groups (a) and aromatic pyrroloxyl groups (b).
Table 6.
N-N distances for biradicals 7 and 8 measured experimentally from the molecular structure (with nitroxyl ring a) and determined via DFT-calculations from DI (with nitroxyl rings a and b).
| Biradical 7 | Biradical 8 | |||||
|---|---|---|---|---|---|---|
| System | D [cm−1] | Interspin distance [Å] | N-N-distance [Å] | D [cm−1] | Interspin distance [Å] | N-N-distance [Å] |
| Experimental resultsa | −0.000341 ± 0.000005 | 19.73 ± 0.14 | - | −0.000115 ± 0.000006 | 28.3 ± 0.5 | 27.84 ± 0.01b |
| DFT model (a) | −0.000335 | 19.8 | 19.11b | −0.000109 | 28.8 | 28.46c |
| DFT model (b) | −0.000455 | 17.9 | 18.93b | −0.000357 | 19.4 | 28.29c |
Available only for systems 7a and 8a.
N-N-distance from X-ray diffraction, from ref. 5
From optimized structure.
We then took the computations one more step forward and performed an in silico experiment in which we substituted the spin-carrying non-aromatic oxypyrrolin groups by aromatic pyrroloxyl groups, in order to see the effect of delocalization. The results of these calculations are also given in table 6.
For biradical 7b, the interspin distance derived from DI and the point dipole approximation is compared to the results obtained for the non-aromatic ring. The change in the inferred distance is about 2 Å, whereas the N-N-distance in the molecule only differs by about 0.2 Å. This change in the point dipole interspin distance is due to the delocalization of the SOMOs in the aromatic ring.
For biradical 8b, the substitution of the non-aromatic ring by the aromatic nitroxyl ring changes the results even more drastically. The calculated DI -value increases by a factor of three. The derived point dipole interspin distance shows a decrease of almost 10 Å (from 28.8 Å down to 19.4 Å), although the N-N-distance only shrinks by about 0.2 Å. This dramatic effect is not only due to the delocalization of the SOMOs onto the ring fragment, but also because of a very small amount of delocalization onto the bridging linker, which is a large conjugated system. This small delocalization occurs despite the presence of the ester groups, which separate the conducting bridge from the ring fragments, and is responsible for the drastic effect on the derived interspin distance.
Thus, as soon as the nitroxyl group becomes aromatic, significant delocalization effects take place and the point-dipole derived distances have to be treated with caution.
5.9 Separation of the Heisenberg isotropic exchange interaction and the anisotropic dipolar interaction
For most systems where distance measurements are done by DEER, it is assumed that the Heisenberg isotropic exchange coupling constant J is negligible.2, 3 But if J is of the same order of magnitude as the anisotropic dipolar splitting DI , problems can arise for the measurement of distances.52 Under these circumstances the splittings of the energy levels are functions of both DI and J. (Nevertheless the effects of J and DI can in principle be separated by careful analysis of the full ‘dipolar’ splitting pattern. Exchange interactions through 8 – 12 bonds in spin-labeled metal complexes are large enough to impact CW EPR line shapes at distances of up to ca. 14 Å.53)
We therefore asked for what situations are J and DI of the same order of magnitude? In order to address this question we carried out calculations of the isotropic exchange coupling constant J for: (a) the fully optimized structure, (b) two aromatic nitroxyl rings connected by a saturated linker and (c) two non-aromatic nitroxyl rings connected by a saturated linker. For (b) and (c) the J- and DI -values were calculated as a function of increasing linker length. Generally it is assumed that the isotropic exchange interaction decreases exponentially with increasing spin-spin distance following the equation J = A · exp(− β · R), where R is the spin-spin distance.54, 55 For our system we thus performed a least squares fit of the calculated J-values with the NO-NO-distance to yield the proportional factor β.
The D-value for the fully relaxed structures was already discussed above and has a magnitude of 0.020 cm−1. The exchange coupling constant J for both fully relaxed structures is 37.52 cm−1 and −32.41, respectively (antiferromagnetic coupling), i.e. the J-value is three orders of magnitude higher than the D-value and thus the EPR signals do not depend on the Heisenberg exchange coupling.
The magnitude of the calculated DI - and J-values for the aromatic nitroxyl ring are shown in the logarithmic plot in Fig. 16 as a function of intervening linker length. The D-value shows the already discussed R−3-behaviour. The J-value apparently shows an exponential dependence on the linker length. Up to the C4-linker the J-value is much larger than DI , we are in the strong exchange limit, the observed EPR signals will be only within the S = 1 level manifold. Only for the C6- and the C8-linker we are in the intermediate exchange limit, DI and J are on the same order of magnitude and the observed EPR signals will depend on both. From the C10-linker on the weak exchange limit is reached, the isotropic exchange coupling is much smaller than the anisotropic dipolar splitting and the interpretation of the dipolar splitting is predicted to become simple again.
Figure 16.

Comparison of the dipolar splitting DI and the Heisenberg exchange coupling constant J as a function of the NO-NO-midpoint-distance for the saturated linker with the aromatic nitroxyl ring (left figure) and with the non-aromatic nitroxyl ring (right figure). Results are given for the magnitude of DI (triangles) and for the magnitude of J (circles). A least squares fit was done for the J-values (black line, extrapolation for higher NO-NO-distances as dashed line).
For the saturated linker with the non-aromatic nitroxyl ring the absolute DI - and J-values are shown in Fig. 16. The D-values are lower than for the aromatic ring for comparable linker length, but show the same R−3-behaviour. As expected, the J-value shows also here an exponential dependence on the linker length. The exchange coupling constant is much lower than with the aromatic rings for comparable linker lengths since the spins are mainly localized in the NO-bond for the non-aromatic rings. For the non-aromatic ring DI and J are on similar scales up to the C6-linker, here we are in the intermediate exchange regime. From the C8-linker on we are in the weak exchange limit, J is much smaller than DI and can be assumed to be negligible in the EPR signals. Thus the linker length where J- and D-values are on similar scales and where thus the splittings of the energy levels are functions of both values is shorter by about 3 Å for the non-aromatic rings than for the aromatic rings.
Finally we performed a least squares fit to study the correlation between the isotropic exchange interaction and the NO-NO-distance, where we fitted J to the equation J = A · exp(− β · R). We found β to be approximately 1 Å−1 for both sets (0.90 for the aromatic nitroxyl ring and 0.95 for the non-aromatic nitroxyl ring). This value may be compared with estimates of β ≈ 0.7 Å−1 for the electron transfer matrix element HAB for covalent linkages.56 Relationships between both quantities have been discussed.57, 58
6. Conclusions
In the present work a detailed experimental and theoretical analysis of the point dipole approximation for the evaluation of interspin distances was presented. The focus of the investigation has been to understand under which circumstances this central approximation is likely to fail. This is of critical importance in methods for determining interspin distances by electron paramagnetic resonance.
The bottom line of the present analysis is the following: delocalization of the spin density strongly affects the D-value and leads to errors in the interspin distances obtained using the point dipole model. Distances calculated using the point dipole approximation usually are underestimated because delocalization leads to tails of the spin density that are closer together than the majority of the spin density. Spins at closer distances are much more strongly weighted by the dipole-dipole interaction operator than larger distances, so even small delocalization effects lead to errors in derived distances of 1–2 Ångströms. These effects become particularly dramatic for aromatic nitroxides or if there are bridging groups between the two spin carrying fragments that are (partially) unsaturated. In the latter case the D-value not only deviates from the expected R−3 behavior but also becomes significantly dependent on the relative orientation of the two spin-carrying fragments. Any type of point dipole model is bound to fail under such circumstances and derived distances from such models have substantial errors. Thus, whenever the spin label or the bridge that separates the two paramagnetic centers is significantly unsaturated one is well advised to avoid the point dipole approximation. If there is sufficient structural information available, quantum chemical calculations can be of great help in the analysis of actual experiments.
Supplementary Material
Table 4.
Overview of the different ZFS terms.
| Designation | ZFS calculation level | see equation | |
|---|---|---|---|
| DI | Explicit quantum chemical calculation | (1) | |
| DDPD | Distributed point dipole model | (5) | |
| DII | DCOG | Center of gravity approximation | (8) |
| DPD | Point dipole approximation | (8) a | |
with R(F1) and R(F2) being the N-O midpoints.
Acknowledgments
CR and FN gratefully acknowledge financial support from the SFB 624 (‘Templates in chemistry’, University of Bonn) and the SFB 813 (‘Chemistry with Spin Centers’, University of Bonn). The work done in the USA was supported by the National Institutes of Health EB002807 (GRE and SSE), GM056481 (JPYK), and DA023473 (GMR). The Center for in vivo EPR imaging is supported by NIH grant P41 EB002034 (GRE, SSE, GMR). The X-ray crystallography was done by Dr. Peter Y. Zavalij (U. of Maryland). We appreciate the exploratory spectroscopy and computations early in this project by Eugene D. Barth, Colin Mailer, and Michael K. Bowman.
LITERATURE
- 1.Borbat PP, Davis JH, Butcher SE, Freed JH. J Am Chem Soc. 2004;126:7746–7747. doi: 10.1021/ja049372o. [DOI] [PubMed] [Google Scholar]
- 2.Jeschke G, Polyhach Y. Phys Chem Chem Phys. 2007;9:1895–1910. doi: 10.1039/b614920k. [DOI] [PubMed] [Google Scholar]
- 3.Eaton GR, Eaton SS. In: Specialist Periodical Report: Electron Paramagnetic Resonance. Gilbert BC, editor. Vol. 21. 2008. pp. 59–75. [Google Scholar]
- 4.Larsen RG, Singel DJ. J Chem Phys. 1993;98:5134–5146. [Google Scholar]
- 5.Pannier M, Veit S, Godt A, Jeschke G, Spiess HW. J Magn Reson A. 2000;142:331–340. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- 6.Bertrand P, More C, Guigliarelli B, Fournel A, Bennett B, Howes B. J Am Chem Soc. 1994;116:3078–3086. [Google Scholar]
- 7.Neese F. In: Specialist Reports on EPR Spectroscopy. Gilbert B, editor. Vol. 20. Royal Society of Chemistry; London: 2007. pp. 73–95. [Google Scholar]
- 8.Sinnecker S, Neese F, Lubitz W. J Biol Inorg Chem. 2005;10(3):231–238. doi: 10.1007/s00775-005-0633-9. [DOI] [PubMed] [Google Scholar]
- 9.van Gastel M, Stein M, Brecht M, Schröder O, Lendzian F, Bittl R, Ogata H, Higuchi Y, Lubitz W. J Biol Inorg Chem. 2006;11:41–51. doi: 10.1007/s00775-005-0048-7. [DOI] [PubMed] [Google Scholar]
- 10.Neese F. J Chem Phys. 2003;118:3939. [Google Scholar]
- 11.Kababya S, Nelson J, Calle C, Neese F, Goldfarb D. J Am Chem Soc. 2006;128(6):2017–2029. doi: 10.1021/ja056207f. [DOI] [PubMed] [Google Scholar]
- 12.Kossmann SBK, Neese F. Molec Phys. 2007;105(15–16):2049–2071. [Google Scholar]
- 13.Ray K, Begum A, Weyhermüller T, Piligkos S, van Slageren J, Neese F, Wieghardt K. J Am Chem Soc. 2005;127:4403–4415. doi: 10.1021/ja042803i. [DOI] [PubMed] [Google Scholar]
- 14.Neese F. J Chem Phys. 2007;127:164112. doi: 10.1063/1.2772857. [DOI] [PubMed] [Google Scholar]
- 15.Sinnecker S, Neese F. J Phys Chem A. 2006;110(44):12267–12275. doi: 10.1021/jp0643303. [DOI] [PubMed] [Google Scholar]
- 16.Shoji M, Koizumi K, Hamamoto T, Taniguchi T, Takeda R, Kitagawa Y, Kawakami T, Okumura M, Yamanaka S, Yamaguchi K. Polyhedron. 2005;24(16–17):2708–2715. [Google Scholar]
- 17.Takeda R, Mitsuo S, Yamanaka S, Yamaguchi K. Polyhedron. 2005;24(16–17):2238–2241. [Google Scholar]
- 18.Neese F. J Am Chem Soc. 2006;128:10213–10222. doi: 10.1021/ja061798a. [DOI] [PubMed] [Google Scholar]
- 19.Pederson MR, Khanna SN. Phys Rev B. 1999;60(13):9566–9572. [Google Scholar]
- 20.McWeeny R. Methods of Molecular Quantum Mechanics. Academic press; London: 1992. [Google Scholar]
- 21.Harriman JE. Theoretical Foundations of Electron Spin Resonance. Academic press; New York: 1978. [Google Scholar]
- 22.Ramasseul R, Rassat A. Bull Soc Chim Fr. 1970;72:4330. [Google Scholar]
- 23.Radner F, Rassat A, Hervsall CJ. Acta Chem Scand. 1996;50:146. [Google Scholar]
- 24.Hanson GR, Gates KE, Noble CJ, Griffin M, Mitchell A, Benson S. J Inorg Biochem. 2004;98:903–916. doi: 10.1016/j.jinorgbio.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Sheldrick GM. SADABS. University of Göttingen; Göttingen, Germany: 1996. [Google Scholar]
- 26.Sheldrick GM. SHELXS97. University of Göttingen; Göttingen, Germany: 1997. [Google Scholar]
- 27.Sheldrick GM. SHELXL97. University of Göttingen; Göttingen, Germany: 1997. [Google Scholar]
- 28.Bruker AXS, I. SAINT. Bruker AXS Inc.; Madison, Wisconsin, USA: 2001. [Google Scholar]
- 29.McWeeny R, Mizuno Y. Proc Roy Soc (London) 1961;A259:554. [Google Scholar]
- 30.Kahn O. Molecular Magnetism. VCH Publishers; New York: 1993. [Google Scholar]
- 31.Bencini A, Gatteschi D. EPR of Exchange Coupled Systems. Springer; Heidelberg: 1990. [Google Scholar]
- 32.Bertrand P, Camensuli P, More C, Guigliarelli B. J Am Chem Soc. 1996;118:1426–1434. [Google Scholar]
- 33.Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Weinhold F. NBO, Version 5.0. Univ. of Wisconsin; Madison, WI: 2001. [Google Scholar]
- 34.Wells GB, Makinen MW. J Am Chem Soc. 1988;110(19):6343–6352. [Google Scholar]
- 35.Neese F. ORCA - an ab initio, Density Functional and Semiempirical Program Package. 2.5–20. Universität Bonn; Bonn, Germany: 2007. [Google Scholar]
- 36.Perdew JP. Phys Rev B. 1986;34(10):7406–7406. [PubMed] [Google Scholar]
- 37.Perdew JP. Phys Rev B. 1986;33(12):8822–8y824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 38.Becke AD. Phys Rev A. 1988;38(6):3098–31y00. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 39.Schäfer A, Huber C, Ahlrichs R. J Chem Phys. 1994;100(8):5829–5835. [Google Scholar]
- 40.Barone Vy. In: Reycent Advances in Density Functional Methods. Chong DP, editor. Part I. World Scientific; Singapore: 1996. [Google Scholar]
- 41.Pipek J, Mezey PG. J Chem Phys. 1989;90:4916–4926. [Google Scholar]
- 42.Rega N, Cossi M, Barone V. J Chem Phys. 1996;105(24):11060–11067. [Google Scholar]
- 43.Beckey AD. J Chem Phys. 1993;98(7):5648–5652.y. [Google Scholar]
- 44.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 45.Neese F. Coord Chem Rev. 2008;253:526–563. [Google Scholar]
- 46.Yamaguchi K, Takahara Y, Fueno T. In: Applied Quantum Chemistry, V. Smith VH, editor. Reidel; Dordrecht: 1986. p. 155. [Google Scholar]
- 47.Soda T, Kitagawa Y, Onishi T, Takano Y, Shigeta Y, Nagao H, Yoshika Y, Yamaguchi K. Chem Phys Lett. 2000;319:223. [Google Scholar]
- 48.Pontillon Y, Caneschi A, Gatteschi D, Grand A, Ressouche E, Sessoli R, Schweizer J. Chem Eur Jy. 1999;5(12):3616–3624. [Google Scholar]
- 49.Schatzschneider U, Rentschler E. J Mol Struct Theochem. 2003;638:163–168. [Google Scholar]
- 50.Yao M, Inoue H, Yoshioka N. Chem Phys Lett. 2005;402:11–16. [Google Scholar]
- 51.Altenbach C, Oh KJ, Trabanino RJ, Hideg K, Hubbell WL. Biochemistry. 2001;40:15471–15482. doi: 10.1021/bi011544w. [DOI] [PubMed] [Google Scholar]
- 52.Jeschke G, Spiess HW. Lecture Notes in Physics. 2006;684:21–63. [Google Scholar]
- 53.Eaton GR, Eaton SS. Accts Chem Res. 1988;21(3):107–13. [Google Scholar]
- 54.Likhtenshytein GI. 2000;19:309–345. [Google Scholar]
- 55.Coffman RE, Buettner GR. J Phys Chem. 1979;83:2387–2392. [Google Scholar]
- 56.Moser CC, Page CC, Farid R, Dutton PL. J Bioenerg Biomembr. 1995;27:263–274. doi: 10.1007/BF02110096. [DOI] [PubMed] [Google Scholar]
- 57.Gamelin DR, Bominaar EL, Kirk ML, Wieghardt K, Solomon EI. J Am Chem Soc. 1996;118:8085–8097. [Google Scholar]
- 58.Bominaar EL, Achim C, Borshch SA, Girerd JJ, Münck E. Inorg Chem. 1997;36(17):3689–3701. doi: 10.1021/ic961298q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.