

Abstract
Functionalized styrenes are extremely useful building blocks for organic synthesis and for functional polymers. One of the most general syntheses of styrenes involves the combination of an aryl halide with a vinyl organometallic reagent under catalysis by palladium or nickel complexes. This Feature Article provides the first comprehensive summary of the vinylation methods currently available along with a critical comparison of the efficiency, cost and scope of the methods.
Introduction
The carbon–carbon double bond is arguably the most diversifiable functional group in organic chemistry.1 The variety of reactions available to functionalize olefins spans the range of reductive (hydrogenation, hydroboration, hydrosilylation etc.), oxidative (epoxidation, aziridination, dihydroxylation, halogenation, etc.), isohypsic (hydroamination, hydration, hydroformylation, etc.) and constructive transformations (cycloadditions). In addition, the scope and utility of olefin metathesis (the exchange of a double bond substituent) continues to grow.2
Styrenes, a subclass of α-olefins in which the alkene bears a single aryl substituent, are useful building blocks for fine chemical synthesis and the polymer industry.3,4 Moreover, these substrates are often workhorses for the optimization of new synthetic methods, often those involving catalytic, asymmetric transformations.5 Hence, the development of efficient, mild, selective, and high-yielding methods for the preparation of styrenes will continue well into the future.
Classically, the installation of a terminal double bond occurs by one of the following strategies: (1) elimination of activated leaving groups, (2) carbonyl olefination (by phosphorus, silicon, or titanium-based reagents), or (3) the partial reduction of a terminal alkyne. A more recent development involves palladium-catalyzed, cross-coupling reactions that employ, as precursors, independent aryl and vinyl units. The features that distinguish each of these approaches include the number of bonds formed, the nature of the precursors needed and the reactions that connect them (Fig. 1).
Fig. 1.

Methods used to form a terminal alkene.
The utility of each of the three approaches can be evaluated by considering the ease of access and stability of the required substrates, as well as the functional group tolerance of the key olefin-forming event. For case A (eliminations), both carbon atoms already must be present, which shifts the problem to the often non-trivial introduction of a functionalized ethyl group. The precursors are generally stable, as leaving group activation is required to effect elimination. Because the reaction conditions for eliminations can involve elevated temperatures and strong bases, only a limited subset of functional groups are compatible. Carbonyl olefination reactions (B) require an aldehyde and ylide starting materials. Aldehydes are readily available precursors and phosphorus ylides are equally accessible. On the other hand both aldehydes and ylides are reactive functions. Most importantly, carbonyl olefination is generally associated with poor atom economy. Finally, each of these disconnections (A and B) is a two-step sequence. The cross-coupling strategy (C), avoids these concerns. In general, the required aryl (or vinyl) halide substrates are commercially available. Aryl halides are inert to most synthetic transformations and can be carried through a multiple reaction sequence as a placeholder for a vinyl group. Further, with the recent development of milder reaction conditions and expanded scope of electrophiles (vide infra) the functional group tolerance of vinylation reactions (Scheme 1) is superior to methods A or B. Therefore, these new methods offer significant strategic advantages over the classical preparations.
Scheme 1.

Transition metal-catalyzed, vinylation reaction.
The cross-coupling disconnection can be further subdivided into four pairwise combinations of aryl and vinyl units (Scheme 2): (1) vinylmetallic donor and aryl halide (or pseudohalide), (2) arylmetallic donor and vinyl halide, (3) aryl halide and vinyl halide (with a reductant), and (4) arylmetallic donor and vinylmetallic donor (with an oxidant). Of these, the first two follow the normal cross-coupling strategy (donor/acceptor) and are therefore the most easily adapted. The latter two are inherently less efficient because the reactants are not oxidation state matched and require stoichiometric amounts of either reductants or oxidants. Moreover, the additional complication of cross, vs. homocoupling products is introduced.
Scheme 2.

Cross-coupling disconnections.
Despite the vast number of newly-developed, transition metal-catalyzed, cross-coupling reactions,6 only a small fraction of these accommodates the attachment of a simple vinyl unit. At first glance, the coupling of a vinyl group appears to be no different than that of more elaborate alkenyl groups. However, the coupling of a vinyl group and an aryl group presents significant differences. The first consideration is cost and atom efficiency.7 Unlike larger and more complex donors and acceptors, the vinyl unit is almost always smaller (lower molecular weight) than the non-transferable group (−SnBu3, −B(OR)2, −BF3, −SiR3, or Br, I). Therefore, the relative size of the non-transferable group is much more pertinent to the overall reaction efficiency in comparison to the alkenyl- or arylmetallic congeners.
The second consideration is the reactivity of the educts and products under the reaction conditions. The vinylmetallic donor (or acceptor) can react in two ways, either in the desired cross-coupling reaction (Scheme 3), or alternatively participate in a Heck reaction8 that leaves the MLn unit intact. Stewart and Whiting,9 and Jeffery10 have independently capitalized upon this disparate reactivity to develop two sets of conditions that are selective for either of these pathways using vinylboronic esters or vinyltrimethylsilane, respectively, and their results will be discussed later.
Scheme 3.

Competitive reaction pathways in vinylation.
In addition, the products of the reaction, by definition, contain a terminal vinyl group, either as a styrenyl or dienyl unit that can serve as substrates for subsequent Heck reactions. Therefore, a successful vinylation reaction must display high selectivity for the primary vinylation process over a secondary Heck process. Finally, the polymerization of the styrenyl and dienyl products is known to occur in the presence of bases and transition-metal catalysts, especially at elevated temperatures. Therefore, mild conditions must be employed to achieve high yields of the desired products.
In the past decade, a number of new vinylation methods have been developed that have dramatically increased the scope of this reaction. Thus, the purpose of this Feature Article is to provide a comprehensive overview of vinylation methods with an emphasis on these recent advances and to evaluate the relative merits of each. The presentation will follow the organization outlined in Scheme 2, beginning with the coupling of vinylmetallic donors and aryl halides. The discussion of this strategy will be organized by the nature of the metal/metalloid on the vinyl donor, following their location in Groups 2–14 in the Periodic Table. The scope and limitations for each of these methods will be discussed, and where applicable, we will specifically illustrate the strategies used to address the aforementioned challenges of the introduction of a vinyl group.
Ethylene (a special case)
Although the reaction of an aryl halide and ethylene catalyzed by a transition metal is not, by definition, a cross-coupling reaction,11 omission would be inappropriate. Ethylene is the ideal vinyl source for a vinylation reaction, as only one hydrogen atom is lost from the ethylene group. Accordingly, the successful incorporation of a vinyl unit using ethylene generates as waste only HX (from an aryl halide). In practice, ethylene was initially demonstrated to be an effective vinyl donor by Heck in 1968.12,13 Subsequent optimization by Heck allowed the conversion of a range of substituted aryl bromides into the corresponding styrenes in moderate yields14 (Table 1). These reactions are generally performed in an autoclave at pressures up to 200 psi. Elevated pressures are required to obtain high yields by suppressing the formation of the symmetrical stilbene 3, formed when the product and starting material participate in a secondary Heck reaction (entries 1–3). The reactions are commonly conducted in acetonitrile with palladium acetate, tri-2-tolylphosphine and triethylamine. The aryl bromides can bear functional groups such as nitro, carboxylic acids, anilines and anilide groups (entries 4–8). A double vinylation of o-dibromobenzene provided one of the highest yields (entry 9). Despite the high atom efficiency, this process has found little use in laboratory scale vinylation reactions, most likely because of the need for specialized equipment.15
Table 1.
Reactions of aryl bromides with ethylene
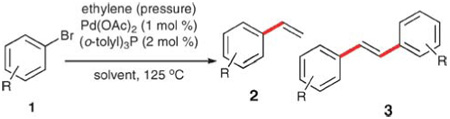 | ||||||
|---|---|---|---|---|---|---|
| Entry | R | Ethylene/psi | Solvent | t/h | Yield of 2 (%) |
Yield of 3 (%) |
| 1 | 2-CH3 | 20 | CH3CN | 20 | 54 | 34 |
| 2 | 2-CH3 | 100 | CH3CN | 7 | 83 | 10 |
| 3 | 2-CH3 | 120 | CH3CN | 18 | 86 | 4 |
| 4 | 2-NO2 | 120 | CH3CN | 2 | 55 | 5 |
| 5 | 4-NHAc | 120 | DMF | 23 | 59 | 20 |
| 6 | 2-NH2 | 200 | CH3CN | 30 | 45 | |
| 7 | a | 200 | CH3CN | 66 | 52 | |
| 8 | 3-COOH | 200 | CH3CN | 4 | 51 | 12 |
| 9 | 2-Br | 200 | CH3CN | 15 | 78 | |
3-Bromopyridine.
Vinylation using vinylmetallic donors
Vinylmagnesium reagents
Vinylmetallic reagents derived from the elements of Groups 1 and 2 have not been widely employed as coupling partners for vinylations. In fact, vinyllithium and vinylmagnesium bromide are most often used for the preparation of milder, more functional group tolerant vinylmetallic donors. Nevertheless, vinylmagnesium bromide (4) can be used directly in cross-coupling reactions with aryl iodides or bromides. Because organomagnesium reagents are strongly basic and highly nucleophilic, they have limited functional group tolerance. Nonetheless, Bumagin and Luzikova have reported two substrates, 3-iodobenzoic acid and 4-bromophenol, that do participate in the vinylation reaction under catalysis by palladium (Scheme 4).16 The removal of the acidic proton in each of these compounds requires the use of a second equivalent of the Grignard reagent and thus the method is inefficient.
Scheme 4.

Vinylation using vinylmagnesium bromide (4).
Vinylboron reagents
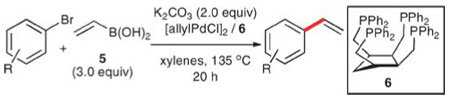
The challenges associated with the high nucleophilicity and basicity of the Grignard reagents have been addressed by the use of less electropositive metals. For this reason, boronic acids and their derivatives have ascended to a preeminent position in the kingdom of transition-metal-catalyzed, cross-coupling processes.17 Curiously, however, the use of boron-based reagents in vinylation reactions is less conspicuous.
Vinylboronic acid
Vinylboronic acid (5) was first prepared by Matteson in 1960, but an uncontrollable polymerization in the final step precluded its isolation and required the conversion to a dibutyl ester.18–20 This instability has obviously hindered the development of vinylation reactions using Suzuki–Miyaura protocols. Despite this limitation, Doucet, Santelli, and co-workers have reported the successful vinylation of aryl bromides using freshly prepared 5.21,22 These reactions employ allylpalladium chloride dimer ([allylPdCl]2) and cis-1,2,3,4-tetrakis(diphenylphosphino)cyclopentane (Tedicyp, 6), potassium carbonate and 3.0 equiv. of 5 in refluxing xylenes for 20 h, to provide various styrenes in yields ranging from 22 to 100% (Table 2). A wide range of aryl bromides, including both electron-rich (entries 1 and 2) and electron-deficient (entries 3–7) substrates, are successfully vinylated and a range of functional groups are tolerated including dimethylamino, nitro, cyano and formyl groups. High conversions are observed using this catalyst at loading as low as 0.1 mol%; lower catalyst loading affords poor results (entry 4). Although the amount of catalyst required for these reactions is indeed low, this advantage is lost in view of the long reaction times (20 h) at high temperatures (135 °C). The requirement that 5 must be freshly prepared before each use and that 3.0 equiv. of 5 are needed for complete consumption of the bromide significantly detracts from the utility of this method.
Table 2.
Vinylation of aryl bromides using vinylboronic acid (5)
 | |||
|---|---|---|---|
| Entry | R | Pd (mol%) | Yield (%) |
| 1 | 4-t-Bu | 0.4 | 87 |
| 2 | 4-NMe2 | 1.0 | 89 |
| 3 | 4-MeC(O) | 0.1 | 85 |
| 4 | 4-MeC(O) | 0.01 | 24 |
| 5 | 4-HC(O) | 0.4 | 100 |
| 6 | 4-CN | 0.4 | 100 |
| 7 | a | 0.4 | 80 |
| 8 | 2-MeC(O) | 0.1 | 84 |
| 9 | 2-HC(O) | 1.0 | 85 |
| 10 | 2-CN | 0.4 | 87 |
| 11 | 2,4,6-Me3 | 0.4 | 88 |
| 12 | 2,4,6-i-Pr3 | 5.0 | 22 |
| 13 | b | 0.1 | 87 |
2-Bromo-6-methoxynaphthalene
3-Bromoquinoline.
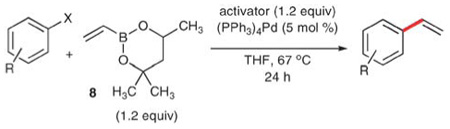
Vinylboronic esters
To ameliorate the instability of 5, a variety of derivatives have been introduced, including boronic esters,23 cyclic boroxanes,24 and potassium trifluoroborates,25 all of which effectively transfer a vinyl group in high yield while offering increased stability of the reagents. Stewart and Whiting first demonstrated that pinacol vinylboronate ester 7, under carefully optimized reaction conditions, selectively reacts with aryl iodides and bromides to provide either the corresponding styrene (Scheme 5, path a) or the alkenyl boronate (Scheme 5, path b).9 Temperature plays a major role in affecting this selectivity, such that lower temperatures favor path a over path b.
Scheme 5.

Competitive reactions of pinacol vinylboronic esters.
Whiting and co-workers subsequently showed that the dioxaborinane 8 efficiently delivers a vinyl group to aryl halides selectively and in good yield.23 This reagent, in conjunction with various activators (potassium hydroxide, potassium tert-butoxide and silver oxide) provides good yields of styrenes in reactions with aryl iodides (Table 3, entries 1–5). Whereas each of the three activators does facilitate the coupling with aryl iodides, silver oxide does not work in reactions that employ aryl bromides. With potassium tert-butoxide, however, aryl bromides can be successfully vinylated, albeit in modest yields in most cases (entries 6–10). Potassium hydroxide also provides some of the styrene products, but was not very effective. Aryl chlorides are unreactive toward 8 regardless of activator. The authors report that they observed the styrene product exclusively in preference to the alkenyl-borinane product.
Table 3.
Yields (%) in vinylation reactions of aryl electrophiles with 8
 | |||||
|---|---|---|---|---|---|
| Activator |
|||||
| Entry | X | R | t-BuOK | KOH | Ag2O |
| 1 | I | H | 62 | 73 | 51 |
| 2 | I | 4-Me | 75 | 68 | 83 |
| 3 | I | 4-MeO | 95 | 66 | 90 |
| 4 | I | 4-CF3 | 87 | 75 | 76 |
| 5 | I | a | 65 | 74 | 96 |
| 6 | Br | H | 56 | 36 | 0 |
| 7 | Br | 4-Me | 52 | 35 | 0 |
| 8 | Br | 4-MeO | 65 | 28 | 0 |
| 9 | Br | 4-CF3 | 71 | 50 | 11 |
| 10 | Br | b | 41 | 31 | 39 |
| 11 | Cl | H | 0 | 0 | 0 |
1-Iodonaphthalene.
1-Bromonaphthalene.
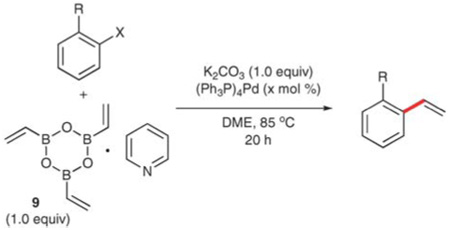
Cyclic vinylboroxane
O’Shea and co-workers have developed a cyclic boroxine as an alternative vinylboronic acid equivalent.24 En route to the development of a novel indole synthesis, the authors modified an earlier Matteson procedure 26 to prepare the stable cyclic anhydride, 2,4,6-trivinyl-cyclotriboroxane (9) as a 1 : 1 complex with pyridine. The trimer participates in palladium-catalyzed, cross-coupling reactions with a variety of ortho-substituted aryl halides to afford 68–84% yields of the corresponding styrenes (Table 4). 1,2-Dimethoxyethane is used as solvent and water is added to generate the vinylboronic acid in situ. As little as 1 (and up to 5) mol% of (Ph3P)4Pd and 1.0 equiv. of the trimer are employed, thus transferring only one of the three vinyl groups. Both aryl iodides (entries 2 and 3) and aryl bromides participate in the reaction and afford similar yields. Substrates bearing an ortho-nitrogen substituent (entries 1–3, 7–9) provide good yields (73–80%) of the corresponding styrenes. Even the presence of the sterically bulky Boc group (entry 9) does not inhibit the reaction, although more catalyst (5 mol%) is required to achieve a satisfactory yield. Additionally, none of the nitrogen-containing functional groups adds electron density from the ring; no electron-rich amine substrates are demonstrated. The presence of a nitrogen atom is not a requirement for successful coupling, but electron-rich substrates require higher catalyst loadings (entries 10–12). Aryl chlorides provide only traces of the desired products under these reaction conditions.
Table 4.
Preparation of ortho-substituted styrenes using boroxane 9
 | ||||
|---|---|---|---|---|
| Entry | X | R | Pd (mol%) | Yield (%) |
| 1 | Br | NO2 | 1 | 74 |
| 2 | I | NO2 | 1 | 77 |
| 3 | I | NHAc | 1 | 73 |
| 4 | Br | CN | 1 | 79 |
| 5 | Br | CHO | 1 | 77 |
| 6 | Br | Ph | 1 | 84 |
| 7 | Br | 2-NHBoc-5-F | 1 | 78 |
| 8 | Br | NHBoc | 1 | — |
| 9 | Br | NHBoc | 5 | 80 |
| 10 | Br | Me | 1 | — |
| 11 | Br | Me | 5 | 68 |
| 12 | Br | OMe | 5 | 70 |
Potassium vinyltrifluoroborate
The introduction of potassium organotrifluoroborates as donors in palladium-catalyzed, cross-coupling reactions by Genet and co-workers have provided a practical solution to the challenges associated with boron-based vinylations.25a Potassium vinyltrifluoroborate (10) is easily prepared from vinylmagnesium bromide by sequential treatment with trimethylborate and potassium hydrogen difluoride.25 The potassium salt is air-stable and can be stored for extended periods of time. Genet and co-workers first reported the coupling of 10 with aryldiazonium tetrafluoroborates using a palladium catalyst in methanol at room temperature.25a In these reactions, the electrophile scope includes arenediazonium salts bearing carboalkoxy, keto, nitro and carboxylic acid substituents (entries 3–7). Moreover, complete chemoselectivity for coupling of the diazonium moiety is observed in substrates bearing aryl iodide, bromide, and triflate moieties (entries 8–10). Only 1.2 equiv. of the potassium salt are required in these fast reactions (10–120 min), and good to excellent yields are obtained (Table 5).
Table 5.
Vinylation reactions of aryldiazonium salts
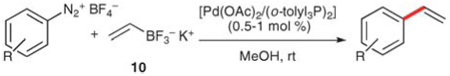 | |||
|---|---|---|---|
| Entry | R | t/min | Yield (%) |
| 1 | 4-OMe | 20 | 81 |
| 2 | 2-Me | 20 | 78 |
| 3 | 4-COOEt | 120 | 88 |
| 4 | 2-COOEt | 20 | 70 |
| 5 | 4-COOH | 15 | 72 |
| 6 | 3-C(O)Ph | 10 | 81 |
| 7 | 4-NO2 | 15 | 84 |
| 8 | 4-Br | 30 | 69 |
| 9 | 4-OTf | 60 | 75 |
| 10 | 3-I | 60 | 76 |
Although the aryldiazonium salts are easily prepared from the corresponding anilines (comprising a large pool of available substrate precursors), this additional step detracts from the overall efficiency of the method. To address this limitation, Molander and Rivero extended the scope of these coupling reactions to engage aryl bromides, aryl triflates (OTf) and activated chlorides.25b These reactions are carried out with triethylamine as the base in refluxing 2-propanol.
More recently, Molander and Brown have further optimized the reaction conditions for a wide range of aryl electrophiles by using cesium carbonate in THF–water, 9 : 1).25c Under these conditions, potassium vinyltrifluoroborate provides high yields of the corresponding styrenes for electron-deficient (entries 1–5), electron rich (entries 6–10), and somewhat sterically-hindered (entries 11–15) aryl bromides (Table 6). Electron-deficient substrates react significantly faster than electron-rich substrates, and numerous heterocyclic substrates are competent in the vinylation reaction. Additionally, the triflate derived from 4-hydroxyacetophenone is converted to the corresponding styrene in an 82% yield. Under the standard conditions, 2-bromomesitylene is not completely converted to the styrene and further optimization was required. Among many ligands tested, dicyclohexyl(2-(2′,6′-diisopropoxy)biphenyl)phosphine (RuPhos)27 provides a preparatively useful ratio of the desired product and the stilbene product (arising from a secondary Heck reaction). The employment of the RuPhos ligand also facilitates the coupling of an activated aryl chloride, 4-chloroacetophenone, to provide the corresponding styrene in 65% yield. The reaction setup (refluxing THF–H2O in a sealed tube (85 °C)) is not ideal for large-scale processes, but is comparable to the conditions developed for alternative vinylboron donors. The broad substrate scope and functional group tolerance, however, clearly highlight the advantages of this method.
Table 6.
Vinylation using potassium vinyltrifluoroborate
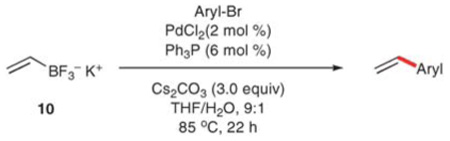 | ||
|---|---|---|
| Entry | Aryl bromide | Yield (%) |
| 1 | 4-Bromobenzonitrile | 83 |
| 2 | 4-Bromoacetophenone | 85 |
| 3 | 4-Bromobenzotrifluoride | 64 |
| 4 | Methyl 4-bromobenzoate | 87 |
| 5 | 4-Bromonitrobenzene | 84 |
| 6 | 4-Bromoanisole | 72 |
| 7 | 4-Bromoacetanilide | 78 |
| 8 | 4-Bromotoluene | 76 |
| 9 | 4-Bromobenzyl alcohol | 82 |
| 10 | N,N-Dimethyl 4-bromoaniline | 93 |
| 11 | 2-Bromotoluene | 82 |
| 12a | 2-Bromomesitylene | 81 |
| 13 | 1-Bromonaphthalene | 82 |
| 14 | 2-Bromobenzonitrile | 82 |
| 15 | 2-Bromoanisole | 71 |
6% RuPhos used.
Although the implementation of organoborane reagents in vinylation reactions was hampered by the instability of vinylboronic acid, the development of vinylboronic esters, cyclic vinylboroxane and potassium vinyltrifluoroborate has addressed this instability and propelled these reagents to the forefront of vinylation reactions.
Vinylaluminium and vinylgallium reagents
Although less commonly employed than vinylboron reagents, vinylaluminium reagents have also been used for the preparation of substituted styrenes. Schumann and co-workers have described examples of complexed vinylaluminium reagents (11) that participate in palladium-catalyzed, cross-coupling reactions with aryl bromides and chloroarene-cobalt complexes under relatively mild conditions (Scheme 6).28 This reaction has a limited scope, but requires no external activation and provides an alternate entry to styrene-Co(CO)3 complexes.
Scheme 6.

Vinylation reactions using vinylaluminium complex 11.
Oshima and co-workers have shown that vinylgallium dichlorides (derived from the hydroalumination of alkynes and transmetalation to gallium trichloride) are capable of transferring a vinyl group to aryl iodides (and in some cases, aryl bromides).29 The standard reaction conditions require no external activation and are conducted in THF/DMSO at refluxing temperatures (Scheme 7). The electrophile scope demonstrates excellent functional group compatibility and the substrates are transformed to the desired products in good to excellent yields.
Scheme 7.

Vinylation using vinylgallium reagent 12.
Vinylsilicon reagents
Whereas vinyl donors derived from Groups 1, 2, and to a lesser extent Group 13 owe their reactivity to the polarization of the carbon–M bond, vinyl donors derived from Group 14 have less ionic character in this bond. Successful strategies to activate Group 14 derived donors include the use of Lewis basic additives and the incorporation of heteroatom substituents on the metal center. Indeed, of all of the cross-coupling donors, those based on organosilanes possess the largest structural variation in the donor substituent. This feature has allowed a wide array of activation protocols and reaction conditions to be developed. Classically, activation of silicon-based donors involved the use of fluoride additives, but recent advances have introduced the use of other activators for a number of oxygenated silicon moieties. Vinylsilicon donors have been developed that incorporate each of these strategies, and will be presented in order of increasing number of heteroatom substituents on silicon.
Alkylsilanes
Vinyltrimethylsilane
The first use of vinyltrimethylsilane (13) in vinylation reactions was described by Hallberg and Westerlund in 1982.30 In this report, four aryl iodides were coupled using 13 in the presence of palladium acetate, triethylamine and triphenylphosphine in DMF, to provide the corresponding styrenes in modest yields (51–60%). Later reports by the same group31 and Kikukawa et al.32 demonstrated that these transformations actually occur by a Heck-type process, i.e. a carbopalladation of 13, followed by the loss of silicon. The first use of 13 in a cross-coupling reaction was reported by Hiyama and Hatanaka in 1988.33 To successfully effect a vinylation with 13 requires the use of tris(diethylamino)sulfonium difluorotrimethylsilicate (TASF), to activate the silicon moiety.34 Interestingly, Jeffery has employed phase transfer conditions that allow formation of either of the two products.10 Unlike in Whiting’s study (with vinylboronic esters), wherein the temperature influenced the product distribution, the key to the selection of pathways here is the presence or absence of fluoride in the reaction mixture (Table 7). When tetrabutylammonium acetate is used, the (E)-β-styryltrimethylsilane (14) is obtained in good yield. In contrast, the combination of fluoride (either potassium or tetrabutylammonium) with tetrabutylammonium chloride is selective for production of styrene (2). The application of these vinylation conditions to a small set of aryl iodides proved to be general, affording styrenes in 78–86% yields.
Table 7.
Cross coupling vs. Heck reaction with 13
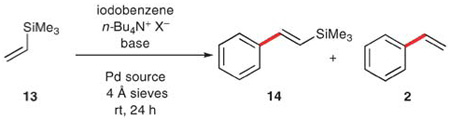 | ||||
|---|---|---|---|---|
| n-Bu4X (equiv.) | Base (equiv.) | Pd source | Solvent | 14 : 2 |
| n-Bu4NOAc (2.5) | — | Pd(OAc)2 | DMF | 99 : 1 |
| n-Bu4NCl (2.5) | KOAc (2.5) | Pd(OAc)2 | DMF | 90 : 10 |
| n-Bu4NCl (1) | TBAF (1.3) | Pd(dba)2 | Toluene | 12 : 84 |
| n-Bu4NCl (2) | KF (3) | Pd(dba)2 | Toluene | 8 : 92 |
1-Methyl-1-vinylsiletane
The recognition by Hiyama and Hatanaka that trialkylsilanes require activation by fluoride to facilitate cross-coupling of the alkenyl group stimulated Denmark and co-workers to develop more reactive trialkylsilanes, namely silacyclobutanes to enhance their fluorophilicity.35 Empirically, this class of compounds readily participates in cross-coupling reaction under considerably milder conditions. 1-Methyl-1-vinylsiletane, 15, (easily prepared from the combination of vinylmagnesium bromide and the commercially available 1-chloro-1-methylsiletane) reacts with aryl and vinyl iodides at room temperature using Pd(dba)2 and TBAF to provide high yields of the corresponding coupling products (Table 8).36 The reaction shows excellent functional group tolerance, as substrates bearing carboalkoxy, keto, nitro, cyano, and hydroxyl groups are transformed to styrenes in excellent yields. Reactions with electron-deficient substrates are generally rapid (entries 1–5, 7 and 8), and employ low catalyst loadings. The presence of electron donating groups (entries 4 and 13) and/or ortho-substituents (entries 10–14) does not decrease the yield but does slow the reaction and requires the use of more catalyst, fluoride and 15. Triphenylarsine is added to stabilize the palladium catalyst in slower reactions (entries 9, 11–13). Additionally, vinylation of 4-bromoacetophenone, could be effected albeit at 40 °C.
Table 8.
Vinylation using 1-methyl-1-vinysiletane (15)
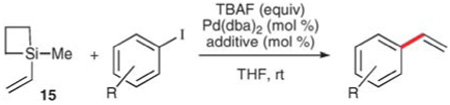 | ||||||
|---|---|---|---|---|---|---|
| Entry | R | 15 (equiv.) |
TBAF (equiv.) |
Pd(dba)2 (mol%) |
t/h | Yield (%) |
| 1 | 4-COOEt | 1.2 | 2.0 | 1 | 1 | 93 |
| 2 | 4-C(O)Me | 1.2 | 2.0 | 1 | 1 | 85 |
| 3 | 4-C(O)Mea | 1.2 | 3.0 | 2.5b | 0.5c | 75 |
| 4 | 4-NO2 | 1.2 | 2.0 | 1 | 1 | 90 |
| 5 | 4-CN | 1.2 | 2.0 | 1 | 1 | 87 |
| 6 | 4-OMe | 1.5 | 4.5 | 5d | 4 | 74 |
| 7 | 3-NO2 | 1.2 | 2.0 | 1 | 1 | 92 |
| 8 | 3-COOEt | 1.2 | 2.0 | 3 | 1 | 90 |
| 9 | 3-CH2OH | 1.2 | 2.0 | 5d | 7.5 | 79 |
| 10 | 2-NO2 | 1.2 | 2.0 | 1 | 1.5 | 86 |
| 11 | 2-COOEt | 1.2 | 3.0 | 5d | 14 | 85 |
| 12 | 2-Me | 1.2 | 3.0 | 5d | 16 | 70 |
| 13 | 2-OMe | 1.5 | 4.5 | 5d | 10 | 75 |
| 14 | e | 1.2 | 3.0 | 5 | 4 | 76 |
4-Bromoacetophenone used as substrate.
[allylPdCl]2 (2.5 mol%) used as catalyst.
40 °C.
AsPh3 (10 mol%) added to the reaction.
1-Iodonaphthalene used as substrate.
Silanes with oxygen substituents
Divinyltetramethyldisiloxane (DVDS)
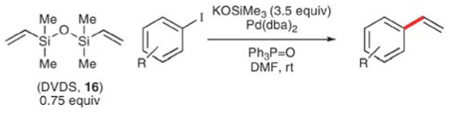
The striking facility of the cross-coupling reactions of alkenylsiletanes stimulated a thorough analysis of the reaction mechanism.37 Treatment of 15 with TBAF·3H2O causes immediate ring opening to reveal an n-propylsilanol in situ. This discovery suggested that alkenyldimethylsilanols are likely intermediates and control experiments verified that this little-known class of silicon compounds are excellent substrates for palladium-catalyzed, cross-coupling reactions.38 Unfortunately, silanols are not well suited for simple vinylation reactions because the parent, vinyldimethylsilanol, readily dimerizes to form divinyltetra-methyldisiloxane (DVDS, 16).
However, DVDS itself can be used as a donor for vinylation reactions. Denmark and Butler have shown that potassium trimethylsilanolate (KOSiMe3) is capable of activating DVDS toward vinylation reactions through a “silanolate exchange” in DMF to generate two equivalents of potassium vinyldimethylsilanolate (17) and one equivalent of the innocuous hexamethyldisiloxane in situ (Scheme 8).39 Therefore, each of the two vinyl groups on DVDS is available for transfer, increasing the efficiency of these reactions.
Scheme 8.

Silanolate exchange of DVDS and KOSiMe3.
The in situ generated vinyldimethylsilanolate reacts with a range of aryl iodides at room temperature (Table 9). Good yields are obtained in all cases, and some functional groups are tolerated (entries 2, 5 and 6). The reactions are generally fast (< 3 h), although 2-iodoanisole requires 14 h. The successful vinylation of ethyl 4-iodobenzoate is notable, as the ester survives the reaction even in the presence of potassium trimethylsilanolate, which is capable of cleaving esters to the corresponding acids.40
Table 9.
Vinylation of aryl iodides using DVDS (16)
 | ||||
|---|---|---|---|---|
| Entry | R | Pda (mol%) | t/h | Yield (%) |
| 1 | 4-OMe | 5 | 1.5 | 80 |
| 2 | 4-COOEt | 2.5 | 0.5 | 81 |
| 3 | 4-OBn | 5 | 1.5 | 74 |
| 4 | 4-t-Bu | 5 | 4 | 69 |
| 5 | 4-CN | 2.5 | 0.5 | 81 |
| 6 | 3-NO2 | 2.5 | 1 | 76 |
| 7 | 2-OMe | 5 | 14 | 68 |
| 8 | b | 5 | 2.5 | 80 |
1.0 equiv. Ph3PO per Pd(dba)2.
1-Iodonaphthalene.
Avoiding the requirement of fluoride-based activators is of great value, as fluoride reagents are generally expensive,41 capable of etching glass reaction vessels, and are incompatible with silicon-based protecting groups. In contrast, the combination of DVDS and KOSiMe3 does not suffer from these limitations; both reagents are inexpensive and widely available.42 Thus, “fluoride-free” activation significantly enhances the scope and utility of these reactions.43
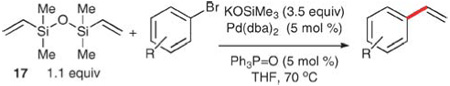
Aryl bromides also succumb to the vinylation conditions with only minor modifications.39 A simple increase in reaction temperature (to 70 °C) and a solvent change to THF is sufficient to engage a number of aryl bromides (Table 10). This modification has a significantly narrower substrate scope than the aryl iodides, although good yields are obtained in some cases (entries 1 and 3–5). Substrates bearing strongly electron-withdrawing substituents (entries 2, 6, and 7) generally give diminished yields due to competing polymerization of the products under the reaction conditions.
Table 10.
Vinylation of aryl bromides using DVDS (17)
 | |||
|---|---|---|---|
| Entry | R | t/h | Yield (%) |
| 1 | 4-OMe | 1 | 71 |
| 2 | 4-Cl | 3 | 55 |
| 3 | 4-PhC(O) | 3 | 93 |
| 4 | a | 3 | 91 |
| 5 | b | 3 | 70 |
| 6 | 4-NMe2C(O) | 3 | 34 |
| 7 | 2-CF3 | 3 | 52 |
3-Bromoquinoline.
2-Bromo-6-methoxynaphthalene.
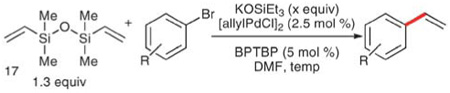
To improve the scope of this reaction with aryl bromides, a second, milder protocol was developed that employs potassium triethylsilanolate (KOSiEt3) in place of KOSiMe3. The superiority of KOSiEt3 is related to its increased steric bulk that allows for silanolate exchange without concomitant attack at the palladium center. The combination of di-tert-butyl(2-biphenyl)phosphine (BPTBP, 18) and [allylPdCl]2 in DMF allows for successful vinylation reactions at much lower temperatures than the initial modification (Table 11). Thus, electron-rich (Table 10, entries 1–2 and 7), electron-deficient (entries 3–5 and 9–12), and sterically encumbered aryl bromides (entries 7–9) are vinylated at or just above room temperature. The functional group tolerance is significantly increased, as amino, amido, carboalkoxy, and silyloxy groups all participate and are converted to the corresponding styrene in good yields. Unfortunately, a bulkier ester (tert-butyl) is required as cleavage of an ethyl ester is observed over the longer reaction times (entry 5). A divinylation of 1,4-dibromobenzene can also be accomplished, albeit in significantly diminished yields, likely due to competing polymerization of the divinylbenzene product (entry 12).
Table 11.
Vinylation of aryl bromides using DVDS (17)
 | |||||
|---|---|---|---|---|---|
| Entry | R | KOSiEt3 (equiv.) | T°/C | t/h | Yield (%) |
| 1 | 4-OMe | 4a | 35 | 4 | 74 |
| 2 | 4-NMe2 | 4a | 40 | 24 | 70 |
| 3 | 4-Cl | 3 | 40 | 3 | 50 |
| 4 | 4-Me2NC(O) | 3 | 40 | 2 | 69 |
| 5 | 4-CO2tBu | 3 | 40 | 2 | 60 |
| 6 | 3-TBSOCH2 | 4a | 40 | 24 | 64 |
| 7 | 2-OMe | 4a | 40 | 24 | 72 |
| 8 | 2,4,6-Me3 | 4a | 40 | 24 | 99 |
| 9 | b | 3 | 25 | 12 | 80 |
| 10 | c | 3 | 25 | 4 | 82 |
| 11 | d | 3 | 25 | 2 | 79 |
| 12 | 4-Br | 6e | 40 | 2 | 48 |
2.0 equiv. of DVDS used.
1-Bromonaphthalene.
2-Bromo-6-methoxynaphthalene.
3-Bromoquinoline.
2.6 equiv. of DVDS used.
Polyvinylsiloxanes
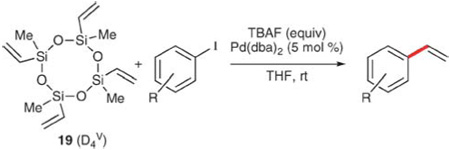
In addition to DVDS, a number of other, stable vinyldimethylsilanol surrogates are commercially available. Polyvinylsiloxanes are inexpensive because of their ubiquitous application in polymer chemistry,44 and have recently been conscripted as vinyl donors in palladium-catalyzed, cross-coupling reactions.45 These reagents are not suitably activated for transmetalation using KOSiMe3 and therefore require fluoride-based reagents for successful coupling. Of these polyvinylsiloxanes, 1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane (D4V), 19, is superior for the vinylation of aryl iodides, due to cost- and vinyl group transfer efficiencies.46,47 As little as 0.3 equiv. of the tetramer is required, thus each of the vinyl groups is available for transfer. Using TBAF and Pd(dba)2 in THF, aryl iodides are converted to the corresponding styrenes in good yields at room temperature (Table 12). The reaction shows good functional group compatibility (entries 1–6), including keto, carboalkoxy, hydroxyl, and nitro groups. Moderate steric encumbrance and the presence of electron-donating or free hydroxyl groups slow the reaction considerably (entries 4, 6–8), but do not significantly affect the yield. Electron-deficient substrates generally react very quickly, and in some cases even exothermically.
Table 12.
Vinylation of aryl iodides using polyvinylsiloxane 19
 | |||||
|---|---|---|---|---|---|
| Entry | R | 19 (equiv.) | TBAF (equiv.) | t/min | Yield (%) |
| 1 | 4-C(O)Me | 0.3 | 2.0 | 10 | 88 |
| 2 | 4-COOEt | 0.3 | 2.0 | 10 | 85 |
| 3a | 4-COOEt | 0.3 | 2.0 | 60 | 83 |
| 4b | 4-OMe | 0.375 | 3.0 | 360 | 63 |
| 5 | 3-NO2 | 0.3 | 2.0 | 10 | 87 |
| 6 | 3-CH2OH | 0.3 | 2.0 | 480 | 59 |
| 7b | 2-OMe | 0.375 | 3.0 | c | 72 |
| 8 | 2-COOMe | 0.3 | 2.0 | 480 | 83 |
| 9 | d | 0.3 | 2.0 | 180 | 64 |
Pd(dba)2 (1 mol%) used.
Ph3As (10 mol%) added to the reaction.
24 h.
1-Iodonaphthalene.
The reaction of 19 with aryl bromides has also been developed 48 but requires the use of phosphine 18 as a ligand for palladium at elevated temperatures (50 °C). Additionally, a slight increase in the loading of D4V is needed to suppress the formation of the symmetrical stilbene. With these adaptations, a wide range of bromides participates in the reaction, including electron-rich (Table 13, entries 6–9 and 13), electron deficient (entries 1–5 and 11), and sterically-encumbered substrates (entries 7, 9, 11 and 12). Bromides containing nitrogen functions (entries 12–14) afford good to excellent yields of the corresponding styrenes, although substrates bearing free −OH and −NH2 groups give lower yields. This method has also been recently shown to work with vinyl halides in the synthesis of Diels–Alder precursors.49
Table 13.
Vinylation of aryl bromides using 19
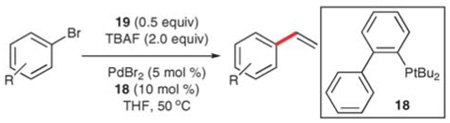 | |||
|---|---|---|---|
| Entry | R | t/h | Yield (%) |
| 1 | 4-C(O)Me | 3 | 91 |
| 2 | 4-COOEt | 5 | 83 |
| 3 | 2-COOEt | 5 | 86 |
| 4 | a | 3 | 71 |
| 5 | b | 3 | 81 |
| 6 | 2-Et | 17 | 75 |
| 7 | 2,4,6-Me3 | 48 | 72 |
| 8 | 4-OMe | 10 | 86 |
| 9 | 2-OMe | 20 | 80 |
| 10 | 4-CH2OH | 14 | 54 |
| 11 | 2-NO2 | 2 | 85 |
| 12 | 4-NHAc | 12 | 77 |
| 13 | 2-NMe2 | 24 | 74 |
| 14 | c | 3 | 89 |
1-Bromonaphthalene.
2-Bromonaphthalene.
3-Bromoquinoline.
Vinyltrialkoxysilanes
Vinyltrimethoxysilane (20) (also of utility in the synthesis of silicon-based polymers) has recently been employed as a donor in vinylation reactions. Nolan and co-workers reported the vinylation of 4-bromo- and 4-chloroacetophenone with 20 and N-heterocyclic carbene ligand 21 in combination with palladium acetate.50 With 2.0 equiv. of TBAF, 100% conversion to 4-vinylacetophenone is observed in each case (Scheme 9). More recently, Clarke also reported the vinylation of 4-chloroacetophenone with 20 using ligand 22 under microwave irradiation.51 To effect this transformation, 10 equiv. of TBAF and 2.0 equiv. of 20 are required to provide a 95% yield of 4-vinylacetophenone after 18 min.
Scheme 9.

Vinylation using vinyltrimethoxysilane (20).
Although both of these methods employ inexpensive52 vinyltrimethoxysilane, TBAF is required as an activator and therefore the methods suffer from the drawbacks discussed earlier. Recently, Alacid and Najera have developed vinylation conditions using 20 or vinyltriethoxysilane (23) that do not require fluoride activation.53 The authors found that both 20 and 23 could engage in cross-coupling with aryl halides in the presence of sodium hydroxide in water at 120 °C, using either conventional or microwave (µW) heating (Table 13). Both palladium acetate and palladacycle 24 are able to effect the reaction (Table 14, entries 1 and 2). Better results are obtained for reactions with 25 mol% of tetrabutylammonium bromide (TBAB) (entry 3 cf. 2), and in those cases, the catalyst loading could be decreased to 0.01 mol% (entry 5), although most reactions require at least 0.1 mol%. The reaction conditions are general for a moderate scope of aryl iodides and aryl bromides. The reactions that employ conventional heating provide similar (and sometimes superior) yields, although those reactions are significantly slower (entry 10 cf. 11). Activated aryl chlorides participate in the reaction, but provide diminished yields of the corresponding styrenes (entries 13 and 14). The functional group tolerance is limited, likely due to the use of aqueous hydroxide at elevated temperatures, although ketone-bearing and pyridine substrates are coupled in high yield (entries 1–3,12 and 13).
Table 14.
Vinylation using 20 and 23 in NaOH–H2O
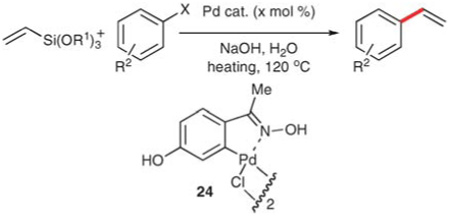 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | X | Pd cat (mol%) |
Heat source |
t/min | Yield (%) |
| 1 | Me | 4-MeC(O) | Br | Pd(OAc)2 (0.5) | µW | 10 | 97 |
| 2 | Et | 4-MeC(O) | Br | 24 (0.5) | µW | 10 | 90 |
| 3 | Me | 4-MeC(O) | Br | 24 (0.1)a | µW | 10 | 99 |
| 4 | Me | 4-MeO | I | 24 (0.1) | µW | 10 | 93 |
| 5 | Me | 4-MeO | I | 24 (0.01)a | Δ | 240 | 89 |
| 6 | Me | 3,5-(MeO)2 | I | Pd(OAc)2 (0.1)a | µW | 15 | 83 |
| 7 | Me | 4-MeO | Br | 24 (1)a | µW | 10 | 97 |
| 8 | Me | b | Br | 24 (1)a | µW | 20 | 92 |
| 9 | Me | 4-Cl | Br | 24 (1)a | µW | 15 | 71 |
| 10 | Me | c | Br | Pd(OAc)2 (0.5)d | Δ | e | 89 |
| 11 | Me | c | Br | Pd(OAc)2 (0.5)a | µW | 10 | 89 |
| 12 | Me | f | Br | 24 (1)a | µW | 15 | 97 |
| 13 | Me | 4-MeC(O) | Cl | 24 (2)a | µW | 25 | 71 |
| 14 | Me | 4-PhC(O) | Cl | 24 (2)a | µW | 25 | 65 |
TBAB (25 mol%) added.
1-Bromonaphthalene.
2-Bromo-6-methoxynaphthalene.
TBAB (200 mol%).
1 day.
3-Bromopyridine.
A number of silicon-based reagents have addressed the challenges of developing a mild and effcient vinylation reaction. Fluoride activation is needed with trialkylsilanes, polyvinylsiloxanes, and trialkoxysilanes. The recently introduced methods that employ non-fluoride activators have considerably enhanced the utility of these reagents. Vinyltrialkoxysilanes can be activated by aqueous hydroxide at high temperatures, whereas the combination of DVDS and KOSiMe3 generates the vinyldimethylsilanolate in situ. Both of these methods are able to engage a range of aryl bromides in good yield without the need for fluoride.
Vinyltin reagents
Vinyltributyltin
Vinyltributyltin54 (25) is the most well-known and the most commonly used vinylmetallic donor. This reagent possesses a number of advantages including air and moisture stability compared to the other vinyl donors, and longstanding precedent of reactivity.55 It is worth noting that although the vinyl group is transferred in good yield and vinyl efficiency, with each two-carbon transfer, one equivalent of tributyltin halide (Bu3SnBr, MW = 270), is discarded. This analysis implies, that for many substrates, the waste stream has a greater molecular weight than the product. In addition to the poor atom economy, one of the main drawbacks to the use of organotin reagents is their toxicity, specifically the byproducts generated in the reaction.56
In 1986, Scott and Stille described the first successful cross-coupling of enol triflates using 25.57 Less than a year later, a second report detailed the vinylation of a wide range of aryl bromides. In the reactions with aryl bromides, no external activation is required, thus simplifying the reaction protocol and facilitating a broad substrate scope and functional group tolerance (Table 15). Aryl bromides bearing nitro, formyl, 1,3-dicarbonyl, keto and carboalkoxy groups in the para position are all tolerated (entries 2, 4 and 6–9). 1,4-Dibromobenzene can undergo a mono- (entry 5) or divinylation (12 h, 73% yield) by using 1 or 2.2 equiv. of 25, respectively.
Table 15.
Vinylation of aryl bromides with vinyltributyltin (25)
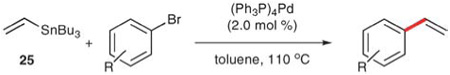 | |||
|---|---|---|---|
| Entry | R | t/h | Yield (%) |
| 1 | 4-OMe | 24 | 76 |
| 2 | 4-NO2 | 4 | 80 |
| 3 | 4-CHO | 3 | 78 |
| 4 | a | 1 | 83 |
| 5 | 4-Br | 1 | 63 |
| 6 | b | 2 | 72 |
| 7 | 4-AcO | 8 | 62 |
| 8 | 4-MeC(O) | 4 | 82 |
| 9 | c | 1 | 85 |
2-Bromo-6-methoxynaphthalene.
4-Bromoacetylacetone.
4′-Bromothiophenone.
Fu and co-workers have introduced an improvement that allows aryl bromides to be vinylated at room temperature.58a By the use of the bulky, electron-rich ligand tri-tert-butylphosphine in combination with Pd2(dba)3 a wide range of bromides undergo cross-coupling with 25 in 66–92% yield (Table 16). In general, the substrate scope is good and electron-deficient (entries 1, 2 and 5), electron-rich (entries 3, 4 and 8), and sterically hindered substrates (entries 5–9) are vinylated with similar degrees of success.
Table 16.
Vinylation of aryl bromides using 25
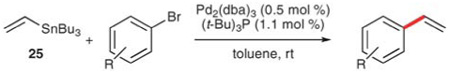 | ||
|---|---|---|
| Entry | R | Yield (%) |
| 1 | 4-MeC(O) | 88 |
| 2 | a | 88 |
| 3 | 4-PhO | 85 |
| 4 | 4-OH | 85b |
| 5 | 2-COOEt | 92 |
| 6 | 2-Ph | 76 |
| 7 | c | 91 |
| 8 | 2,4-(OMe)2 | 89 |
| 9 | d | 66 |
2-Bromonaphthalene.
Reaction carried out in Et2O.
1-Bromonaphthalene.
9-Bromoanthracene.
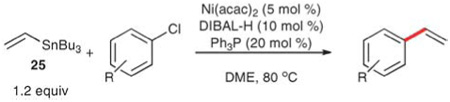
Shirakawa and Hiyama extended the scope of this reaction to include aryl chlorides by using nickel catalysis.59 These reactions require the preformation of a nickel hydride complex derived from Ni(acac)2, Ph3P and DIBAL-H. Aryl chlorides are converted to the corresponding styrenes at 80 °C in dioxane in 9–96 h using this catalyst. Reaction yields range from 37–91% (Table 17) and electron-deficient substrates afford higher yields in shorter reaction times (entries 1–4). Substrates bearing sulfur- and nitrogen-containing substituents also undergo cross-coupling albeit in diminished yields. In general, steric encumbrance does not affect the yield or reaction rate significantly (entries 1 vs. 2 and 6 vs. 8), although 2-bromobenzonitrile reacts more slowly and in poorer yield than does 4-bromobenzonitrile. This tendency was confirmed by competition experiments, which showed that the relative rate of the vinylation of 2-bromobenzonitrile vs. 4-bromobenzonitrile was significantly lower than similar comparisons with other functional groups.
Table 17.
Vinylation of aryl chlorides with 25 using nickel catalysis
 | |||
|---|---|---|---|
| Entry | R | t/h | Yield (%) |
| 1 | 4-PhC(O) | 23 | 91 |
| 2 | 2-PhC(O) | 23 | 80 |
| 3 | 4-CHO | 9 | 86 |
| 4 | 4-CN | 10 | 78 |
| 5 | 2-CN | 96 | 37 |
| 6 | 4-OMe | 91 | 51 |
| 7 | 3-OMe | 40 | 69 |
| 8 | 2-OMe | 30 | 66 |
| 9 | 4-NH2 | 67 | 40 |
| 10 | 2-NH2 | 67 | 51 |
| 11 | 4-SMe | 37 | 65 |
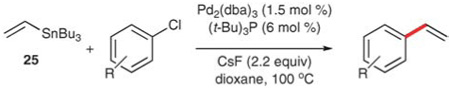
Littke and Fu have developed a procedure for the vinylation of aryl chlorides using palladium catalysis.58b Two aspects of the reaction conditions are crucial to the success of the method. First, the reactions require the use of the bulky, electron-rich tri-tert-butylphosphine as described above. Second, a fluoride source is needed, presumably to facilitate the transmetalation from tin to palladium. Cesium fluoride is optimal for this catalyst/ligand system. Using these conditions, aryl chlorides can be converted to the corresponding styrenes in good to excellent yield at either 80 °C or 100 °C in dioxane (Table 18). Amino and keto groups are tolerated (entries 1 and 4), and sterically encumbered substrates react in good yield (entry 5).
Table 18.
Vinylation of aryl chlorides with 25 using palladium catalysis
 | ||
|---|---|---|
| Entry | R | Yield (%) |
| 1a | 4-MeC(O) | 87 |
| 2 | 4-n-Bu | 80 |
| 3 | 4-OMe | 90 |
| 4 | 4-NH2 | 61 |
| 5 | 2,5-Me2 | 84 |
Reaction temperature: 80 °C.
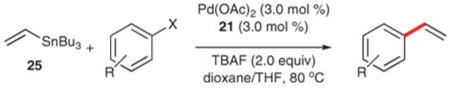
Nolan and co-workers confirmed the beneficial aspects of fluoride additives in Stille couplings by observing hypercoordinate aryl- and vinylstannate intermediates with 19F-NMR spectroscopy.60 These observations translated to preparative utility, as Nolan is able to vinylate aryl bromides using vinyltributyltin and TBAF. These reactions occur at lower temperatures (80 °C) than previously reported by Stille, again using their NHC ligands, although reaction times for unactivated bromides were rather long (Table 19, entries 1–4). Whereas 4-chloroacetophenone is vinylated under similar reaction conditions in good yield (entry 5), less activated substrates did not provide satisfactory yields (entries 6 and 7).
Table 19.
Vinylation of aryl halides with 25 and NHC ligands
 | ||||
|---|---|---|---|---|
| Entry | R | X | t/h | Yield (%) |
| 1 | 4-MeC(O) | Br | 3 | 92 |
| 2 | 4-OMe | Br | 48 | 69 |
| 3 | 2,4,6-Me3 | Br | 48 | 25 |
| 4 | 4-Me | Br | 48 | 98 |
| 5 | 4-MeC(O) | Cl | 3 | 83 |
| 6 | 4-MeO | Cl | 24 | 15 |
| 7 | 4-Me | Cl | 12 | 41 |
Charette and co-workers have recently developed recyclable triarylphosphonium-supported tin reagents.61 The application of these reagents under the conditions described above provides equivalent, in some cases superior, results (Scheme 10). More importantly, the tin byproducts are removed by precipitation and filtration, thus minimizing the toxic waste stream.
Scheme 10.

Vinylation reaction with a supported tin reagent.
Vinylation using vinyl acceptors
A complementary approach to all of the previous examples involves reversing the roles of reactive partners, namely the combination of a vinyl halide or pseudohalide with an aryl organometallic donor. This approach is used less often and is hampered by the diffculty in handling the low boiling vinyl halides, (although these reagents have been used in the palladium-catalyzed vinylation of ketone enolates62). Additionally, the highest boiling congener, vinyl iodide, is unstable to light, heat, and bases.63 Nevertheless, this combination was described in Kumada’s seminal report, namely the nickelcatalyzed, cross-coupling of phenylmagnesium bromide and vinyl chloride (Scheme 11).64 Because vinyl chloride is a gas (bp −13.4 °C), the reaction is run in a sealed tube. Additionally, as in Bumagin’s work, the substrate scope is limited, because of the nucleophilic and basic nature of the phenyl-magnesium bromide.65
Scheme 11.

Vinylation using vinyl chloride.
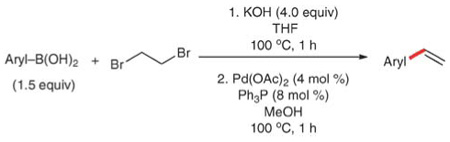
Three methods have been developed recently to further enable this disconnection and circumvent these challenges. The first method, developed by Lando and Monteiro, employs the reaction of arylboronic acids and dibromoethane.66 The coupling of these reagents in methanol provides the corresponding styrenes in moderate to excellent yields (Table 20). The reaction involves the dehydrobromination of 1,2-dibromoethane using 4 equiv. of KOH to generate vinyl bromide in a separate step prior to the introduction of the arylboronic acid and the catalyst (Pd(OAc)2). The reaction tolerates a free carboxylic acid (entry 6) and a range of halogen substituents (entries 3–5). Electron rich (entries 1 and 7) and sterically encumbered substrates (entries 7–9) provide the corresponding styrenes in similar yield. Because the roles of the donor and the acceptor are reversed in this strategy, the nucleophilicity of the arylboronic acids plays a role in these reactions, and, not surprisingly, the electron-deficient (and thus less nucleophilic) substrates (entries 3–6) afford lower yields. Although this method addresses the challenge of using vinyl bromide by generating it in situ, the loss of two moles of HBr significantly lowers its atom efficiency. Unfortunately, 1.5 equiv. of the more expensive arylboronic acid are required, thus making this a less economical process.
Table 20.
Vinylation of arylboronic acids with 1,2-dibromoethane
 | ||
|---|---|---|
| Entry | Aryl | Yield (%) |
| 1 | 4-Methoxyphenyl | 94 |
| 2 | 4-Tolyl | 86 |
| 3 | 4-Trifluoromethylphenyl | 58 |
| 4 | 4-Fluorophenyl | 62 |
| 5 | 4-Chlorophenyl | 58 |
| 6 | 4-Benzoyl | 69 |
| 7 | 2-Methoxyphenyl | 89 |
| 8 | 2-Mesityl | 63 |
| 9 | 1-Naphthyl | 100 |
| 10 | 2-(6-Methoxynaphthyl) | 72 |
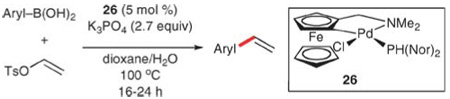
The second strategy, developed by Skrydstrup and co-workers, employs vinyl tosylate (derived from the fragmentation of tetrahydrofuran) and arylboronic acids.63 The combination of these two addends with potassium phosphate and the commercially available catalyst SK-CCO2-A (26) provides corresponding styrenes in 60–99% yields (Table 21). Electron-rich (entries 4, 5 and 7) and electron deficient (entries 2, 8 and 9) substrates are transformed in similar yields. Excellent functional group tolerance is observed, and includes cyano, acetamide, aldehyde and thio ether groups (entries 2–4 and 8–9). Additionally, the use of aryl- and heteroarylboronic esters (pinacol and neopentyl glycol) increases the substrate scope to include pyridyl and quinolyl precursors, and even those bearing secondary amines.
Table 21.
Vinylation of arylboronic acids with vinyl tosylate
 | ||
|---|---|---|
| Entry | Aryl | Yield (%) |
| 1 | 1-Naphthyl | 99 |
| 2 | 4-Cyanophenyl | 89 |
| 3 | 3-Acetamidophenyl | 89 |
| 4 | 4-Thiomethoxyphenyl | 90 |
| 5 | 3′-(1,3-Benzodioxolyl) | 85 |
| 6 | 2-(6-Methoxynaphthyl) | 60 |
| 7 | 1-Dibenzofuryl | 89 |
| 8 | 2-Formylphenyl | 63 |
| 9 | 4-Formylphenyl | 90 |
Dunet and Knochel have also described a method that implements this strategy in the coupling of arylcyanocuprates and vinyl nonaflates (n-C4F9SO3) to provide styrenes in 64–72% yields.67
A third strategy, involving the coupling of vinyl acetates and aryl halides catalyzed by a cobalt(II) complex, has been reported by Gosmini and co-workers.68 This example represents the third motif described in the introduction, wherein both vinyl and aryl groups are introduced as acceptors, and 10 equiv. of manganese (per aryl chloride) is used to reduce the cobalt catalyst and complete the catalytic cycle. Aryl bromides and chlorides are converted to the corresponding styrenes in 37–81% yield (Scheme 12).
Scheme 12.

Vinylation of aryl halides using a cobalt catalyst.
Conclusions
Despite the overwhelming impact of palladium-catalyzed, cross-coupling reactions in organic synthesis, the seemingly straightforward installation of a vinyl group has been, by comparison, overlooked. A direct, quantitative comparison of the different methods presented in this review is compiled in Table 22. The analysis presents a number of different criteria that are relevant to the comparison of the methods. The most critical feature is the “nominal atom efficiency” of the vinylmetallic donors which is the percentage of the reagent molecular weight that is transferred. The “actual atom efficiency” takes into account the stoichiometry of the reactions. Additional comparison can be made if the additives needed are also taken into consideration (e.g. including the number of grams of TBAF or Cs2CO3 needed per vinyl group). Inspection of the data in the table reveals that no method is very efficient. From a purely atom efficiency perspective, it is clear that the vinylsilicon reagents (19–26% of molecular weight transferred) and 10 (20% of molecular weight transferred) are superior, whereas vinylaluminium and vinyltin donors are the least efficient. Interestingly, when the activators required for each of these reactions is included in the calculation, none are especially efficient. The vinylgallium, and -magnesium reagents that do not require activators are the most efficient, (16 and 11%, respectively). Those reactions that require TBAF and Cs2CO3 activation are most affected by this calculation.
Table 22.
Efficiency analysis for vinylmetallic donors
| Vinyl reagent |
Vinyl donor | MW | Equiv. requireda |
Nominal atom efficiencyb (%) |
Actual atom efficiencyc (%) |
Actual atom efficiency (+ activator)d (%) |
$ mol −1e | $ per mol vinyl transferredf |
Electrophile scope |
|---|---|---|---|---|---|---|---|---|---|
| 4 | VinylMgBr | 131.25 | 2 | 21 | 11 | 11c | 61 | 132 | Poor |
| 5 | VinylB(OH)2 | 71.87 | 3 | 38 | 13 | 5.5 (K2CO3) | — | — | Good |
| 8 | VinylB(OR)2 | 154.01 | 1.2 | 17 | 15 | 8.4 (KOt-Bu) | 3578 | 4293 | Moderate |
| 9 | (VinylBO)3·Pyr | 240.67 | 1 | 11 | 11 | 7 (K2CO3) | 4615 | 4615 | Good |
| 10 | VinylBF3K | 133.95 | 1 | 20 | 20 | 2.4 (Cs2CO3) | 1958 | 1958 | Excellent |
| 11 | VinylAl(OR)NR | 338.2 | 1 | 8 | 8 | 8 | — | — | Moderate |
| 12 | VinylGaCl2 | 167.67 | 1 | 16 | 16 | 16 | — | — | Excellent |
| 13 | VinylSiMe3 | 100.27 | 1.3 | 27 | 21 | 9 (KF) | 615 | 799 | Moderate |
| 15 | Vinylsiletane | 112.24 | 1.2 | 24 | 20 | 3.6 (TBAF) | — | — | Excellent |
| 16 | DVDS | 186.40 | 0.75 | 14 | 19 | 4.6 (KOSiMe3) | 236 | 177 | Good |
| 19 | D4V | 344.66 | 0.3 | 8 | 26 | 3.4 (TBAF) | 345 | 103 | Excellent |
| 20 | VinylSi(OMe)3 | 148.23 | 2 | 18 | 9 | 6.8 (NaOH) | 29 | 58 | Good |
| 25 | VinylSnBu3 | 317.10 | 1.08 | 9 | 8 | 8 | 3675 | 3969 | Excellent |
Equivalents of vinyl donor required to effect vinylation.
MW C2H3/MW donor.
Nominal efficiency/equiv. required.
MW C2H3/((MW donor X equiv.) + (MW activator X equiv.)).
Based upon 2007–2008 Aldrich catalog.
($ mol−1)/required equivalents.
Another important criterion is the relative cost of the commercially available reagents. In this comparison, the silicon-based reagents ($29–615 mol−1) are considerably more attractive than the corresponding vinylboron and vinyltin donors ($1958–4615 mol−1). In many cases, these cost and atom efficiencies will greatly impact a synthetic strategy and therefore must be considered when evaluating alternative vinylation methods.
In this Feature Article, we have described advances that have enhanced the substrate scope and utility of the palladium-catalyzed vinylation reaction. The three main developments that have been highlighted are: (1) the preparation of specifically-tuned vinyl donors, stable for storage but reactive under palladium (or nickel) catalysis, (2) the incorporation of newly developed ligands that facilitate various components of the catalytic cycle and allow for reactions to occur under milder conditions, and (3) the elimination of toxic reagents and by-products from the reactions. Collectively, the methods that arose from these developments provide access to a wide range of styrene derivatives from multiple classes of aryl electrophiles. These methods encompass considerable overlap, thus affording many options (vinyl donors and conditions) for a specific vinylation. From the perspective of scope and utility, the current state of the art is deemed acceptable. However, as clearly highlighted in Table 22, considerable room for improvement remains, particularly to provide solutions that are more amenable to scalable processes. Thus, future work should focus upon the development of more cost- and atom-efficient vinyl donors to address these limitations.
Biographies
Scott E. Denmark was born in Lynbrook, New York on 17 June 1953. He obtained an S.B. degree from MIT in 1975 (working with Richard H. Holm and Daniel S. Kemp) and his D.Sc.Tech. (under the direction of Albert Eschenmoser) from the ETH Zürich in 1980. That same year he began his career at the University of Illinois. He was promoted to associate professor in 1986, to full professor in 1987 and since 1991 he has been the Reynold C. Fuson Professor of Chemistry. His research interests include the invention of new synthetic reactions, exploratory organoelement chemistry and the origin of stereocontrol in fundamental carbon–carbon bond forming processes. Professor Denmark is currently on the Board of Editors of Organic Syntheses and has served on many editorial advisory boards (including Chemical Communications). He is Co-Editor of Topics in Stereochemistry and was an Associate Editor of Organic Letters. In 2008 he became Editor in Chief and President of Organic Reactions.
Christopher R. Butler was born in Peoria, IL on 22 October 1978. He obtained a B.A. degree from Illinois Wesleyan University in 2000 (working with Ram S. Mohan and Jeffery A. Frick). He then worked as a research associate in Medicinal Chemistry for Johnson and Johnson, PRD in La Jolla, California. In the fall of 2003, he began his graduate studies at the University of Illinois (under the direction of Scott E. Denmark). His thesis work has focused up the development of vinylation reactions using organosilicon reagents. After completing his Ph.D., he will resume his medicinal chemistry career at Pfizer in Groton, CT.
Footnotes
Dedicated to the memory of Prof. Makoto Kumada, a pioneer in cross-coupling chemistry.
Notes and references
- 1.(a) Patai S, editor. The Chemistry of Alkenes. vol. 1. New York: Wiley; 1964. [Google Scholar]; (b) Zabicky J, editor. The Chemistry of Alkenes. vol. 2. New York: Wiley; 1970. [Google Scholar]; (c) Patai S, editor. Supplement A: The Chemistry of Double-Bonded Functional Groups. vol. 2. New York: Wiley; 1989. Parts 1 and 2. [Google Scholar]; (d) Patai S, editor. The Chemistry of Double-Bonded Functional Groups. vol. 2. New York: Wiley; 1997. Parts 1 and 2. [Google Scholar]
- 2.Grubbs RH, editor. Handbook of Metathesis. Weinheim: Wiley-VCH; 2003. [Google Scholar]
- 3.(a) Agbossou F, Carpentier J-F, Mortreaux A. Chem. Rev. 1995;95:2485–2506. [Google Scholar]; (b) Crudden CM, Hleba YB, Chen AC. J. Am. Chem. Soc. 2004;126:9200–9201. doi: 10.1021/ja049761i. [DOI] [PubMed] [Google Scholar]; (c) RajanBabu TV, Nomura N, Jin J, Nandi M, Park H, Sun X. J. Org. Chem. 2003;68:8431–8446. doi: 10.1021/jo035171b. [DOI] [PubMed] [Google Scholar]
- 4.(a) Scheirs J, Priddy DB, editors. Modern Styrenic Polymers: Polystyrenes and Styrenic Copolymers. Chichester, UK: John Wiley & Sons; 2003. [Google Scholar]; (b) Hirao A, Loykulnant S, Ishizone T. Prog. Polym. Sci. 2002;27:1399–1471. [Google Scholar]; (c) Schellenberg J, Tomotsu N. Prog. Polym. Sci. 2002;27:1925–1982. [Google Scholar]
- 5.Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis. vol. I-III. Heidelberg: Springer-Verlag; 1999. [Google Scholar]
- 6.(a) de Meijere A, Diedrich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Weinheim: Wiley-VCH; 2004. [Google Scholar]; (b) Tsuji J, editor. Palladium Reagents and Catalysts. Chichester: Wiley; 1995. [Google Scholar]; (c) Negishi E, de Meijere A, editors. Handbook of Organopalladium Chemistry for Organic Synthesis. Chichester: Wiley; 2002. [Google Scholar]
- 7.(a) Trost BM. Angew. Chem., Int. Ed. 1995;34:259–281. [Google Scholar]; (b) Sheldon RA. Pure Appl. Chem. 2000;72:1233–1246. [Google Scholar]
- 8.Heck RF. Org. React. 1982;27:345–390. [Google Scholar]
- 9.Stewart SK, Whiting A. J. Organomet. Chem. 1994;482:293–300. [Google Scholar]
- 10.Jeffery T. Tetrahedron Lett. 1999;40:1673–1676. [Google Scholar]
- 11.Echavarren AM, Cárdenas DJ. ch. 1. In: de Meijere A, Diedrich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Weinheim: Wiley-VCH; 2004. [Google Scholar]
- 12.Heck RF. J. Am. Chem. Soc. 1968;90:5518–5526. [Google Scholar]
- 13.A similar process was reported using Pd(0) as a precatalyst instead of Pd(II)Mori K, Mizoroki T, Ozaki A. Bull. Chem. Soc. Jpn. 1973;46:1505–1508.
- 14.Plevyak JE, Heck RF. J. Org. Chem. 1978;43:2454–2456. [Google Scholar]
- 15.For an electrocatalytic process with nickel, see:Rollin R, Meyer G, Troupel M, Fauvarque J-F, Perichon J. J. Chem. Soc., Chem. Commun. 1983:793–794.
- 16.Bumagin NA, Luzikova EV. J. Organomet. Chem. 1997;532:271–273. [Google Scholar]
- 17.(a) Hall DG. Boronic Acids. Weinheim: Wiley-VCH; 2005. [Google Scholar]; (b) Suzuki A, Brown H. Organic Syntheses via Boranes. vol. 3. Milwaukee: Aldrich Chemical Company; 2003. [Google Scholar]; (c) Suzuki A. ch. 2. In: Diedrich F, Stang P, editors. Metal-Catalyzed Cross-coupling Reactions. Weinheim: Wiley-VCH; 1998. [Google Scholar]; (d) Miyaura N. ch. 2. In: de Meijere A, Diedrich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Weinheim: Wiley-VCH; 2004. [Google Scholar]; (e) Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457–2383. [Google Scholar]
- 18.Matteson DS. J. Am. Chem. Soc. 1960;66 4228-4233. [Google Scholar]
- 19.Braun J, Normant H. Bull. Soc. Chim. Fr. 1966:2557–2664. [Google Scholar]
- 20.In refs. 18, 19 and 25 the authors report that vinylboronic acid is unstable and cannot be isolated, however, ref. 24 describes its preparation using the method described in ref. 18 and state it can be isolated, albeit with “very poor reproducibility and did indeed on several attempts polymerize”.
- 21.Peyroux E, Berthiol F, Doucet H, Santelli M. Eur. J. Org. Chem. 2004:1075–1082. [Google Scholar]
- 22.It is unclear how the authors circumvent the polymerization of vinylboronic acid.
- 23.Lightfoot AP, Twiddle SJR, Whiting A. Synlett. 2005:529–531. [Google Scholar]
- 24.(a) Kerins F, O’Shea DF. J. Org. Chem. 2002;67:4968–4971. doi: 10.1021/jo020074o. [DOI] [PubMed] [Google Scholar]; (b) Cottineau B, Kessler A, O’Shea DF. Org. Synth. 2006;83:45–48. [Google Scholar]
- 25.(a) Darses S, Michaud G, Genet J-P. Eur. J. Org. Chem. 1999:1875–1883. [Google Scholar]; (b) Molander GA, Rivero MR. Org. Lett. 2002;4:107–109. doi: 10.1021/ol0169729. [DOI] [PubMed] [Google Scholar]; (c) Molander GA, Brown AR. J. Org. Chem. 2006;71:9681–9686. doi: 10.1021/jo0617013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matteson DS. J. Org. Chem. 1962;27:3712. [Google Scholar]
- 27.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 28.Schumann H, Kaufmann J, Schmalz H-G, Bottcher A, Gotov B. Synlett. 2003:1783–1788. [Google Scholar]
- 29.Mikami S, Yorimitsu H, Oshima K. Synlett. 2002:1137–1139. [Google Scholar]
- 30.Hallberg A, Westerlund C. Chem. Lett. 1982:1993–1994. [Google Scholar]
- 31.Karabelas K, Hallberg K. J. Org. Chem. 1986;51:5286–5290. [Google Scholar]
- 32.Kikukawa K, Ikenaga K, Kono K, Toritani K, Wada F, Matsuda T. J. Organomet. Chem. 1984;270:277–282. [Google Scholar]
- 33.Hatanaka Y, Hiyama T. J. Org. Chem. 1988;53:918–920. [Google Scholar]
- 34.Noyori R, Nishida I, Sakata J. J. Am. Chem. Soc. 1981;103:2106. [Google Scholar]
- 35.Denmark SE, Choi JY. J. Am. Chem. Soc. 1999;121:5821–5822. [Google Scholar]
- 36.Denmark SE, Wang Z. Synthesis. 2000:999–1002. [Google Scholar]
- 37.Denmark SE, Sweis RF, Wehrli D. J. Am. Chem. Soc. 2004;126:4865–4875. doi: 10.1021/ja037234d. [DOI] [PubMed] [Google Scholar]
- 38.(a) Denmark SE, Sweis RF. Acc. Chem. Res. 2002;35:835–846. doi: 10.1021/ar020001r. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Ober MH. Aldrichimica Acta. 2003;36:75–85. [Google Scholar]; (c) Denmark SE, Sweis RF. Chem. Pharm. Bull. 2002;50:1531–1541. doi: 10.1248/cpb.50.1531. [DOI] [PubMed] [Google Scholar]; (d) Denmark SE, Sweis RF. ch. 4. In: de Meijere A, Diedrich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Weinheim: Wiley-VCH; 2004. [Google Scholar]; (e) Denmark SE, Regens CS. Acc. Chem. Res. 2008 doi: 10.1021/ar800037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Denmark SE, Butler CR. J. Am. Chem. Soc. 2008;130:3690–3704. doi: 10.1021/ja7100888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laganis ED, Chenard BL. Tetrahedron Lett. 1984;25:5831–5834. [Google Scholar]
- 41.TBAF ($777 mol−1, Aldrich cat #86872).
- 42.DVDS ($52 mol−1, Gelest cat #SID4613.0); KOSiMe3 ($89 mol−1, Aldrich cat #324868).
- 43.Denmark SE, Sweis RF. J. Am. Chem. Soc. 2001;123:6439–6440. doi: 10.1021/ja016021q. [DOI] [PubMed] [Google Scholar]
- 44.Hammouch SO, Beinert GJ, Herz JE. Polymer. 1996;37:3353. [Google Scholar]
- 45.Denmark SE, Wang Z. J. Organomet. Chem. 2001;621:372–375. [Google Scholar]
- 46.Denmark SE, Butler CR. e-EROS Encyclopedia of Reagents for Organic Synthesis. 2007 [Google Scholar]
- 47.1,3,5,7-Tetramethyl-1,3,5,7-tetravinylcyclotetrasiloxane ($67 mol−1, Gelest, cat # SIT7900.0).
- 48.Denmark SE, Butler CR. Org. Lett. 2006;8:63–66. doi: 10.1021/ol052517r. [DOI] [PubMed] [Google Scholar]
- 49.Chen R, Lee V, Adlington RM, Baldwin JE. Synthesis. 2007:113–117. [Google Scholar]
- 50.Lee HM, Nolan SP. Org. Lett. 2000;2:2053–2055. doi: 10.1021/ol005956t. [DOI] [PubMed] [Google Scholar]
- 51.Clarke ML. Adv. Synth. Catal. 2005;347:303–307. [Google Scholar]
- 52.Vinyltriethoxysilane ($10.5 mol−1, Gelest cat # SIV9112.0).
- 53.(a) Alacid E, Najera C. Adv. Synth. Catal. 2006;348:2085–2091. [Google Scholar]; (b) Alacid E, Najera C. J. Org. Chem. 2008;73:2315–2322. doi: 10.1021/jo702570q. [DOI] [PubMed] [Google Scholar]
- 54.Cragg ST. In: Patty’s Toxicology. Bingham E, Cohrssen B, Powell CH, editors. Hoboken: Wiley; 2001. [Google Scholar]
- 55.(a) Stille JK. Angew. Chem., Int. Ed. Engl. 1986;25:508–523. [Google Scholar]; (b) Farina V, Krishnamurthy V, Scott W. J. Org. React. 1997;50:1–652. [Google Scholar]; (c) Mitchell TN. ch. 4. In: Diedrich F, Stang P, editors. Metal-Catalyzed Cross-coupling Reactions. Weinheim: Wiley-VCH; 1998. [Google Scholar]; (d) Mitchell TN. ch. 3. In: de Meijere A, Diedrich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Weinheim: Wiley-VCH; 2004. [Google Scholar]
- 56.Thoonen SHL, Deelman BJ, van Koten G. J. Organomet. Chem. 2004;689:2145–2157. [Google Scholar]
- 57.Scott WJ, Stille JK. J. Am. Chem. Soc. 1986;108:3033–3040. [Google Scholar]
- 58.(a) Littke AF, Schwarz L, Fu GC. J. Am. Chem. Soc. 2002;124:6343–6348. doi: 10.1021/ja020012f. [DOI] [PubMed] [Google Scholar]; (b) Littke AF, Fu GC. Angew. Chem., Int. Ed. 1999;38:2411–2413. doi: 10.1002/(sici)1521-3773(19990816)38:16<2411::aid-anie2411>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 59.Shirakawa E, Yamasaki K, Hiyama T. Synthesis. 1998:1544–1549. [Google Scholar]
- 60.Grasa GA, Nolan SP. Org. Lett. 2001;3:119–122. doi: 10.1021/ol006827f. [DOI] [PubMed] [Google Scholar]
- 61.Poupon J-C, Marcoux D, Cloarec J-M, Charette AB. Org. Lett. 2007;9:3591–3594. doi: 10.1021/ol701447q. [DOI] [PubMed] [Google Scholar]
- 62.Chieffi A, Kamikawa K, Ahman J, Fox JM, Buchwald SL. Org. Lett. 2001;3:1897–1900. doi: 10.1021/ol0159470. [DOI] [PubMed] [Google Scholar]
- 63.Gøgsig TM, Søjberg LS, Lindhardt (nee Hansen) AT, Jensen KL, Skrydstrup T. J. Org. Chem. 2008;73:3404–3410. doi: 10.1021/jo7027097. [DOI] [PubMed] [Google Scholar]
- 64.Tamao K, Sumitani K, Kumada M. J. Am. Chem. Soc. 1972;94:4374–4376. [Google Scholar]
- 65.Nevertheless, this process is used in the industrial scale preparation of 4-(tert-butoxy)styrene. Jpn. Pat., H01-106835, 1989. We thank a referee for making us aware of this application.
- 66.Lando VR, Monteiro A. Org. Lett. 2003;5:2891–2894. doi: 10.1021/ol034948k. [DOI] [PubMed] [Google Scholar]
- 67.Dunet G, Knochel P. Synlett. 2006:407–410. [Google Scholar]
- 68.Amatore M, Gosmini C, Perichon J. Eur. J. Org. Chem. 2005:989–992. doi: 10.1021/jo060855f. [DOI] [PubMed] [Google Scholar]