

Abstract
Cardiac myxomas are the most common primary tumors of the heart, although little is known about their etiology. Mutations of the protein kinase A (PKA) regulatory subunit gene PRKAR1A cause inherited myxomas in the setting of the Carney complex tumor syndrome, providing a possible window for understanding their pathogenesis. We recently reported that cardiac-specific knockout of this gene causes myxomatous changes in the heart, although the mice die during gestation from cardiac failure. In this review, we discuss these findings and place them in the larger understanding of how PKA dysregulation might affect cardiac function and cause myxomagenesis.
Introduction
Cardiac myxoma is the most common primary tumor affecting the heart, accounting for nearly half of cardiac neoplasms (Reynen 1995). Its name derives from its appearance as a cell-poor, myxoid neoplasm with a mucopolysaccharide-rich extracellular matrix. Myxomas usually arise from the inter-atrial septum and more frequently involve the left side of the heart. Although cardiac myxomas are histopathologically benign, they cause a chronic inflammatory-like syndrome, and can cause catastrophic morbidity due to embolism or intracardiac obstruction.
Inherited myxomas are observed in the setting of the Carney complex (CNC), a dominantly inherited syndrome characterized by cardiac and extracardiac myxomas in the setting of spotty skin pigmentation, endocrine overactivity, and schwannomas (Carney et al. 1985). Approximately 30% to 60% of CNC patients will develop cardiac myxomas (Bertherat 2006), usually at much younger ages than the sporadic tumors. In this setting, the myxomas can be seen in any chamber of the heart, can be multiple, and can recur after initial resection. As a cardinal feature of CNC, cardiac myxomas are responsible for the death of more than 50% of patients, either from tumors themselves, or from post-surgical complications (Stratakis et al. 2001).
Molecular genetic analysis has revealed that mutations in the PRKAR1A gene, encoding the type 1A regulatory subunit of the cAMP-dependent protein kinase (PKA), are the cause of CNC in at least 50% of cases (Kirschner et al. 2000a; Kirschner et al. 2000b). In tumors from CNC patients, PKA activity is elevated (Bertherat 2006; Kirschner et al. 2000a), a finding also seen in mouse cells lacking the Prkar1a protein (Nadella and Kirschner 2005).
The cAMP-PKA Signaling Pathway
The cAMP-PKA signaling pathway regulates numerous important biological processes under both physiological and pathological conditions, including diabetes, heart failure, and cancer. At the cellular level, cAMP-PKA signal transduction is critically involved in numerous cellular processes, including the regulation of metabolism, gene expression and cell growth. At the structural level, PKA is composed of two catalytic (C) and two regulatory (R) subunits which form a tetrameric holoenzyme, R2C2. In the presence of cAMP, the holoenzyme dissociates into an R2(cAMP)4 dimer and two free (active) C subunits, which can phosphorylate a diverse number of target proteins both in the cytoplasmic and the nuclear compartments, including enzymes, structural proteins, and transcription factors.
In higher organisms, there are two isoforms of PKA, Type 1 and Type 2, based on the pattern of enzymatic elution in anion-exchange chromatography. The isoform specificity is determined by the identity of the regulatory subunits in the holoenzyme, and humans (and mice) have two genes encoding Type I regulatory subunits (PRKARIA and PRKARIB) and two encoding Type 2 subunits (PRKAR2A and PRKAR2B). The R subunits differ in molecular weight, tissue specificity, subcellular distribution, and expression pattern during development and cell cycle (ChoChung et al. 1995). PRKAR1A is ubiquitously expressed, whereas PRKAR1B is expressed primarily in brain, testis and lymphocytes. Similarly, PRKAR2A has ubiquitous expression, while PRKAR2B is expressed mainly in brain, adipose, and some endocrine tissues. The catalytic subunits are encoded by three functionally equivalent genes in humans (PRKACA, PRKACB, PRKACG), whereas mice only have two of these (Prkaca and Prkacb) (Foss et al. 1994). The catalytic subunits exhibit differential tissue expression, although most tissues show a predominance of the Cα catalytic subunit (Beebe et al. 1990). The type 1 PKA isoforms appear to have a higher affinity for cAMP, making them the major source of regulated cytoplasmic PKA activity within the cell. (Tasken et al. 1997). Type 2 PKA appears to exist more commonly anchored in multiprotein complexes, suggesting that it may a play a role in producing localized cAMP-dependent effects in specific cellular subdomains (Ruehr et al. 2004).
In addition, cAMP-dependent responses may be mediated by PRKX, an X-chromosome (located at Xp22.3) encoded protein that appears to function as a novel catalytic subunit of Type I PKA (Klink et al. 1995; Zimmermann et al. 1999). In mice, Prkx is ubiquitously and highly expressed in the central nervous system and in the heart during early developmental stages, but the status of older embryos is unclear. In humans, low levels of PRKX mRNA were detected in mRNA from embryonic heart, but no protein was detected by Western blotting of either fetal or adult heart (Li et al. 2005). Thus, the role of PRKX in cardiovascular function is unknown. A related kinase on the Y-chromosome, PRKY, (located at Yp11.2 has been studied even less.
PKA in Cardiac Function and Development
The cAMP-PKA pathway is known to play a critical role in mediating the effects of adrenergic stimulation on the heart (Rockman et al. 2002). The classic paradigm of cAMP-PKA-mediated signal transduction in cardiomyocytes involves ligand-dependent activation of the β-adrenoceptors (β-AR), including β1– and β2– ARs, which triggers the interaction of guanine nucleotide-binding protein (G- proteins) with adenylyl cyclases (ACs), and then ACs catalyze the conversion of ATP to second messenger cAMP. The increase in intracellular cAMP level in turn activates PKA. PKA then phosphorylates cardiac target proteins such as Ca2+ channels, phospholamban, troponin I and C. These biochemical responses result in increases in contractility, rate of cardiac relaxation, heart rate, and impulse conduction (Wang and Dhalla 2000). The physiological functions and regulation of the key components of the β-adrenergic receptor-mediated PKA signal pathway in cardiac function have been extensively studied in human patients and by genetic manipulation in mice (Wang and Dhalla 2000). The cAMP/PKA system can also be activated by other G-protein-coupled receptors in the heart, including multiple isoforms of the adenosine receptor (Dobson and Fenton 1997).
In addition to its role as a mediator of normal physiology, PKA protein level and activity are enhanced in the failing heart (Wang et al. 1999). In studies of human patients with heart failure, the increased PKA phosphorylation of ryanodine receptor (RyR2) resulted in leakage of Ca2+ from the sarcoplasmic reticulum, contributing to impaired contractility and ventricular arrhythmias causing sudden cardiac death (Marks 2003). Taken together, these data demonstrate that PKA is a critical regulator of cardiac function.
Ablation of Prkar1a from the Heart Leads to Cardiac Failure
Very little information regarding the role of PRKAR1A in cardiac development is available. Unlike other PKA-R subunit knockouts, mice lacking Prkar1a do not survive embryogenesis and die before embryonic day 9.5 (e9.5) (Amieux et al. 2002; Kirschner et al. 2005). Amieux et al. demonstrated that ablation of Prkar1a leads to enhanced PKA activity and a developmental failure of the mesoderm (Amieux et al. 2002). Because the mesoderm gives rise to essential structures, including the embryonic heart tube, the Prkar1a knockout (KO) embryos cannot be used to study cardiac development.
To circumvent this generalized developmental failure, we utilized a tissue-specific knockout approach to delete Prkar1a from the developing heart. Using the promoter of the α-myosin heavy chain (αMHC) to drive expression of the cre recombinase, we generated mice lacking Prkar1a in the developing heart (Prkar1a-CKO mice) (Yin et al. 2008). The αMHC promoter is active at low levels by e10.5 (Ng et al. 1991), which is after the major cardiac specification events have taken place. As might be predicted, these animals developed normal cardiac structures, with clearly identifiable atria, ventricles, and pericardial structures. However, cardiac failure became evident at e11.5, with marked thinning of ventricular walls characterized by both hypoplasia of the compact layer and severely reduced trabeculation. These ventricular changes were associated with atrial dilation, heart failure, and embryonic demise before e12.5.
Examination of the hearts demonstrated that the Prkar1a-CKO animals had reduced cardiomyocyte proliferation which could be detected at e10.5, when the hearts appeared structurally normal. Ablation of Prkar1a was not associated with changes in the abundance of the other regulatory subunits, indicating a lack of compensation. In concordance with the reduced levels of the R1a regulatory subunit, hearts from Prkar1a-CKO mice exhibited enhancement of both free and total PKA activity, similar to the observation in null cells in vitro (Nadella and Kirschner 2005; Yin et al. 2008).
Because one of the well-known targets of PKA is the transcription factor cAMP-response element binding (CREB) protein, we sought to determine the effects of Prkar1a ablation on the expression of cardiac-specific proteins. In Prkar1a-CKO hearts, there was significant downregulation of all cardiac transcription factors investigated, including Nkx2–5, Gata4, serum response factor (Srf), and myocardin (Myocd). As has been thoroughly reviewed by other authors (Bruneau 2002; Nemer 2008; Olson 2004; Srivastava 2006), cardiogenesis occurs by means of an intricate transcriptional program requiring the precise expression of cardiac-specific genes directed by a complex array of transcription factors. A detailed discussion of this process is beyond the scope of the present review, but some general comments are worthwhile. Through inductive interactions in the developing heart field, the production of transcription factors such as Nkx2-5, Myocd, Gata family members (likely Gata4, -5, and -6) and possibly Srf, begins in the cardiac crescent as early as e7.5 in the mouse embryo (Buckingham et al. 2005). Although it is dispensable for establishment of cardiac lineage (Lyons et al. 1995; Tanaka et al. 1999), Nkx2-5 is required for proper left ventricular chamber specification (Yamagishi et al. 2001). In human, mutations in NKX2-5 are associated with septal and conduction defects (Benson et al. 1999; Schott et al. 1998). The analysis of Nkx2-5-deficient embryos indicates that it plays a crucial role in transcriptional regulation of several cardiac-specific genes and for the embryonic myocardium to differentiate beyond the stage of heart looping (reviewed by (Akazawa and Komuro 2005). Moreover, mice lacking Nkx2-5 fail to express the eHAND transcription factor in the heart, suggesting that eHAND and its downstream targets depend on Nkx2-5 in the left ventricle (Biben and Harvey 1997).
Both transcript and protein of Gata4 are found during formation of the heart tube at E8.0 in endocardium, myocardium and precardiac mesoderm (Heikinheimo et al. 1994). The importance of Gata4 in heart development is evidenced by the Gata4 knockout mouse, which develop bilateral heart tubes (cardia bifida), and have a reduced number of cardiac myocytes (Kuo et al. 1997; Molkentin et al. 1997). Similar to NKX2-5, mutations in GATA4 cause atrial and ventricular septal defects in human (Garg et al. 2003), and GATA4 and the T-box transcription factor TBX5 also form a complex to regulate downstream genes, such as myosin heavy chain. Moreover, Srf is a MADS-Box family transcription factor, which broadly expressed in the embryo and essential for mesoderm formation. Srf interacts with Nkx2-5, GATA factors and myocardin (Myocd) to synergistically activate various transcriptional target genes (Bruneau 2002).
Because the cre transgene is not expressed until after cardiac differentiation is underway, the excess PKA activity caused by ablation of Prkar1a must interfere with the actions of these transcription factors that are necessary for the maintenance of the developing heart, rather than its initial specification. To look at the downstream effects of this interference, we examined the expression of structural proteins required for cardiac function, including members of the actin and myosin families. All of the structural proteins exhibited lower message levels, and significant reductions were observed for β-MHC and for actins A1 and A2. The effects of excess PKA activity resulting from Prkar1a ablation may either be direct effects on transcription (via CREB) or may be due to interference with other transcription factors, as has previously been demonstrated to be the case for Srf (Bruneau 2002; Davis et al. 2003; Nguyen et al. 2004). The fact that transcription factors function in combination and hierarchical networks (Bruneau 2002; Durocher et al. 1997) may explain why small reductions in individual factors, as observed here, causes synergistic and deleterious effects during cardiogenesis. This hypothesis is corroborated by our data: reductions in both Gata4 and Nkx2–5 lead to significant reductions in Nppa. Also, as mentioned, reduction in Nkx2-5 may cause loss of eHAND/HAND1, which works in concert for the expression of the structural proteins. In addition, we observed consistent downregulation of the Slc8a1 (Ncx1) which is required for Ca2+ extrusion (Schulze et al. 1993) and plays a pivotal role in the regulation of intracellular Ca2+ homeostasis in cardiac myocytes.
The importance of proper regulation of transcription is underscored by the similarity among other mouse knockouts that have targeted various transcription factors. For example, the phenotype of embryonic demise resulting from a thinning of the myocardium has previously been observed by mice lacking either Srf (Miano et al. 2004; Parlakian et al. 2004) or Gata4 (Oka et al. 2006; Zeisberg et al. 2005). In Srf knockout hearts, increased apoptosis caused the decrease in cell number, whereas in our model, the defect appeared to lie in reduced proliferation. This observation is likely due to a direct antiproliferative effect of elevated PKA activity which has been well described in many cell systems, including cultured smooth muscle myocytes (Indolfi et al. 2001).
One of the striking aspects of the cardiac phenotype exhibited in the Prkar1a-CKO hearts was the disorganization of the sarcomeres. Z disks were easily seen, but they were not properly assembled, and myofilaments appeared loose and disarrayed. Defective expression of the Z-disk actins Acta1 and Acta2 likely underlies this ultrastructural defect. In addition, the significantly diminished expression of actins and Myh7 (β-MHC) may result in a deficiency in both thin (actin) and thick (myosin) myofilament components and disruption of both M lines and I bands. PKA is also known to phosphorylate myosin associate proteins such as the myosin binding protein C (MyBP-C) and Troponin I (cTnI) (Kentish et al. 2001; Yang et al. 2001), although the role of potential altered phosphorylation of these proteins in the observed phenotype has not yet been investigated. Most likely, the disorganized Z disks and myofilaments contribute to poor cardiac contraction and heart failure, causing demise of the growing embryo.
Prkar1a Ablation as a Cause of Cardiac Myxoma Development
Although cardiac myxomas are common in CNC patients, no cardiac tumors have been observed in mice heterozygous for Prkar1a mutations, made as the exact genetic model for human patients (Kirschner et al. 2005; Veugelers et al. 2004). These observations shed doubt on the notion that haploinsufficiency of Prkar1a is sufficient for myxomagenesis, but suggest that the mouse lifespan may be too short for the development of additional genetic hits (at the Prkar1a locus or elsewhere) that cause these tumors. In general, Prkar1a-CKO mice did not survive long enough to develop bona fide myxomas. However, analysis of Prkar1a-CKO mice showed that approximately 50% of Prkar1a-CKO hearts exhibited stroma-rich lesions suggestive of human cardiac myxomas. At dissection of e11.5 mice, typical myxoid material was frequently noted in the endocardial cushion. This structure is composed of a subset of cells that undergo remodeling to form the valvular structures and membranous septa of the mature heart after cardiac looping (Person et al. 2005). This observation may help explain the observation that myxomas arise predominantly in the interatrial septum (Reynen 1995).
Histologically, the majority of Prkar1a-CKO hearts exhibited the characteristic features of cardiac myxomas, including the presence of small numbers of polygonal or stellate cells surrounded by abundant loose stroma rich in acid mucopolysaccharides. Most strikingly, they also exhibit epithelium- or gland-like changes, which are rare but diagnostic features of human cardiac myxomas (Goldman et al. 1987; Johansson 1989). Finally, the tumors showed evidence of nuclear polymorphism and cellular atypia, as is ordinarily seen only in neoplastic cells. Thus, the evidence is convincing that this mouse model develops early phases in the development of cardiac myxoma.
There has been considerable debate regarding the histogenesis of cardiac myxoma. Various tumor cells have been identified in cardiac myxomas, including endothelial, fibroblastic, and muscle cells along with the typical stromal cells and glands (Abenoza and Sibley 1986). Most authors favor that myxomas arise from the endocardium (Fine et al. 1968) and are derived from subendocardial multipotent mesenchymal cells (Ferrans and Roberts 1973; Lie 1989). Our findings support the hypothesis that cardiac myxomas originate from primitive pluripotent mesenchymal cells, such as those arising from epithelial mesenchymal transition (EMT) in the endocardial cushion (Kinsella and Fitzharris 1980; Person et al. 2005). This tissue origin also may shed insight on why PKA activation, with its anti-proliferative effect in differentiated cardiomyocytes, may cause the formation of tumors. Specifically, it is known, from our work and the work of others, that the effects of PKA activation are cell- and differentiation-state specific. In this model, PKA activation in these pluripotent cells may lead to cell division, whereas the overall effect in the heart is anti-proliferative.
Integration of the Current Data with Prior Studies
The Prkar1a-CKO model demonstrates that Prkar1a is required for proper cardiac development and function. Depletion of Prkar1a causes excess PKA activity, with resultant downregulation of the transcriptional activity of cardiac transcription factors and their downstream targets, including key cardiac structural proteins and proteins involved in calcium handling. The similarities of the Prkar1a-CKO model with other cardiac transcription factor knockouts suggest that there is a common downstream mechanism for these pathways in the production of a functioning heart (Figure 1).
Figure 1. Model for the interactions of Prkar1a with other genes sharing a cardiac phenotype.
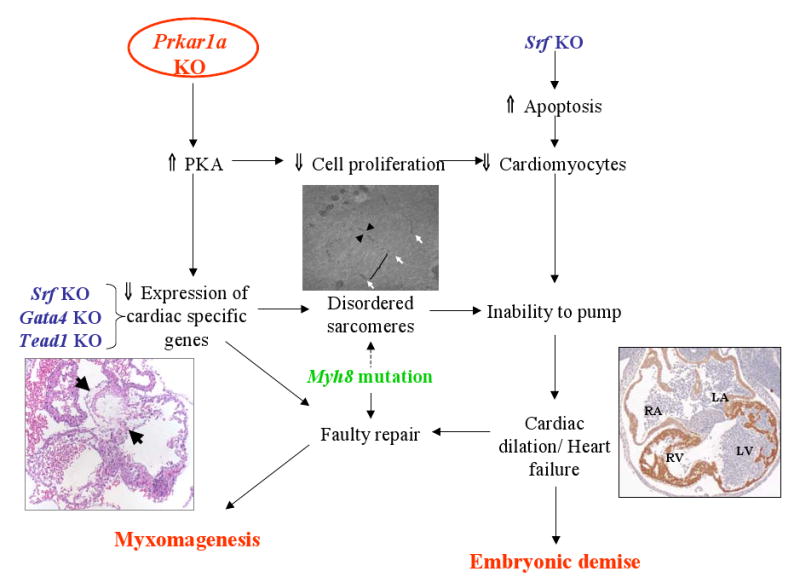
Prkar1a KO (red text) causes reduction of cardiac-specific genes and a reduced number of cardiomyocytes with aberrant sarcomeres (Middle figure: white arrow: residual Z-disk; arrowhead: residual I-band)), eventually leading to a thin-walled, dilated heart (Right figure). At the end, these changes cause myxomagenesis (Left figure, arrows), and embryonic demise. These same pathways may be affected by knockout of other cardiac-specific transcription factors (blue text). A mechanism to account for myxomagenesis associated with mutation of MYH8 is also proposed in the same scheme. See text for details and references.
Intriguingly, Prkar1a-CKO mutants developed atypia and myxoma-like changes in the myocardium. Neither of these findings has been reported in conventional Prkar1a knockouts or in any other genetically modified mouse, suggesting that Prkar1a-CKO mice may serve as the first good model with which to study the formation of cardiac myxomas. Inherited cardiac myxomas have also been observed in a single family with the trismus-pseudocamptodactyly syndrome (sometimes called a CNC variant), caused by inactivating mutations in MYH8, encoding perinatal myosin (Veugelers et al. 2004). The fact that ablation of PRKAR1a/Prkar1a causes disruption of myosin expression suggests that the two mechanisms may also be related. Although PKA is not known to directly phosphorylate myosins, its effects on myosin-associated proteins is well described and has important functional consequences (Kentish et al. 2001; Yang et al. 2001), providing a plausible link between the two genetic defects. Although there are no data to indicate that other transcription factor knockouts produce the predicted myxomatous changes, it would be interesting to reexamine the hearts of those mice.
Regardless of the outcome, examining the interrelationships of these proteins both for cardiac development and for myxomas formation should provide for an interesting set of future investigations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abenoza P, Sibley RK. Cardiac myxoma with glandlike structures. An immunohistochemical study. Arch Pathol Lab Med. 1986;110:736–9. [PubMed] [Google Scholar]
- Akazawa H, Komuro I. Cardiac transcription factor csx/nkx2-5: Its role in cardiac development and diseases. Pharmacol Ther. 2005;107:252–268. doi: 10.1016/j.pharmthera.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Amieux PS, Howe DG, Knickerbocker H, Lee DC, Su T, Laszlo GS, et al. Increased basal cAMP-dependent protein kinase Activity inhibits the formation of mesoderm-derived structures in the developing mouse embryo. J Biol Chem. 2002;277:27294–27304. doi: 10.1074/jbc.M200302200. [DOI] [PubMed] [Google Scholar]
- Beebe SJ, Oyen O, Sandberg M, Froysa A, Hansson V, Jahnsen T. Molecular-cloning of a tissue-specific protein-kinase (c-gamma) from human testis - representing a 3rd isoform for the catalytic subunit of cAMP-dependent protein-kinase. Mol Endocrinol. 1990;4:465–475. doi: 10.1210/mend-4-3-465. [DOI] [PubMed] [Google Scholar]
- Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang YZ, Riggs S, et al. Mutations in the cardiac transcription factor Nkx2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–1573. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertherat J. Carney complex (cnc) Orphanet Journal of Rare Diseases. 2006;1 doi: 10.1186/1750-1172-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biben C, Harvey RP. Homeodomain factor Nkx2-5 controls left/right asymmetric expression of bhlh gene eHAND during murine heart development. Genes Dev. 1997;11:1357–1369. doi: 10.1101/gad.11.11.1357. [DOI] [PubMed] [Google Scholar]
- Bruneau BG. Transcriptional regulation of vertebrate cardiac morphogenesis. Circ Res. 2002;90:509–519. doi: 10.1161/01.res.0000013072.51957.b7. [DOI] [PubMed] [Google Scholar]
- Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6:826–835. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VLW. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 1985;64:270–283. doi: 10.1097/00005792-198507000-00007. [DOI] [PubMed] [Google Scholar]
- ChoChung YS, Pepe S, Clair T, Budillon A, Nesterova M. cAMP-dependent protein kinase: Role in normal and malignant growth. Crit Rev Oncol Hematol. 1995;21:33–61. doi: 10.1016/1040-8428(94)00166-9. [DOI] [PubMed] [Google Scholar]
- Davis A, Hogarth K, Fernandes D, Solway J, Niu JX, Kolenko V, et al. Functional significance of protein kinase A activation by endothelin-1 and ATP: Negative regulation of Srf-dependent gene expression by PKA. Cell Signal. 2003;15:597–604. doi: 10.1016/s0898-6568(02)00148-1. [DOI] [PubMed] [Google Scholar]
- Dobson JG, Jr, Fenton RA. Adenosine A2 receptor function in rat ventricular myocytes. Cardiovasc Res. 1997;34:337–47. doi: 10.1016/s0008-6363(97)00023-0. [DOI] [PubMed] [Google Scholar]
- Durocher D, Charron F, Warren R, Schwartz RJ, Nemer M. The cardiac transcription factors Nkx2-5 and Gata-4 are mutual cofactors. EMBO J. 1997;16:5687–96. doi: 10.1093/emboj/16.18.5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrans VJ, Roberts WC. Structural features of cardiac myxomas. Histology, histochemistry, and electron microscopy. Hum Pathol. 1973;4:111–46. doi: 10.1016/s0046-8177(73)80051-6. [DOI] [PubMed] [Google Scholar]
- Fine G, Morales A, Horn RC., Jr Cardiac myxoma. A morphologic and histogenetic appraisal. Cancer. 1968;22:1156–62. doi: 10.1002/1097-0142(196811)22:6<1156::aid-cncr2820220611>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Foss KB, Landmark B, Skalhegg BS, Tasken K, Jellum E, Hansson V, et al. Characterization of in-vitro-translated human regulatory and catalytic subunits of cAMP-dependent protein-kinases. Eur J Biochem. 1994;220:217–223. doi: 10.1111/j.1432-1033.1994.tb18617.x. [DOI] [PubMed] [Google Scholar]
- Garg V, Kathiriyra IS, Barnes R, Schluterman MK, King IN, Butler CA, et al. Gata4 mutations cause human congenital heart defects and reveal an interaction with Tbx5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- Goldman BI, Frydman C, Harpaz N, Ryan SF, Loiterman D. Glandular cardiac myxomas. Histologic, immunohistochemical, and ultrastructural evidence of epithelial differentiation. Cancer. 1987;59:1767–75. doi: 10.1002/1097-0142(19870515)59:10<1767::aid-cncr2820591015>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Heikinheimo M, Scandrett JM, Wilson DB. Localization of transcription factor Gata-4 to regions of the mouse embryo involved in cardiac development. Dev Biol. 1994;164:361–373. doi: 10.1006/dbio.1994.1206. [DOI] [PubMed] [Google Scholar]
- Indolfi C, Stabile E, Coppola C, Gallo A, Perrino C, Allevato G, et al. Membrane-bound protein kinase A inhibits smooth muscle cell proliferation in vitro and in vivo by amplifying cAMP-protein kinase A signals. Circ Res. 2001;88:319–324. doi: 10.1161/01.res.88.3.319. [DOI] [PubMed] [Google Scholar]
- Johansson L. Histogenesis of cardiac myxomas. An immunohistochemical study of 19 cases, including one with glandular structures, and review of the literature. Arch Pathol Lab Med. 1989;113:735–41. [PubMed] [Google Scholar]
- Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, et al. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res. 2001;88:1059–65. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- Kinsella MG, Fitzharris TP. Origin of cushion tissue in the developing chick heart: Cinematographic recordings of in situ formation. Science. 1980;207:1359–60. doi: 10.1126/science.7355294. [DOI] [PubMed] [Google Scholar]
- Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000a;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- Kirschner LS, Kusewitt DF, Matyakhina L, Towns WH, Carney JA, Westphal H, et al. A mouse model for the Carney complex tumor syndrome develops neoplasia in cyclic AMP-responsive tissues. Cancer Res. 2005;65:4506–4514. doi: 10.1158/0008-5472.CAN-05-0580. [DOI] [PubMed] [Google Scholar]
- Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA. Genetic heterogeneity and spectrum of mutations of the Prkar1a gene in patients with the carney complex. Hum Mol Genet. 2000b;9:3037–3046. doi: 10.1093/hmg/9.20.3037. [DOI] [PubMed] [Google Scholar]
- Klink A, Schiebel K, Winkelmann M, Rao E, Horsthemke B, Ludecke HJ, et al. The human protein-kinase gene Pkx1 on Xp22.3 displays Xp/Yp homology and is a site of chromosomal instability. Hum Mol Genet. 1995;4:869–878. doi: 10.1093/hmg/4.5.869. [DOI] [PubMed] [Google Scholar]
- Kuo CT, Morrisey EE, Anandappa R, Sigrist K, Lu MM, Parmacek MS, et al. Gata4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11:1048–1060. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- Li W, Yu ZX, Kotin RM. Profiles of Prkx expression in developmental mouse embryo and human tissues. J Histochem Cytochem. 2005;53:1003–1009. doi: 10.1369/jhc.4A6568.2005. [DOI] [PubMed] [Google Scholar]
- Lie JT. The identity and histogenesis of cardiac myxomas. A controversy put to rest. Arch Pathol Lab Med. 1989;113:724–6. [PubMed] [Google Scholar]
- Lyons I, Parsons LM, Hartley L, Li RL, Andrews JE, Robb L, et al. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- Marks AR. A guide for the perplexed - towards an understanding of the molecular basis of heart failure. Circulation. 2003;107:1456–1459. doi: 10.1161/01.cir.0000059745.95643.83. [DOI] [PubMed] [Google Scholar]
- Miano JM, Ramanan N, Georger MA, Bentley KLD, Emerson RL, Balza RO, et al. Restricted inactivation of serum response factor to the cardiovascular system. Proc Natl Acad Sci U S A. 2004;101:17132–17137. doi: 10.1073/pnas.0406041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor Gata4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- Nadella KS, Kirschner LS. Disruption of protein kinase A regulation causes immortalization and dysregulation of D-type cyclins. Cancer Res. 2005;65:10307–10315. doi: 10.1158/0008-5472.CAN-05-3183. [DOI] [PubMed] [Google Scholar]
- Nemer M. Genetic insights into normal and abnormal heart development. Cardiovascular Pathology. 2008;17:48–54. doi: 10.1016/j.carpath.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Ng WA, Grupp IL, Subramaniam A, Robbins J. Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart. Circ Res. 1991;68:1742–50. doi: 10.1161/01.res.68.6.1742. [DOI] [PubMed] [Google Scholar]
- Nguyen GH, French R, Radhakrishna H. Protein kinase A inhibits lysophosphatidic acid induction of serum response factor via alterations in the actin cytoskeleton. Cell Signal. 2004;16:1141–1151. doi: 10.1016/j.cellsig.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, et al. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- Olson EN. A decade of discoveries in cardiac biology. Nat Med. 2004;10:467–474. doi: 10.1038/nm0504-467. [DOI] [PubMed] [Google Scholar]
- Parlakian A, Tuil D, Hamard G, Tavernier G, Hentzen D, Concordet JP, et al. Targeted inactivation of serum response factor in the developing heart results in myocardial defects and embryonic lethality. Mol Cell Biol. 2004;24:5281–5289. doi: 10.1128/MCB.24.12.5281-5289.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. 2005;243:287–335. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- Reynen K. Cardiac myxomas. N Engl J Med. 1995;333:1610–7. doi: 10.1056/NEJM199512143332407. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- Ruehr ML, Russell MA, Bond M. A-kinase anchoring protein targeting of protein kinase A in the heart. J Mol Cell Cardiol. 2004;37:653–65. doi: 10.1016/j.yjmcc.2004.04.017. [DOI] [PubMed] [Google Scholar]
- Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, et al. Congenital heart disease caused by mutations in the transcription factor Nkx2-5. Science. 1998;281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- Schulze D, Kofuji P, Hadley R, Kirby MS, Kieval RS, Doering A, et al. Sodium/calcium exchanger in heart muscle: Molecular biology, cellular function, and its special role in excitation-contraction coupling. Cardiovasc Res. 1993;27:1726–34. doi: 10.1093/cvr/27.10.1726. [DOI] [PubMed] [Google Scholar]
- Srivastava D. Making or breaking the heart: From lineage determination to morphogenesis. Cell. 2006;126:1037–1048. doi: 10.1016/j.cell.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: Diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001;86:4041–4046. doi: 10.1210/jcem.86.9.7903. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Chen Z, Bartunkova S, Yamasaki N, Izumo S. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development. 1999;126:1269–1280. doi: 10.1242/dev.126.6.1269. [DOI] [PubMed] [Google Scholar]
- Tasken K, Skalhegg BS, Tasken KA, Solberg R, Knutsen HK, Levy FO, et al. Structure, function, and regulation of human cAMP-dependent protein kinases. Signal Transduction in Health and Disease. 1997;31:191–204. doi: 10.1016/s1040-7952(97)80019-5. [DOI] [PubMed] [Google Scholar]
- Veugelers M, Wilkes D, Burton K, McDermott DA, Song Y, Goldstein MM, et al. Comparative Prkar1a genotype-phenotype analyses in humans with Carney complex and Prkar1a haploinsufficient mice. Proc Natl Acad Sci U S A. 2004;101:14222–14227. doi: 10.1073/pnas.0405535101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Liu XL, Arneja AS, Dhalla NS. Alterations in protein kinase A and protein kinase C levels in heart failure due to genetic cardiomyopathy. Can J Cardiol. 1999;15:683–690. [PubMed] [Google Scholar]
- Wang X, Dhalla NS. Modification of beta-adrenoceptor signal transduction pathway by genetic manipulation and heart failure. Mol Cell Biochem. 2000;214:131–155. doi: 10.1023/a:1007131925048. [DOI] [PubMed] [Google Scholar]
- Yamagishi H, Yamagishi C, Nakagawa O, Harvey RP, Olson EN, Srivastava D. The combinatorial activities of Nkx2.5 and dHAND are essential for cardiac ventricle formation. Dev Biol. 2001;239:190–203. doi: 10.1006/dbio.2001.0417. [DOI] [PubMed] [Google Scholar]
- Yang Q, Hewett TE, Klevitsky R, Sanbe A, Wang X, Robbins J. PKA-dependent phosphorylation of cardiac myosin binding protein C in transgenic mice. Cardiovasc Res. 2001;51:80–8. doi: 10.1016/s0008-6363(01)00273-5. [DOI] [PubMed] [Google Scholar]
- Yin ZR, Jones GN, Towns WH, Zhang X, Abel ED, Binkley PF, et al. Heart-specific ablation of Prkar1a causes failure of heart development and myxomagenesis. Circulation. 2008;117:1414–1422. doi: 10.1161/CIRCULATIONAHA.107.759233. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Ma Q, Juraszek AL, Moses K, Schwartz RJ, Izumo S, et al. Morphogenesis of the right ventricle requires myocardial expression of Gata4. J Clin Invest. 2005;115:1522–1531. doi: 10.1172/JCI23769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann B, Chiorini JA, Ma YL, Kotin RM, Herberg FW. Prkx is a novel catalytic subunit of the cAMP-dependent protein kinase regulated by the regulatory subunit type I. J Biol Chem. 1999;274:5370–5378. doi: 10.1074/jbc.274.9.5370. [DOI] [PubMed] [Google Scholar]