

Abstract
A-kinase anchoring protein (AKAP) 79/150 is a scaffold protein found in dendritic spines that recruits the cAMP-dependent protein kinase (PKA) and protein phosphatase 2B–calcineurin (CaN) to membrane-associated guanylate kinase (MAGUK)-linked AMPA receptors (AMPARs) to control receptor phosphorylation and synaptic plasticity. However, AKAP79/150 may also coordinate regulation of AMPAR activity with spine structure directly through MAGUK binding and membrane-cytoskeletal interactions of its N-terminal targeting domain. In cultured hippocampal neurons, we observed that rat AKAP150 expression was low early in development but then increased coincident with spine formation and maturation. Overexpression of human AKAP79 in immature or mature neurons increased the number of dendritic filopodia and spines and enlarged spine area. However, RNA interference knockdown of AKAP150 decreased dendritic spine area only in mature neurons. Importantly, AKAP79 overexpression in immature neurons increased AMPAR postsynaptic localization and activity. Neither the AKAP79 PKA nor CaN anchoring domain was required for increasing dendritic protrusion numbers, spine area, or AMPAR synaptic localization; however, an internal region identified as the MAGUK binding domain was found to be essential as shown by expression of a MAGUK binding mutant that formed mainly filopodia and decreased AMPAR synaptic localization and activity. Expression of the AKAP79 N-terminal targeting domain alone also increased filopodia numbers but not spine area. Overall, these results demonstrate a novel structural role for AKAP79/150 in which the N-terminal targeting domain induces dendritic filopodia and binding to MAGUKs promotes spine enlargement and AMPAR recruitment.
Introduction
Dendritic spines are minute protrusions on neuronal dendrites that mediate most excitatory synaptic connections in the brain (Yuste and Bonhoeffer, 2004; Matus, 2005). Spines contain a specialized structure below the postsynaptic membrane called the postsynaptic density (PSD) that is a network of scaffold and signaling proteins linked to NMDA receptors (NMDARs), AMPARs, and filamentous actin (F-actin) (Kim and Sheng, 2004; Tada and Sheng, 2006). Spine structure regulation is important for synaptic development and function, and spine abnormalities are characteristics of many human neurological and psychiatric disorders. In early development, synapses contain NMDARs but few AMPARs with filopodia and stubby spines predominating, whereas mature neurons contain mostly thin or large mushroom-shaped spines (Yuste and Bonhoeffer, 2004; Matus, 2005). Developmental maturation of spines is regulated by actin polymerization and recruitment of PSD scaffolds and AMPARs (Lamprecht and LeDoux, 2004; Yuste and Bonhoeffer, 2004; Matus, 2005; Tada and Sheng, 2006). Spine size correlates with AMPAR number such that increases in spine size and synaptic strength occur together during development (Matsuzaki et al., 2001). Importantly, NMDAR-dependent long-term potentiation (LTP) that increases synaptic AMPAR activity also rapidly stimulates new spine formation and existing spine enlargement, whereas long-term depression (LTD) that decreases synaptic AMPAR activity causes spine shrinkage and elimination (Lamprecht and LeDoux, 2004; Matsuzaki et al., 2004; Nägerl et al., 2004; Okamoto et al., 2004; Zhou et al., 2004).
PSD scaffold proteins play key roles in modifying synaptic structure and function. In particular, membrane-associated guanylate kinase (MAGUK) scaffold proteins regulate spine numbers, spine size, and AMPAR synaptic strength during development and plasticity (Migaud et al., 1998; El-Husseini et al., 2000; Schnell et al., 2002; Rumbaugh et al., 2003; Stein et al., 2003; Ehrlich and Malinow, 2004; Nakagawa et al., 2004b; Béïque et al., 2006; Elias et al., 2006; Schlüter et al., 2006; Cuthbert et al., 2007; Ehrlich et al., 2007; Kim et al., 2007; Xu et al., 2008). Another scaffold that binds MAGUKs and is implicated in AMPAR regulation during plasticity is A-kinase anchoring protein (AKAP) 79/150 (Colledge et al., 2000; Gomez et al., 2002; Tavalin et al., 2002; Smith et al., 2006; Lu et al., 2007, 2008; Tunquist et al., 2008). AKAP79/150 is localized to PSDs through its N-terminal targeting domain that binds phosphatidylinositol-4,5,-bisphosphate (PIP2), F-actin, and cadherin adhesion molecules (Dell'Acqua et al., 1998; Gomez et al., 2002; Gorski et al., 2005). AKAP79/150 anchors protein kinase A (PKA) and calcineurin (CaN) to promote regulation of neuronal AMPAR phosphorylation and activity during LTP and LTD (Tavalin et al., 2002; Hoshi et al., 2005; Lu et al., 2007, 2008; Tunquist et al., 2008). In particular, negative regulation of AKAP79/150 and AMPAR spine localization occurs concomitantly with AMPAR dephosphorylation, actin depolymerization, and spine shrinkage during LTD; therefore, AKAP79/150 may also control spine structural plasticity (Gomez et al., 2002; Gorski et al., 2005; Smith et al., 2006; Horne and Dell'Acqua, 2007). Here, we demonstrate that AKAP79/150 regulates dendritic spine formation and morphology in developing hippocampal neurons through its targeting and MAGUK binding domains but surprisingly not its PKA and CaN anchoring domains.
Materials and Methods
Mammalian cDNA expression vectors.
The pEC/G/YFPN1 (Clontech) vectors encoding C-terminal cyan/green/yellow fluorescent protein (C/G/YFP) fusions of PSD95, AKAP79 wild type (WT), and AKAP79 mutants (1–153, 1–315, Δ109–315, ΔCaN, and ΔPKA) were described previously (Dell'Acqua et al., 2002; Gomez et al., 2002; Oliveria et al., 2003; Gorski et al., 2005). pSilencer and pSilencer–sh150RNAi constructs were provided by John Scott (University of Washington, Seattle, WA) and have been characterized previously for efficient knockdown of rat AKAP150 while allowing rescue with human AKAP79 [not recognized by the short-hairpin RNA interference (shRNAi)] in cultured rat hippocampal neurons in our laboratory and others (Hoshi et al., 2005; Oliveria et al., 2007). For additional evidence showing effective knockdown of AKAP150 by pSilencer–sh150RNAi in both day in vitro 0 (DIV0)–DIV12 and DIV14–DIV16 neurons, see supplemental Figure 2 (available at www.jneurosci.org as supplemental material).
Primary culture and transfection of rat hippocampal neurons.
Neurons were cultured as described previously (Gomez et al., 2002; Smith et al., 2006). Briefly, the hippocampus was dissected from postnatal day 0 (P0) to P2 Sprague Dawley rats and dissociated in papain. For imaging and electrophysiology experiments, neurons were plated at medium density (150,000–225,000 cells/ml) on glass-bottom 35 mm plates or glass coverslips coated with poly-d-lysine and laminin (Discovery Labware; BD Biosciences). For immunoblotting analysis of total protein expression, neurons were plated at high density (500–750,000 cells/ml) and grown 4–20 d in vitro. For chronic Amaxa nucleofector transfection, P0–P2 neurons were resuspended at 600,000–900,000 cells per transfection, electroporated with 4–12 μg of each cDNA or the sh150RNAi construct, plated at medium density, and grown to DIV11–DIV13. For acute Lipofectamine 2000 transfection (Invitrogen), medium-density neurons grown to DIV14 were transfected with 4–12 μg of each cDNA and/or the sh150RNAi construct and then grown to DIV16.
Immunocytochemistry.
Cultured hippocampal neurons were fixed and immunostained as described previously (Smith et al., 2006). Briefly, cells were fixed in 3.7% formaldehyde/PBS, permeabilized in 0.2% Triton X-100 in PBS, and blocked overnight in PBS/10% BSA at 4°C. For immunostaining with NR1, cells were washed in cold PBS, followed by fixing in 4% paraformaldehyde at 4°C for 4 min. Cells were permeabilized using 100% methanol for 2 min at −20°C and incubated overnight in PBS/10% BSA at 4°C. Primary antibodies were incubated for 2 h at room temperature in PBS/10% BSA as follows: 1:1000 rabbit anti-AKAP150, 1:200 mouse anti-PSD95 (Affinity BioReagents), 1:100 rabbit anti-GluR1 (Millipore Bioscience Research Reagents), 1:100 rabbit anti-GluR2/3 (Millipore), 1:100 mouse anti-NR1 (BD Biosciences Pharmingen), 1:400 mouse anti-bassoon (Stressgen), and 1:500 rabbit anti-vesicular GABA transporter (vGAT) (Synaptic Systems). After incubation, cells were washed in PBS, followed by PBS/10% BSA and incubated with fluorescent secondary antibody conjugates [goat anti-rabbit- or goat anti-mouse Texas Red (1:250) or Alexa647 (1:500) (Invitrogen)] for 1 h at room temperature. Coverslips were washed in PBS/10% BSA, PBS, and water before mounting on glass slides in ProLong mounting media (Invitrogen).
Fluorescence microscopy and quantitative digital image analysis.
Imaging of neurons was performed on an inverted Zeiss Axiovert 200M with 100× plan-apo/1.4 numerical aperture objective, 175 W xenon illumination (Sutter Instruments), Coolsnap CCD camera (Princeton Instruments), and SlideBook version 4.0–4.2 software (Intelligent Imaging Innovations) as described previously (Gorski et al., 2005; Smith et al., 2006). Living neurons were maintained at 33°C and ∼5% CO2 during the entire imaging period. For detection of indirect immunofluorescence or direct fluorescence of C/G/YFP, three-dimensional z-stack images of x–y planes with 0.5 μm steps were collected. Images were deconvolved to the nearest neighbor to generate confocal x–y sections. Two-dimensional maximum intensity projection images were generated to better represent a complete picture of dendrites and spines and used for quantitative mask analysis (see below).
Quantification of fluorescent images was performed in SlideBook version 4.0. For dendritic spine area, masks were drawn to cover the entire spine head and exclude the spine shaft. Area in pixels was then determined for each spine head and converted to area in square micrometers. Dendritic protrusion (spines and filopodia) density was determined by using the ruler tool to measure a length of dendrite and count the number of dendritic protrusions in that length. Density was then expressed as a measure of protrusions per 10 μm of dendrite. For immunocytochemistry, a dendrite mask was generated by using the segment function to label the G/C/YFP transfected neuron and eliminate areas of the image outside of the transfected neuron. Each fluorescence channel was gated to produce a puncta mask using 1.5× the mean intensity in the dendrite mask, followed by use of the AND function to define puncta within the dendrite mask. Puncta were then separated manually using the eraser tool. For the synaptic colocalized puncta masks (GluR1/PSD95; GluR2/3/PSD95; NR1/GluR1), a new mask was generated after puncta separation using the AND function to combine the dendrite puncta masks of each channel. To determine puncta density, the ruler tool was used as described above, and the number of puncta was counted and expressed as a measure of puncta per 10 μm of dendrite. For all imaging experiments, multiple neurons from two to five separate cultures per condition were analyzed.
Hippocampal protein expression time course assays.
Samples were collected either in vivo by dissecting the whole hippocampus from Sprague Dawley rats at P0–P28 or in vitro by harvesting high-density hippocampal neuron cultures from 6 cm dishes at DIV4–DIV20. Samples were suspended in 1% SDS, 1 mm EDTA, and 10 mm Tris, pH 8, and sonicated. Protein concentrations were determined using the Bio-Rad protein assay kit. Proteins were resolved on Tris-SDS gels and transferred in 20% methanol to nitrocellulose. Primary antibodies were incubated with the membranes for a minimum of 90 min before detection with horseradish peroxidase-coupled secondary antibodies (Bio-Rad) followed by enhanced chemiluminescence (West Pico Chemiluminescent Substrate or West Dura Extended Duration Chemiluminescent Substrate; Pierce-Fisher). Anti-AKAP150 was a gift from Dr. Yvonne Lai (Icos, Bothel, WA). Anti-GluR1, GluR2/3, and tubulin antibodies were from Millipore, and anti-PSD95 was from Affinity BioReagents. Immunoreactivity was detected using an Alpha Innotech imaging system. The ratio of immunoreactivity observed was calculated using the AlphaEase software. After initial antibody detection, the membranes were stripped using Restore stripping buffer (Pierce-Fisher), followed by several washes in Tris-buffered saline plus Tween 20 and a single wash in blotto. A second set of antibodies was applied as described above. Semiquantitative Western blot analysis of protein concentration for each time point was performed as described previously (Smith et al., 2006). Briefly, a five-point standard curve of hippocampal total protein was included on each gel. The integrated density values for each antibody were plotted to generate a standard curve then used determine to unknown protein concentrations. Final values were expressed as a percentage of the protein concentration for the last time point.
Immunoprecipitation of AKAP79–MAGUK complexes from HEK293 cells.
Immunoprecipitations from HEK293 cells were performed as described previously (Gomez et al., 2002; Gorski et al., 2005). Briefly, HEK293 cells were transfected by calcium phosphate at 60–70% confluency with pEGFPN1–AKAP79WT or AKAP79 mutant cDNA with or without pGW–mycPSD95 cDNA (Colledge et al., 2000) at 5–10 μg/construct. Cells were solubilized in a 1% Triton X-100 lysis buffer and incubated overnight at 4°C with 10 μg of anti-GFP or rabbit IgG control. Immunoprecipitation was detected by immunoblotting for endogenous synapse-associated protein 97 (SAP97) using pan-anti-PSD–MAGUK (1:1000; Millipore) or mycPSD95 using 9E10–anti-myc (1:1000; Santa Cruz Biotechnology). Chemiluminescence was detected using an Alpha Innotech imager.
Recordings of miniature EPSCs.
Whole-cell voltage clamp recordings of spontaneous miniature EPSCs (mEPSCs) were performed at room temperature (20–25°C) at −70 mV holding potential. Electrodes were pulled on a horizontal puller (Sutter Instruments) to have resistance of 2–7 MΩ when filled with intracellular solution containing the following (in mm): 130 CsMeSO4, 20 CsCl, 10 HEPES, 0.5 EGTA, 2 Na2-ATP, 0.2 Na3-GTP, and 3 MgCl2, pH 7.25 with CsOH. Coverslips with plated neurons were placed in extracellular solution comprising the following (in mm): 150 NaCl, 3 KCl, 10 HEPES, 10 d-glucose, 0.001 TTX, 0.05 picrotoxin, 2 MgCl2, and 2 CaCl2, pH 7.4 with NaOH. Transfected neurons were identified by G/YFP fluorescence. Tight-seal (≥10 GΩ) whole-cell recordings of membrane currents were amplified and filtered (5 kHz) using an Axopatch 200 (Molecular Devices), digitized at 20 kHz (Digidata; Molecular Devices), and displayed on a personal computer running pClamp (Molecular Devices) with series resistance (<20 MΩ) monitoring; recordings with changes >10% were discarded. Offline detection and analysis of individual mEPSCs interevent interval (frequency) and peak current (amplitudes) were measured with MiniAnalysis Program (Synaptosoft), binned, averaged and plotted as cumulative probability distributions with purpose-written software; typically, 200–3000 events per neuron were analyzed. For these experiments, n is number of neurons, and two to five separate cultures per transfectant were used.
Statistical analysis.
Comparisons with control for group data was performed in Prism (GraphPad Software) using one-way ANOVA, followed by Dunnett's or Bonferroni's post hoc analysis or in Excel (Microsoft Corporation) or Prism for pairwise comparisons with control using Student's t test or Mann–Whitney test as indicated in figure legends. Data are expressed as mean ± SEM error bars in all figures. Cumulative distributions were compared using the Kolmogorov–Smirnoff (K–S) test as indicated in the figure legends. Significance is reported as p < 0.05.
Results
AKAP150 protein expression in hippocampal neurons increases in parallel with PSD95 and AMPAR expression during early postnatal development
AKAP79/150 is a PSD scaffold protein localized to dendritic spines that forms complexes with the NMDAR and AMPAR through binding to the C-terminal Src homology 3 (SH3) and guanylate kinase (GK) domains of MAGUK family scaffold proteins, including PSD95 and SAP97, which in turn link to glutamate receptors through their N-terminal postsynaptic density 95/Discs large/zona occuldens 1 (PDZ) domains (Colledge et al., 2000; Gomez et al., 2002). Human AKAP79 and its rodent ortholog AKAP150 are both protein products of the AKAP5 gene; these proteins are highly homologous other than insertion of an internal repetitive amino acid sequence of unknown function that is found only in rodents and not in other species, including humans. AKAP79/150 is the major AKAP isoform present in PSD fractions and plays a dominant role in controlling postsynaptic spine localization of PKA (Carr et al., 1992; Glantz et al., 1992; Smith et al., 2006; Lu et al., 2007; Tunquist et al., 2008). AKAP79/150–MAGUK complexes are thought to anchor and target PKA and CaN in spines to regulate AMPAR activity through phosphorylation of the GluR1 subunit and control of receptor trafficking (Coghlan et al., 1995; Colledge et al., 2000; Dell'Acqua et al., 2002; Tavalin et al., 2002; Hoshi et al., 2005; Snyder et al., 2005; Smith et al., 2006; Lu et al., 2007, 2008; Tunquist et al., 2008).
Although it is not known exactly how AKAP–MAGUK complexes initially form, AKAP150 and PSD95 begin to colocalize in cultured rat hippocampal pyramidal neurons as spinogenesis occurs between 1 and 2 weeks in culture (Gomez et al., 2002). Similarly, in rodent hippocampus in vivo, spinogenesis also begins ∼1 week after birth (Bonhoeffer and Yuste, 2002; Yuste and Bonhoeffer, 2004). To further evaluate the relationship between PSD95 and AKAP150 during development, we analyzed total protein expression levels in rat hippocampus from P0 to P28. We observed a relatively low level of AKAP150 expression from P0 to P8, followed by a significant increase in expression at P12 compared with P0, and then relatively stable elevated expression levels from P16 to P28 (Fig. 1A,B). This increasing pattern of AKAP150 expression during development approximately paralleled increases in expression observed for PSD95 and AMPAR GluR1 and GluR2/3 subunits between P12 and P28 (Fig. 1A,B). We next examined protein expression levels in cultured dissociated hippocampal neurons to determine whether in vitro development parallels in vivo development. AKAP150, PSD95, GluR1, and GluR2/3 protein expression levels were evaluated in hippocampal neurons grown from DIV4 to DIV20. Similar to the hippocampus in vivo, PSD95 and AKAP150 showed relatively low expression at DIV4–DIV8 and then significantly higher expression at DIV12–DIV20 compared with DIV4 (Fig. 1C,D). Both GluR1 and GluR2/3 expression as a percentage of DIV20 levels were already substantial at DIV4 (Fig. 1C,D). GluR1 then showed a significant increase in expression for DIV8–DIV20 compared with DIV4 levels, but GluR2/3 did not show any significant increases. Thus, AMPAR subunit expression during in vitro development appears to be somewhat accelerated compared with in vivo. Nonetheless, the parallel increases in AKAP150 and PSD95 expression during development observed here and the increase in AKAP150 colocalization with PSD95 during spinogenesis previously observed (Gomez et al., 2002) both suggest that AKAP150 may regulate spine and excitatory synapse development.
Figure 1.

Expression of AKAP150 increases in parallel with PSD95 and AMPAR subunits during early postnatal development in hippocampal neurons both in vivo and in vitro. A, Representative immunoblots illustrating an increase in in vivo protein expression levels of AKAP150, PSD95, GluR1, and GluR2/3 during postnatal development at P0–P28 in the rat hippocampus. Protein samples were taken from whole hippocampal homogenates with 5 μg of protein loaded for each time point. The same immunoblots were stripped and reprobed with anti-tubulin as a control for equal protein loading. B, Graph of immunoreactivity levels calculated from immunoblots as shown as in A normalized to P28 expression levels. For each time point, hippocampal tissue from two to four rats was analyzed in duplicate (P0–P20, n = 8; P24–P28, n = 4). *p < 0.05; **p < 0.01 by t test to P0. C, Representative immunoblots illustrating an increase in in vitro expression levels of AKAP150, PSD95, and GluR1 expression levels in cultured hippocampal neurons at DIV4–DIV20. GluR2/3 starts high at DIV4 and shows a nonsignificant increasing trend (p = 0.08) for DIV8–DIV20. Ten micrograms of protein was loaded for each time point. D, Graph of immunoreactivity levels calculated from immunoblots as in C normalized to DIV20 expression levels. For each time point, neurons from two separate culture preparations were analyzed in duplicate (n = 4). *p < 0.05; **p < 0.01 by t test to DIV4.
Regulation of dendritic spine development by AKAP79 overexpression in hippocampal neurons
As mentioned above, spines, including filopodial spine precursors, undergo dramatic changes in morphology during development, and this change is regulated by actin polymerization and recruitment of PSD proteins. Dynamic regulation of spine morphology also occurs during LTP and LTD; therefore, mechanisms regulating spinogenesis might also be important in synaptic plasticity. AKAP79/150 is an important modulator of NMDAR-dependent LTP and LTD, and its localization to spines and binding to MAGUKs is dynamically regulated coincident with dendritic spine structure in LTD (Gomez et al., 2002; Snyder et al., 2005; Smith et al., 2006; Horne and Dell'Acqua, 2007; Lu et al., 2007, 2008; Tunquist et al., 2008). We therefore wanted to determine whether AKAP79/150 can modulate dendritic spine development and morphology by overexpressing human AKAP79 in rat cultured hippocampal neurons early during spinogenesis (DIV0–DIV12) when there is low endogenous AKAP150 expression (Fig. 1C,D). Neurons were transfected at DIV0 with equal amounts of CFP-tagged AKAP79WT, various AKAP79 mutants, or CFP alone (negative control) with YFP to fill the cytoplasm of dendrites and spines (Fig. 2A). Previous work by others showed that overexpression of PSD95 during this time period leads to an increase in spine number and area (El-Husseini et al., 2000; Prange et al., 2004); therefore, expression of CFP-tagged PSD95 with YFP filler was included as a positive control (Fig. 2A). Using fluorescence microscopy, CFP and YFP were imaged in dendrites of living transfected neurons at DIV11–DIV13 (Fig. 2B) (for larger images of multiple dendrites, see supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and evaluated for dendritic protrusion density and spine head area, including both spines with well formed heads and filopodia in protrusion density counts (Fig. 2C–G). We found that, relative to CFP/YFP controls, both AKAP79WT and PSD95 increased protrusion density per 10 μm of dendrite (Fig. 2B,G) and enlarged spines as seen in cumulative probability plots shifted toward larger spine areas (Fig. 2C) and increased mean spine area values (Fig. 2F).
Figure 2.

Overexpression of AKAP79 in developing DIV12 cultured hippocampal neurons increases dendritic spine size and protrusion density independent of its PKA and CaN anchoring functions. A, Diagram of CFP-tagged AKAP79 WT and deletion mutants expressed in cultured hippocampal neurons for analysis of spine regulation. PSD95–CFP served as a positive control, whereas CFP alone served as a negative control. B, Representative images of dendrites from living cultured hippocampal neurons transfected at DIV0 and imaged at DIV12 expressing the indicated CFP-tagged AKAP79 constructs in A (blue) along with YFP as a cytoplasmic filler (green). Scale bar, 10 μm. C–E, Cumulative distribution plots of the areas of spine heads (in square micrometers) for the indicated constructs analyzed in B showing that neither the PKA or CaN anchoring domains were required for AKAP79-induced spine enlargement, but the region between residues 153 and 315 was required. F, Graph of mean spine areas (in square micrometers) for PSD95 (0.5 ± 0.02; n = 384 spines on 23 neurons), AKAP79WT (0.78 ± 0.02; n = 2507 spines on 58 neurons), ΔCaN (0.69 ± 0.02; n = 948 spines on 19 neurons), ΔPKA (0.65 ± 0.02; n = 636 spines on 24 neurons), and 1–315 (0.69 ± 0.02; n = 1058 spines on 19 neurons) showing increased spine area when compared with Y/CFP control (0.36 ± 0.01; n = 1321 spines on 39 neurons), with 1–153 (0.36 ± 0.01; n = 657 spines on 23 neurons) having no effect on spine size. G, Graph of dendritic protrusion density measured as number of filopodia and spines per 10 μm of dendrite showing that PSD95 (8.7 ± 0.4; n = 57 lengths on 13 neurons), AKAP79 (10.8 ± 0.2; n = 206 lengths on 49 neurons), ΔCaN (10.7 ± 0.2; n = 148 lengths on 29 neurons), ΔPKA (10.8 ± 0.2; n = 169 lengths on 25 neurons), 1–315 (10.5 ± 0.3; n = 135 lengths on 19 neurons, and 1–153 (10.2 ± 0.3; n = 143 lengths on 23 neurons) increased protrusion density when compared with Y/CFP control (6.3 ± 0.2; n = 219 lengths on 43 neurons). In F and G, **p < 0.01 by ANOVA, and data are graphed as mean ± SEM.
Next, we determined what AKAP79 domains were responsible for the observed increases in dendritic spine area and protrusion number. We began by imaging AKAP79 mutants with deletions of the PKA binding site (ΔPKA), the CaN binding site (ΔCaN), both the PKA and CaN binding sites (1–315), and the targeting domain alone (1–153) (Fig. 2A,B). When compared with the C/YFP negative control, the ΔPKA and ΔCaN constructs still showed a significant increase in both dendritic spine area (Fig. 2D,F) and protrusion density (Fig. 2G). This suggested that effects of AKAP79 overexpression on dendritic spines did not require PKA or CaN anchoring. Interestingly, the targeting domain alone (1–153) was sufficient to increase protrusion density (Fig. 2G) but did not increase spine area when compared with controls (Fig. 2E,F). We also noticed that the additional protrusions on neurons expressing (1–153) were mostly filopodial, with few spines showing development of large heads (Fig. 2B). These results indicate that the ability of AKAP79 to increase spine area is located in a region between the end of the targeting domain (153) and the beginning of the CaN binding site (315) that may be involved in MAGUK binding (Tavalin et al., 2002; Oliveria et al., 2003). To extend this analysis, we expressed 1–315, a construct that contains this potential MAGUK binding site but lacks PKA and CaN binding (Fig. 2A), and observed an increase in dendritic spine area and protrusion density similar to AKAP79WT (Fig. 2B,E–G), further implicating the potential MAGUK binding domain as the region of AKAP79/150 responsible for increasing spine size.
Identification of the MAGUK binding domain on AKAP79/150 that is required for regulation of dendritic spine size
To more directly test the hypothesis that the AKAP–MAGUK interaction is important for spine size regulation, we decided to characterize an AKAP79Δ109–315 deletion mutant that removes the potential MAGUK binding site while maintaining N-terminal membrane targeting and C-terminal PKA and CaN anchoring functions (Dell'Acqua et al., 1998, 2002). To this end, the MAGUK binding activities of GFP-tagged AKAP79WT, 1–315, 1–153, Δ109–315, and GFP alone (negative control) were tested by coimmunoprecipitation of endogenous SAP97 (Fig. 3A) or transfected myc-tagged PSD95 (Fig. 3B) from HEK293 cells. Using an antibody against GFP, AKAP79–GFP complexes were immunoprecipitated and immunoblotted to detect SAP97 or mycPSD95 binding. Our results showed that AKAP79WT and 1–315 coimmunoprecipitated SAP97 and mycPSD95, whereas the targeting domain 1–153 and internal deletion mutant Δ109–315 did not (Fig. 3A,B). Neither GFP alone nor nonspecific rabbit IgG immunoprecipitated SAP97 (Fig. 3A) or mycPSD95 (Fig. 3B), further demonstrating specificity.
Figure 3.

AKAP79 residues 153–315 are required for MAGUK binding. Immunoprecipitation (IP) of whole-cell extracts from HEK293 expressing the indicated GFP-tagged AKAP79 WT and mutants or GFP alone (negative control) using anti-GFP (specific) or nonimmune IgG (nonspecific) as indicated. A, Immunoblotting (IB) for SAP97 and AKAP79 (A) or mycPSD95 and AKAP79–GFP (B) showed specific coimmunoprecipitation of endogenous SAP97 or mycPSD95 with AKAP79 constructs containing residues 153–315 but not with those constructs lacking this region. Note that, in B, 1–153–GFP migrates at the same molecular weight as the IgG heavy chain of the precipitating anti-GFP antibody [indicated by (IgG)]; thus, it is difficult to see the precipitating 1–153–GFP in the anti-GFP IP lane.
These biochemical findings indicate that the AKAP79 153–315 region that is important for regulating spine enlargement is also required for MAGUK binding. To confirm that MAGUK binding is important in regulating spine area, we expressed Y/GFP-tagged AKAP79WT, Δ109–315 that lacks MAGUK binding, or YFP/CFP alone as the negative control in cultured hippocampal neurons (Fig. 4A,B) (for larger images of dendrites, see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In contrast to 1–315, Δ109–315 showed an increase in protrusion density (Fig. 4F) but failed to increase spine area as dramatically as AKAP79WT (Fig. 4C,E) and produced mainly filopodia (Fig. 4B), further implicating the MAGUK binding domain in regulation of spine area.
Figure 4.

An AKAP79 internal deletion mutant that lacks the MAGUK binding domain increases protrusion density but does not increase dendritic spine area in DIV12 hippocampal neurons. A, Diagram of YFP-tagged AKAP79WT and the Δ109–315 MAGUK binding site deletion mutant expressed in cultured hippocampal neurons for analysis of spine regulation along with knockdown of endogenous rat AKAP150 by RNAi. YFP/CFP alone and the pSilencer RNAi empty vector served as negative controls. B, Representative images of dendrites from living cultured hippocampal neurons transfected at DIV0 and imaged at DIV12 expressing the indicated YFP-tagged AKAP79 constructs in A (green) along with CFP or YFP as a cytoplasmic filler (blue). Scale bar, 10 μm. C, Cumulative distribution plots of the areas of spine heads (in square micrometers) for the indicated conditions analyzed in B showing that the MAGUK binding region of AKAP79 that is deleted in the Δ109–315 construct was required for AKAP79-induced spine enlargement; D, AKAP150RNAi had no effect in DIV0–D12 neurons. E, Graph of mean spine area (in square micrometers) for Δ109–315 (0.43 ± 0.01; n = 842 spines on 16 neurons) showing only a very small enlargement of spine area compared with AKAP79WT (from Fig. 2) and Y/CFP control (from Fig. 2). AKAP150RNAi (0.33 ± 0.02; n = 496 spines on 11 neurons) and the pSilencer RNAi empty vector control (0.38 ± 0.01; n = 600 spines on 12 neurons) had no effect on spine area. F, Graph of dendritic protrusion density measured as number of filopodia and spines per 10 μm of dendrite showing that AKAP79 (from Fig. 2) and Δ109–315 (10.2 ± 0.3; n = 136 lengths on 29 neurons) increased protrusion density when compared with Y/CFP control (from Fig. 2). Neither AKAP150 knockdown by RNAi (6.4 ± 0.2; n = 89 lengths on 12 neurons) nor pSilencer RNAi empty vector control (6.5 ± 0.2; n = 105 lengths on 15 neurons) had any effect on protrusion density. In E and F, *p < 0.05, **p < 0.01 by ANOVA, and data are graphed as mean ± SEM.
Knockdown of endogenous AKAP150 expression in hippocampal neurons decreases dendritic spine size later but not early in spine development
Finally, we evaluated the effect of chronic knockdown of endogenous AKAP150 on dendritic spine area and protrusion density in immature developing DIV0–DIV12 hippocampal neurons using a previously characterized AKAP150shRNAi plasmid transfected with YFP (Hoshi et al., 2005; Oliveria et al., 2007). Importantly, as published previously, YFP-positive DIV12 neurons cotransfected on DIV0 with AKAP150shRNAi consistently showed greatly reduced AKAP150 dendritic staining compared with neighboring untransfected neurons (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). AKAP150RNAi expressed during DIV0–DIV12 had no effect on dendritic spine area or protrusion density compared with control neurons expressing YFP alone or pSilencer vector with YFP (Fig. 4B,D–F). This lack of effect of AKAP150RNAi on early spine development from DIV0 to DIV12 may be explained by our observation above that AKAP150 is expressed at relatively low levels before DIV12 and, thus, may not be essential for spine formation but be more important for regulating spine maturation and morphology later in development at DIV16–DIV20 when its expression levels are higher (Fig. 1C,D). Consistent with this predicted role for AKAP150 in spine maturation, neurons transfected on DIV14 with YFP and AKAP150shRNAi to acutely knockdown AKAP150 from DIV14 to DIV16 (Fig. 5A) (for AKAP150 staining confirming knockdown, see supplemental Fig. 2B, available at www.jneurosci.org as supplemental material), exhibited a reduction in dendritic spine area (Fig. 5B,C) with no significant change in protrusion numbers (Fig. 5D) compared with control neurons transfected with pSilencer vector and YFP. As an additional control for RNAi specificity, acute expression of AKAP79–YFP, which is resistant to knockdown by AKAP150shRNAi (Hoshi et al., 2005; Oliveria et al., 2007) not only rescued the decrease in spine area seen with RNAi knockdown of AKAP150 in DIV14–DIV16 neurons (Fig. 5B,C) but also increased both spine area (Fig. 5B,C) and protrusion density (Fig. 5D) relative to control neurons. However, these positive effects of acute AKAP79 overexpression on spine size and protrusion numbers in the more mature DIV14–DIV16 neurons were not as large as seen with chronic overexpression in the immature DIV0–DIV12 neurons. These differences could be related to the different developmental ages of the neurons as well as the different time periods of overexpression (2 vs 12 d). Nonetheless, these overexpression results along with the RNAi results suggest that AKAP79/150 is a positive regulator of spine maturation.
Figure 5.

Knockdown of AKAP150 expression by RNAi decreases dendritic spine area in more mature DIV16 cultured hippocampal neurons. A, Representative images of dendrites from living cultured hippocampal neurons transfected at DIV14 and imaged at DIV16 expressing (green) pSilencer RNAi empty vector with YFP (control), AKAP150RNAi with YFP, or AKAP150RNAi with AKAP79WT–YFP. AKAP79WT–YFP was also coexpressed with CFP as cytoplasmic filler (data not shown). Top panels show larger images of multiple dendrites. Bottom panels show magnifications of single dendrites. Scale bars, 10 μm. B, Cumulative distribution plots of the areas of spine heads (in square micrometers) for the indicated conditions analyzed in A showing that AKAP150RNAi decreases spine area, whereas AKAP79WT rescue expression increases spine area compared with the pSilencer control in DIV14–DIV16 neurons. C, Graph of mean spine area (in square micrometers) showing increased spine size for AKAP150RNAi with AKAP79WT (0.67 ± 0.02; n = 874 spines on 14 neurons) and decreased spine size for AKAP150RNAi (0.38 ± 0.01; n = 684 spines on 10 neurons) compared with pSilencer control (0.55 ± 0.01; n = 603 spines on 10 neurons). D, Graph of dendritic protrusion density measured as number of filopodia and spines per 10 μm of dendrite showing that AKAP150RNAi (4.8 ± 0.3; n = 639 lengths on 40 neurons) had no influence on protrusion density compared with pSilencer control (4.8 ± 0.3; n = 619 lengths on 40 neurons), whereas AKAP79WT with AKAP150RNAi led to an increase (6.3 ± 0.3; n = 488 lengths on 41 neurons). **p < 0.01 by ANOVA compared with pSilencer control; ++p < 0.01 by ANOVA compared with AKAP150RNAi. Data are graphed as mean ± SEM.
Regulation of AMPAR recruitment to synapses by AKAP79 expression in hippocampal neurons
As mentioned previously, normal postsynaptic development involves recruitment of F-actin and AMPARs to excitatory synapses containing NMDARs, leading to maturation of dendritic spines and increased synaptic strength (Matus, 2000; Matsuzaki et al., 2001; Lamprecht and LeDoux, 2004). Thus, it is possible that the increase in spine area caused by AKAP79 overexpression may lead to increased AMPAR recruitment and synaptic strength. Accordingly, we performed immunocytochemistry to evaluate effects of chronic AKAP79 expression on AMPAR localization in DIV0–DIV12 transfected neurons. We also evaluated the effect of AKAP79 on PSD95 localization to determine whether the change in spine area and dendritic protrusion number is regulated through increased PSD95 clustering. Our evaluation of AMPAR localization was limited to AKAP79WT, 1–315 that lacks PKA and CaN anchoring but contains MAGUK binding, and Δ109–315 that lacks MAGUK binding but contains PKA and CaN anchoring, compared with YFP alone controls, because these constructs resulted in the most interesting effects on dendritic spine area and protrusion density observed above. AKAP150RNAi was also evaluated as an additional condition. First, the effect of AKAP79 on AMPAR total puncta density was evaluated in neurons immunostained for GluR1 (Fig. 6A) (for larger images of dendrites, see supplemental Fig. 3, available at www.jneurosci.org as supplemental material) or GluR2/3 (Fig. 6B). We found that AKAP79WT and 1–315 increased total GluR1 puncta density, whereas only 1–315 increased GluR2/3 total puncta density along dendrites compared with YFP alone controls (Fig. 6C). In contrast, Δ109–315 had no effect on total GluR1 puncta density but slightly decreased total GluR2/3 total puncta density (Fig. 6C). Consistent with the lack of effect of spine size and density observed above at DIV12, knockdown of endogenous AKAP150 during DIV0–DIV12 had no effect on either GluR1 or GluR2/3 total puncta density when compared with YFP control (Fig. 6C).
Figure 6.

AKAP79 increases AMPA receptor synaptic localization through its MAGUK binding domain. A, Representative images of dendrites from DIV12 neurons expressing the indicated AKAP79–YFP constructs, AKAP150RNAi, or YFP alone (green) immunostained for and PSD95 (blue) and GluR1 (red) or GluR2/3 (red) (B). In A and B, colocalization of GluR subunits with PSD95 can be seen as pink–purple in the merge images. Scale bars, 10 μm. C, Graph of total GluR1 and GluR2/3 puncta density per 10 μm of dendrite length. AKAP79WT increased total GluR1 (10.6 ± 0.3; n = 205 lengths on 40 neurons) with no effect on total GluR2/3 puncta density (11.0 ± 0.4; n = 147 lengths on 37 neurons) when compared with YFP controls (GluR1, 9.0 ± 0.3, n = 150 lengths on 30 neurons; GluR2/3, 10.4 ± 0.4, n = 139 lengths on 28 neurons). 1–315 increased both total GluR1 (10.4 ± 0.4; n = 116 lengths on 18 neurons) and GluR2/3 (12.2 ± 0.6; n = 109 lengths on 18 neurons). Δ109–315 had no effect on total GluR1 (9.1 ± 0.4; n = 114 lengths on 16 neurons) and showed a slight decrease in GluR2/3 (8.5 ± 0.5; n = 110 lengths on 18 neurons). AKAP150RNAi (transfected with YFP) had no effect on total GluR1 (9.1 ± 0.3; n = 129 lengths on 18 neurons) or GluR2/3 (10.9 ± 0.6; n = 96 lengths on 18 neurons). D, Graph of PSD95 colocalized synaptic GluR1 and GluR2/3 puncta density per 10 μm of dendrite length. AKAP79WT increased synaptic GluR1 (8.6 ± 0.3; n = 205 lengths on 40 neurons) and GluR2/3 (6.7 ± 0.3; n = 147 lengths on 37 neurons) puncta density when compared with YFP controls (GluR1, 6.7 ± 0.3, n = 150 lengths on 30 neurons; GluR2/3, 5.9 ± 0.3; n = 139 lengths on 28 neurons). 1–315 also increased synaptic GluR1 (8.7 ± 0.4; n = 116 lengths on 18 neurons) and GluR2/3 (6.9 ± 0.3; n = 109 lengths on 18 neurons). Δ109–315 had no effect on synaptic GluR1 (7.0 ± 0.4; n = 114 lengths on 16 neurons) and decreased synaptic GluR2/3 (5.0 ± 0.4; n = 110 lengths on 18 neurons). AKAP150RNAi had no effect on synaptic GluR1 (7.0 ± 0.3; n = 129 lengths on 18 neurons) or GluR2/3 (5.7 ± 0.3; n = 96 lengths on 18 neurons). E, Graph of total PSD95 puncta per 10 μm showing that AKAP79 increased PSD95 puncta density (13.4 ± 0.3; n = 352 lengths on 77 neurons) compared with YFP control (12.2 ± 0.3; n = 289 lengths on 58 neurons). 1–315 (13.0 ± 0.4; n = 225 lengths on 36 neurons), Δ109–315 (11.5 ± 0.4; n = 224 lengths on 34 neurons), and AKAP150RNAi (12.7 ± 0.3; n = 225 lengths on 36 neurons) had no significant effects. In C–E, *p < 0.05, **p < 0.01 by ANOVA and #p < 0.05 by t test, and data are graphed as mean ± SEM.
Next, we evaluated the effect of AKAP overexpression on AMPAR synaptic puncta density as defined by colocalization with PSD95 puncta. AKAP79 and 1–315 significantly increased GluR1 synaptic puncta density, whereas Δ109–315 had no effect compared with YFP controls (Fig. 6D). Both 1–315 and AKAP79 also increased, whereas Δ109–315 slightly decreased, GluR2/3 synaptic puncta density, although these changes for GluR2/3 were not as large as those seen for GluR1 with AKAP79 and 1–315 (Fig. 6D). AKAP150RNAi had no impact on synaptic GluR1 or GluR2/3 localization (Fig. 6D). These results indicate that the differential effects of AKAP79WT and 1–315 compared with Δ109–315 on spine area correlate with differential effects on AMPAR localization as well. We also evaluated the effect of AKAP79 expression on PSD95 puncta density. Overexpression of AKAP79 slightly increased total PSD95 puncta density along dendrites, whereas 1–315 and Δ109–315 did not show any significant changes in PSD95 puncta density when compared with control. These results suggest that changes in PSD95 localization are unlikely to account for all of the effects of AKAP79 expression on spine area and AMPAR localization (Fig. 6E). Finally, consistent with the AMPAR staining results, AKAP150 RNAi from DIV0 to DIV12 had no effect on PSD95 puncta density compared with control (Fig. 6E).
Regulation of AMPAR synaptic strength by AKAP79 expression in hippocampal neurons
AMPAR localization in spines can be within the synaptic cleft, extrasynaptic, or contained within internal vesicles. Only AMPARs located within the synaptic cleft are activated during synaptic transmission, whereas AMPARs located in extrasynaptic regions are thought to laterally diffuse in and out of the synaptic cleft, and AMPARs contained within internal vesicles are inserted into the membrane as part of normal receptor cycling and turnover and in response to LTP induction (Schnell et al., 2002; Park et al., 2004; Ashby et al., 2006; Boehm et al., 2006; Brown et al., 2007; Ehlers et al., 2007; Yudowski et al., 2007). Although our immunostaining data showed that overexpression of AKAP79 constructs altered AMPAR localization, it could not tell us whether there was also an impact on the number of active AMPARs within the synaptic cleft.
To evaluate AMPAR function in neurons overexpressing AKAP79 constructs, we recorded mEPSCs. Spontaneous AMPAR-mediated mEPSCs were recorded from neurons transfected with YFP-tagged AKAP79WT, Δ109–315, 1–315, or YFP alone (Fig. 7A). Our results showed that AKAP79WT and 1–315 transfected neurons increased mEPSC amplitudes (Fig. 7A,B,D) with no or little effect, respectively, on mEPSC frequency (Fig. 7A,C,E) when compared with neurons expressing YFP alone. In contrast, Δ109–315-expressing cells showed no change in amplitude (Fig. 7A,B,D) but exhibited a dramatic decrease in frequency (Fig. 7A,C,E). These results suggest that the ability of AKAP79 and 1–315 to increase dendritic spine area correlates with increased AMPAR postsynaptic strength. In contrast, the Δ109–315 mutant greatly reduced mEPSC frequency, indicating a probable loss of active synapses relative to the YFP alone control.
Figure 7.

AKAP79 increases AMPA receptor synaptic function through its MAGUK binding domain. A, Sample mEPSC traces recorded from DIV10–DIV12 cultured hippocampal neurons expressing YFP control, AKAP79WT, 1–315, and Δ109–315 reveal increased mEPSC amplitudes for AKAP79WT and 1–315 and decreased mEPSC frequency for Δ109–315. Calibration: top four traces, 40 pA, 100 ms; bottom trace, for Δ109–315, 40 pA, 500 ms to better show decreased frequency but no apparent changes in amplitudes. Cumulative distribution plots of mEPSC amplitudes (picoamperes) (B) and frequency (interevent interval in seconds) (C) for YFP control (yellow; n = 14 neurons), AKAP79WT (black; n = 11 neurons), 1–315 (red; n = 6 neurons), and Δ109–315 (green; n = 7 neurons) measured from transfected hippocampal pyramidal neurons. Analysis by K–S test showed that AKAP79 (p = 1.22 × 10−14) and 1–315 (p < 1.53 × 10−38) significantly increased mEPSC amplitude compared with YFP control cells. Δ109–315 (p = 0.725) had no effect on mEPSC amplitude when compared with control. Analysis of mEPSC frequency by K–S test showed that AKAP79 (p = 0.128) had no effect when compared with YFP control. 1–315 (p = 1.24 × 10−4) showed a very slight but significant decrease in frequency, whereas Δ109–315 (p = 1.44 × 10−25) caused a very large and significant decrease in mEPSC frequency. D, Graph of mean mEPSC amplitudes showing that AKAP79 (15.5 ± 1.9) and 1–315 (15.1 ± 2.5) significantly increased mEPSC amplitude compared with YFP control (11.4 ± 0.8), whereas Δ109–315 (10.2 ± 0.9) had no effect. E, Graph of mean mEPSC frequency showing that AKAP79 (3.3 ± 2.7) and 1–315 (4.4 ± 1.7) had no significant effect when compared with YFP control (2.5 ± 1.2), whereas Δ109–315 (8.2 ± 2.8) showed a significant decrease in frequency. Statistics for D and E, *p < 0.05 by both t test and Mann–Whitney test. Data are graphed as mean ± SEM.
AKAP79 expression does not alter the number or balance of excitatory versus inhibitory presynaptic terminals in hippocampal neurons
Although changes in AMPAR mEPSC amplitudes are usually indicative of postsynaptic modifications, effects on frequency can be either presynaptic or postsynaptic. Thus, we wanted to investigate how Δ109–315 caused a decrease in mEPSC frequency. First, we evaluated the AKAP79 constructs for changes in the number of presynaptic terminals making synapses on dendrites of the transfected neurons using the generic presynaptic marker bassoon and found that none of the AKAP79 constructs had any effect on the total number of presynaptic terminals (Fig. 8A,C). As an additional control, we evaluated whether AKAP79 had any effect on inhibitory synapses identified by staining for the vGAT. Others have shown that overexpression of PSD95 increases the number of excitatory synapses at the expense of inhibitory synapses by altering neuroligin localization through binding to its N-terminal PDZ domains. This PSD95 retrograde communication via neuroligins involves coordinated presynaptic and postsynaptic changes, leading to a large increase in mEPSC frequency and a decrease in the frequency of inhibitory synaptic events (El-Husseini et al., 2000; Prange et al., 2004; Levinson et al., 2005). Although AKAP79 also predominantly localizes to excitatory synapses and interacts with PSD95, no increase in mEPSC frequency was observed for any AKAP79 construct; therefore, changes in inhibitory synapses were not likely to be occurring for AKAP79. Consistent with this prediction, no changes in the density of vGAT-stained synaptic terminals were observed in AKAP79WT-, 1–315-, or Δ109–315-expressing neurons when compared with YFP control (Fig. 8B,D). These results indicate that, whereas AKAP79 regulation of AMPAR localization and activity depends on MAGUK binding, AKAP79 does not use the same mechanisms as PSD95 to enhance AMPAR postsynaptic function. Indeed, the positive effects of PSD95 on presynaptic development and mEPSC frequency only required the N-terminal PDZ domains of PSD95 and not the SH3–GK domains in which AKAP79 binds (El-Husseini et al., 2000; Prange et al., 2004; Levinson et al., 2005).
Figure 8.

An AKAP79 mutant deficient in MAGUK binding increases the number of morphologically silent synapses. A, Representative images of dendrites from DIV12 neurons expressing the indicated AKAP79–YFP constructs or YFP only control (green) and immunostained for bassoon (pink). The localization of many of the bassoon-labeled presynaptic terminals on AKAP–YFP-filled spines can be seen as white in the composite images. B, Representative images of dendrites expressing the indicated AKAP79–YFP constructs or YFP control (green) immunostained for vGAT (pink). Consistent with the majority of inhibitory synapses being located on dendrite shafts and not spines, very little overlap (white) is seen for AKAP–YFP and vGAT fluorescence in the composite images. C, Graph of bassoon puncta per 10 μm showed no difference between AKAP79 (16.2 ± 0.6; n = 102 lengths on 13 neurons), 1–315 (16.0 ± 0.6; n = 100 lengths on 11 neurons), Δ109–315 (16.0 ± 0.6; n = 100 lengths on 12 neurons), and YFP control (16.0 ± 0.5; n = 104 lengths on 12 neurons). D, Graph of vGAT puncta per 10 μm showed no difference between AKAP79 (10.4 ± 0.4; n = 105 lengths on 12 neurons), 1–315 (9.8 ± 0.4; n = 100 lengths on 12 neurons), Δ109–315 (10.3 ± 0.4; n = 100 lengths on 11 neurons), and YFP control (9.4 ± 0.4; n = 100 lengths on 12 neurons). E, Representative images of dendrites expressing the indicated AKAP79–YFP constructs or YFP control (green) immunostained for GluR1 (red) and NR1 (blue). Colocalization of NR1 with GluR1 can be seen as pink–purple in the merge images. Scale bars: A, B, E, 10 μm. F, Graph of total NR1 puncta per 10 μm showed that AKAP79 (16.5 ± 0.6; n = 118 lengths on 15 neurons) and 1–315 (16.9 ± 0.6; n = 99 lengths on 14 neurons) increased total NR1 puncta density compared with YFP control (13.7 ± 0.6; n = 99 lengths on 10 neurons). Δ109–315 (12.7 ± 0.5; n = 145 lengths on 12 neurons) showed no change. G, Graph of NR1 puncta colocalized with GluR1 puncta per 10 μm showed that AKAP79 (8.3 ± 0.4; n = 118 lengths on 15 neurons) and 1–315 (8.4 ± 0.5; n = 99 lengths on 14 neurons) increased NR1/GluR1 colocalized puncta density when compared with YFP control (7.0 ± 0.5; n = 99 lengths on 10 neurons). Δ109–315 (5.5 ± 0.3; n = 145 lengths on 12 neurons) showed a decrease in NR1/GluR1 puncta density. In C,D and F,G, *p < 0.05, **p < 0.01 by ANOVA, and data are graphed as mean ± SEM.
Regulation of morphologically silent synapse numbers by AKAP79 expression in hippocampal neurons
The results from analysis of presynaptic terminal densities suggested that the mechanism for Δ109–315 decreasing mEPSC frequency may be related to postsynaptic alterations instead. One possibility is that Δ109–315 decreases mEPSC frequency through postsynaptic silencing resulting from an increase in morphologically silent synapses that contain NMDARs with no AMPARs. Synapses containing only NMDARs are immature and functionally silent because they are incapable of responding to presynaptic release of glutamate attributable to insufficient removal of the Mg2+ block on NMDARs (Gomperts et al., 1998; Liao et al., 1999, 2001; Voronin and Cherubini, 2004). We therefore costained neurons expressing AKAP79 constructs for the NR1 subunit of the NMDAR and GluR1. Consistent with our previous observation of increased GluR1 localization with PSD95 and increased postsynaptic strength, both AKAP79WT and 1–315 constructs not only increased total NR1 puncta density but also increased the density of NR1/GluR1 colocalized puncta compared with YFP controls (Fig. 8E–G). In contrast, Δ109–315 showed no change in total NR1 puncta numbers and exhibited a decrease in the number of NR1/GluR1 colocalized puncta, indicating an increase in the number of morphologically silent synapses in these neurons, which could contribute to the observed decrease in mEPSC frequency (Fig. 8E–G).
Discussion
Our previous studies demonstrated that AKAP79/150 is targeted to spines through interactions with structural components of the postsynaptic membrane and cytoskeleton, including PIP2 (Dell'Acqua et al., 1998), F-actin (Gomez et al., 2002), cadherins (Gorski et al., 2005), and MAGUKs (Colledge et al., 2000; Gomez et al., 2002). Furthermore, we reported that localization of AKAP79/150–PKA complexes to the PSD and associations with MAGUKs and cadherins are disrupted during NMDAR LTD through CaN and phospholipase C (PLC) signaling pathways that lead to PIP2 cleavage and actin depolymerization in spines (Gomez et al., 2002; Gorski et al., 2005; Smith et al., 2006; Horne and Dell'Acqua, 2007). These AKAP79/150 targeting studies suggested that this protein could play a structural role in spines; however, until now, AKAP79/150 had not been directly implicated in controlling spine structure. Here we show in neonatal cultured hippocampal neurons that AKAP79 overexpression increases dendritic protrusion numbers and spine size. Based on our currents results, we propose a model (Fig. 9) in which the AKAP79 targeting domain (1–153) is sufficient to increase dendritic protrusions that are mainly filopodia and do not form active synapse, whereas the MAGUK binding domain is required for maturation of dendritic protrusions into large, spinous synapses with increased synaptic AMPARs. Importantly, overexpression of an AKAP79 Δ109–315 construct that deletes the MAGUK binding region acts as a dominant negative by preventing normal synapse maturation through a reduction in AMPAR synaptic localization and an increase in NMDAR only silent synapses.
Figure 9.
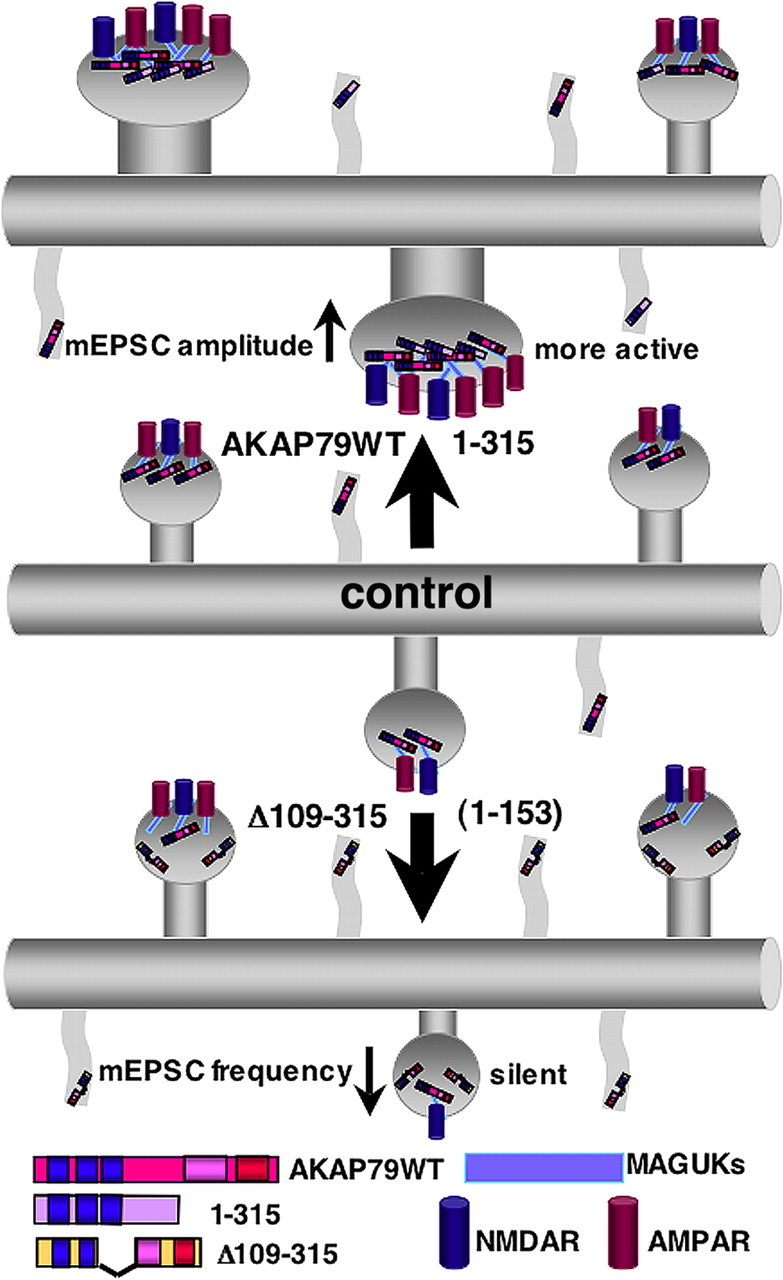
Model for regulation of dendritic spine structure and function by AKAP79/150 binding to MAGUKs. Diagrams of dendrites showing increases in protrusion density, dendritic spine area, AMPAR synaptic localization with MAGUKs and NMDARs, and increased mEPSC amplitude with overexpression of AKAP79WT or 1–315 that bind MAGUK proteins (top) compared with a control dendrite (middle). Alternatively, overexpression of AKAP constructs that lack MAGUK binding such as Δ109–315 and the N-terminal targeting domain (1–153) alone increase protrusion density with no change in dendritic spine area (bottom). Additionally, Δ109–315 decreases synaptic AMPAR localization and mEPSC frequency by increasing the number of NMDAR only silent synapses.
The targeting domain of AKAP79/150 uses three polybasic domains to interact with PIP2, F-actin, and cadherins and is sufficient for dendritic plasma membrane and spine localization (Gomez et al., 2002; Gorski et al., 2005; Horne and Dell'Acqua, 2007). Many other proteins that bind actin, such as cortactin, α-actinin, drebrin, neurabin, spinophilin, and myosin, have also been shown to regulate spine morphology and numbers (Hering and Sheng, 2003; Takahashi et al., 2003; Nakagawa et al., 2004a; Zito et al., 2004; Osterweil et al., 2005; Terry-Lorenzo et al., 2005; Ryu et al., 2006; Schüler and Peti, 2008). In addition, a number of postsynaptic adhesion molecules, such as ephrins, neuroligins, and cadherins, control actin-dependent flipodial dynamics, spine morphology, and synapse development (Togashi et al., 2002; Abe et al., 2004; Yu and Malenka, 2004; Elia et al., 2006; Nuriya and Huganir, 2006; Okuda et al., 2007; Saglietti et al., 2007; Silverman et al., 2007; Kayser et al., 2008; Ochiishi et al., 2008; Xie et al., 2008). F-actin plays a dominant role in regulating AKAP79/150 spine targeting and binding to cadherins and MAGUKs (Gomez et al., 2002; Gorski et al., 2005). Furthermore, PIP2 controls the activity of number of proteins that regulate actin polymerization/depolymerization and filament cross-linking, and AKAP79/150 spine targeting is negatively regulated in coordination with PLC hydrolysis of PIP2 and actin depolymerization in response to NMDAR activation (Horne and Dell'Acqua, 2007). Thus, when it is overexpressed in neurons, the AKAP targeting domain polybasic domains may link to cadherins, cortical F-actin, and membrane PIP2 to control any number of processes that could either promote formation of new filopodia or stabilize existing filopodia.
Consistent with possible stabilization of filopodia by the AKAP targeting domain, AKAP79 overexpression increased protrusion numbers to a much greater extent in immature neurons in which filopodia are common compared with mature neurons in which filopodia are rare and spines predominate. Accordingly, the primary impact of AKAP79 overexpression and AKAP150 knockdown in more mature neurons was to regulate dendritic spine size but not protrusion numbers. In addition, whereas filopodia can serve as spine precursors in immature neurons, the filopodia induced by AKAP79 did not go on to form new functional synapses as seen by no changes in mEPSC frequency or presynaptic marker staining. Indeed, other proteins mentioned above, such as ephrin B receptors, that control filopodia formation and dynamics as well as synaptic adhesion may be required in addition to AKAP79 to convert filopodia to spines with synapses during development (Kayser et al., 2008). Thus, AKAP79/150 expression levels appear to primarily control the maturation of existing synapses but not new synapse formation.
Despite this lack of regulation of basal synapse numbers, the coordinated positive regulation of spine maturation and AMPAR postsynaptic targeting we observed for AKAP79 still support the proposed roles for this AKAP in synaptic plasticity. Interestingly, 8-week-old AKAP150 knock-out mice also have been reported to maintain normal basal hippocampal spine density and synaptic transmission but to exhibit deficits in LTD and learning and memory (Tunquist et al., 2008). Spine morphology and structural plasticity during LTD have not been examined for these AKAP150 null mice. Likewise, two other recent studies in culture preparations found that AKAP79/150 binding to the PSD95 SH3 domain is required for LTD induction and NMDA-induced AMPAR endocytosis but not for maintenance of basal synaptic transmission (Xu et al., 2008; Bhattacharyya et al., 2009). However, consistent with our present findings, one of these studies reported that AKAP150 overexpression increased basal AMPAR surface expression in hippocampal neurons (Bhattacharyya et al., 2009).
Many previous studies have focused on PKA phosphorylation and CaN dephosphorylation of GluR1 subunits and control of AMPAR activity and localization in LTP and LTD (Roche et al., 1996; Lee et al., 1998, 2000, 2003; Banke et al., 2000; Ehlers, 2000; Tavalin et al., 2002; Esteban et al., 2003; Oh et al., 2006; Man et al., 2007). Surprisingly, as most clearly shown by the 1–315 construct, we found no requirement for the AKAP79 PKA or CaN anchoring sites in regulating dendritic protrusion numbers, spine area, AMPAR synaptic localization, or mEPSCs. The lack of AKAP79/150–PKA anchoring requirement for enhancement of AMPAR synaptic localization and activity might have been expected based on recent observations that AKAP150 knock-in mutant mice lacking the PKA anchoring site exhibit normal basal GluR1 phosphorylation and CA3–CA1 synaptic transmission (Lu et al., 2007). Normal basal transmission at CA1 synapses is also seen in GluR1 Ser845Ala, Ser831Ala mutant mice lacking the major PKA, and calcium/calmodulin-dependent kinase II/PKC phosphorylation sites (Roche et al., 1996; Barria et al., 1997; Lee et al., 2003). However, a considerable amount of evidence suggests that AKAP150–PKA anchoring and AMPAR phosphorylation are important in controlling LTP and LTD, even if they may not be for basal transmission. The AKAP150 and GluR1 mutant mice mentioned above exhibit alterations in LTP and LTD and learning and memory depending on developmental age and input from adrenergic pathways (Lee et al., 2003; Hu et al., 2007; Lu et al., 2007, 2008; Tunquist et al., 2008). In addition, previous studies have shown that acute disruption of postsynaptic AKAP–PKA anchoring or inhibition of PKA activity in hippocampal neurons blocks LTD and causes a rundown in synaptic and agonist-evoked AMPAR responses likely through activation of anchored CaN and AMPAR internalization (Rosenmund et al., 1994; Kameyama et al., 1998; Tavalin et al., 2002; Hoshi et al., 2005; Snyder et al., 2005). Thus, we cannot rule out that part of the dominant-negative actions of the MAGUK binding-deficient Δ109–315 mutant could be through sequestering PKA and/or CaN away from endogenous AKAP150–MAGUK–AMPAR complexes to alter AMPAR phosphorylation and/or trafficking.
Although the AKAP–MAGUK binding domain of AKAP79 was required for the enhancement spine maturation and AMPAR activity observed here, AKAP79 is not simply mimicking PSD95 or SAP97. In particular, AKAP79 or 1–315 overexpression increased mEPSC amplitudes but not mEPSC frequency, indicating that the major action of AKAP79–MAGUK binding was to promote additional AMPAR recruitment to spines, as confirmed by our immunostaining data showing increased AMPAR colocalization with PSD95 and NR1. These specific postsynaptic actions of AKAP79 are in contrast to large increases in mEPSC frequency and coordinated presynaptic and postsynaptic changes observed when SAP97 or PSD95 are overexpressed in developing neurons (El-Husseini et al., 2000; Rumbaugh et al., 2003; Prange et al., 2004; Regalado et al., 2006). Thus, our results indicate that AKAP79 requires MAGUK binding to modify postsynaptic development but does not use all the same effectors as MAGUKs, which also trigger significant retrograde regulation of presynaptic development. In conclusion, our data provide strong evidence supporting a novel structural role for AKAP79/150 in regulation of dendritic spine morphology through mechanisms elicited by its N-terminal targeting and internal MAGUK binding domains. In the future, it will be interesting to further explore how these two AKAP79/150 structural domains, in addition to the kinase and phosphatase anchoring domains, contribute to synaptic plasticity mechanisms and learning and memory.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grants NS040701 (M.L.D.) and NS056090 (T.A.B.). H.R.R. was supported by a predoctoral fellowship from the Pacific Mountain Affiliate of the American Heart Association. Thanks to Dr. K. Ulrich Bayer and members of the Dell'Acqua and Bayer laboratories for advice during preparation of this manuscript.
References
- Abe K, Chisaka O, Van Roy F, Takeichi M. Stability of dendritic spines and synaptic contacts is controlled by alpha N-catenin. Nat Neurosci. 2004;7:357–363. doi: 10.1038/nn1212. [DOI] [PubMed] [Google Scholar]
- Ashby MC, Maier SR, Nishimune A, Henley JM. Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci. 2006;26:7046–7055. doi: 10.1523/JNEUROSCI.1235-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Béïque JC, Lin DT, Kang MG, Aizawa H, Takamiya K, Huganir RL. Synapse-specific regulation of AMPA receptor function by PSD-95. Proc Natl Acad Sci U S A. 2006;103:19535–19540. doi: 10.1073/pnas.0608492103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schlüter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer T, Yuste R. Spine motility. Phenomenology, mechanisms, and function. Neuron. 2002;35:1019–1027. doi: 10.1016/s0896-6273(02)00906-6. [DOI] [PubMed] [Google Scholar]
- Brown TC, Correia SS, Petrok CN, Esteban JA. Functional compartmentalization of endosomal trafficking for the synaptic delivery of AMPA receptors during long-term potentiation. J Neurosci. 2007;27:13311–13315. doi: 10.1523/JNEUROSCI.4258-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser ID, Cone RD, Scott JD. Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP 79. J Biol Chem. 1992;267:16816–16823. [PubMed] [Google Scholar]
- Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, Scott JD. Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science. 1995;267:108–111. doi: 10.1126/science.7528941. [DOI] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Cuthbert PC, Stanford LE, Coba MP, Ainge JA, Fink AE, Opazo P, Delgado JY, Komiyama NH, O'Dell TJ, Grant SG. Synapse-associated protein 102/dlgh3 couples the NMDA receptor to specific plasticity pathways and learning strategies. J Neurosci. 2007;27:2673–2682. doi: 10.1523/JNEUROSCI.4457-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua ML, Faux MC, Thorburn J, Thorburn A, Scott JD. Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4, 5-bisphosphate. EMBO J. 1998;17:2246–2260. doi: 10.1093/emboj/17.8.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua ML, Dodge KL, Tavalin SJ, Scott JD. Mapping the protein phosphatase-2B anchoring site on AKAP79. Binding and inhibition of phosphatase activity are mediated by residues 315–360. J Biol Chem. 2002;277:48796–48802. doi: 10.1074/jbc.M207833200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Heine M, Groc L, Lee MC, Choquet D. Diffusional trapping of GluR1 AMPA receptors by input-specific synaptic activity. Neuron. 2007;54:447–460. doi: 10.1016/j.neuron.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci U S A. 2007;104:4176–4181. doi: 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Elia LP, Yamamoto M, Zang K, Reichardt LF. p120 catenin regulates dendritic spine and synapse development through Rho-family GTPases and cadherins. Neuron. 2006;51:43–56. doi: 10.1016/j.neuron.2006.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Glantz SB, Amat JA, Rubin CS. cAMP signaling in neurons: patterns of neuronal expression and intracellular localization for a novel protein, AKAP 150, that anchors the regulatory subunit of cAMP-dependent protein kinase II beta. Mol Biol Cell. 1992;3:1215–1228. doi: 10.1091/mbc.3.11.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci. 2002;22:7027–7044. doi: 10.1523/JNEUROSCI.22-16-07027.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomperts SN, Rao A, Craig AM, Malenka RC, Nicoll RA. Postsynaptically silent synapses in single neuron cultures. Neuron. 1998;21:1443–1451. doi: 10.1016/s0896-6273(00)80662-5. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Gomez LL, Scott JD, Dell'Acqua ML. Association of an A-kinase-anchoring protein signaling scaffold with cadherin adhesion molecules in neurons and epithelial cells. Mol Biol Cell. 2005;16:3574–3590. doi: 10.1091/mbc.E05-02-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Sheng M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci. 2003;23:11759–11769. doi: 10.1523/JNEUROSCI.23-37-11759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne EA, Dell'Acqua ML. Phospholipase C is required for changes in postsynaptic structure and function associated with NMDA receptor-dependent long-term depression. J Neurosci. 2007;27:3523–3534. doi: 10.1523/JNEUROSCI.4340-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, Scott JD. Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat Cell Biol. 2005;7:1066–1073. doi: 10.1038/ncb1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131:160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Kameyama K, Lee HK, Bear MF, Huganir RL. Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron. 1998;21:1163–1175. doi: 10.1016/s0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- Kayser MS, Nolt MJ, Dalva MB. EphB receptors couple dendritic filopodia motility to synapse formation. Neuron. 2008;59:56–69. doi: 10.1016/j.neuron.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Futai K, Jo J, Hayashi Y, Cho K, Sheng M. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron. 2007;56:488–502. doi: 10.1016/j.neuron.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Lamprecht R, LeDoux J. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54. doi: 10.1038/nrn1301. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Levinson JN, Chéry N, Huang K, Wong TP, Gerrow K, Kang R, Prange O, Wang YT, El-Husseini A. Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1beta in neuroligin-induced synaptic specificity. J Biol Chem. 2005;280:17312–17319. doi: 10.1074/jbc.M413812200. [DOI] [PubMed] [Google Scholar]
- Liao D, Zhang X, O'Brien R, Ehlers MD, Huganir RL. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat Neurosci. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
- Liao D, Scannevin RH, Huganir R. Activation of silent synapses by rapid activity-dependent synaptic recruitment of AMPA receptors. J Neurosci. 2001;21:6008–6017. doi: 10.1523/JNEUROSCI.21-16-06008.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 2007;26:4879–4890. doi: 10.1038/sj.emboj.7601884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhang M, Lim IA, Hall DD, Allen M, Medvedeva Y, McKnight GS, Usachev YM, Hell JW. AKAP150-anchored PKA Activity is important for LTD during its induction phase. J Physiol. 2008;586:4155–4164. doi: 10.1113/jphysiol.2008.151662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man HY, Sekine-Aizawa Y, Huganir RL. Regulation of {alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci U S A. 2007;104:3579–3584. doi: 10.1073/pnas.0611698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–758. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- Matus A. Growth of dendritic spines: a continuing story. Curr Opin Neurobiol. 2005;15:67–72. doi: 10.1016/j.conb.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Engler JA, Sheng M. The dynamic turnover and functional roles of alpha-actinin in dendritic spines. Neuropharmacology. 2004a;47:734–745. doi: 10.1016/j.neuropharm.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Futai K, Lashuel HA, Lo I, Okamoto K, Walz T, Hayashi Y, Sheng M. Quaternary structure, protein dynamics, and synaptic function of SAP97 controlled by L27 domain interactions. Neuron. 2004b;44:453–467. doi: 10.1016/j.neuron.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Nuriya M, Huganir RL. Regulation of AMPA receptor trafficking by N-cadherin. J Neurochem. 2006;97:652–661. doi: 10.1111/j.1471-4159.2006.03740.x. [DOI] [PubMed] [Google Scholar]
- Ochiishi T, Futai K, Okamoto K, Kameyama K, Kosik KS. Regulation of AMPA receptor trafficking by delta-catenin. Mol Cell Neurosci. 2008;39:499–507. doi: 10.1016/j.mcn.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Okuda T, Yu LM, Cingolani LA, Kemler R, Goda Y. beta-Catenin regulates excitatory postsynaptic strength at hippocampal synapses. Proc Natl Acad Sci U S A. 2007;104:13479–13484. [Google Scholar]
- Oliveria SF, Gomez LL, Dell'Acqua ML. Imaging kinase–AKAP79–phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol. 2003;160:101–112. doi: 10.1083/jcb.200209127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dell'Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil E, Wells DG, Mooseker MS. A role for myosin VI in postsynaptic structure and glutamate receptor endocytosis. J Cell Biol. 2005;168:329–338. doi: 10.1083/jcb.200410091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Prange O, Wong TP, Gerrow K, Wang YT, El-Husseini A. A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc Natl Acad Sci U S A. 2004;101:13915–13920. doi: 10.1073/pnas.0405939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regalado MP, Terry-Lorenzo RT, Waites CL, Garner CC, Malenka RC. Transsynaptic signaling by postsynaptic synapse-associated protein 97. J Neurosci. 2006;26:2343–2357. doi: 10.1523/JNEUROSCI.5247-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL. Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature. 1994;368:853–856. doi: 10.1038/368853a0. [DOI] [PubMed] [Google Scholar]
- Rumbaugh G, Sia GM, Garner CC, Huganir RL. Synapse-associated protein-97 isoform-specific regulation of surface AMPA receptors and synaptic function in cultured neurons. J Neurosci. 2003;23:4567–4576. doi: 10.1523/JNEUROSCI.23-11-04567.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J, Liu L, Wong TP, Wu DC, Burette A, Weinberg R, Wang YT, Sheng M. A critical role for myosin IIb in dendritic spine morphology and synaptic function. Neuron. 2006;49:175–182. doi: 10.1016/j.neuron.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, Passafaro M. Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron. 2007;54:461–477. doi: 10.1016/j.neuron.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Schlüter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51:99–111. doi: 10.1016/j.neuron.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci U S A. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüler H, Peti W. Structure-function analysis of the filamentous actin binding domain of the neuronal scaffolding protein spinophilin. FEBS J. 2008;275:59–68. doi: 10.1111/j.1742-4658.2007.06171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JB, Restituito S, Lu W, Lee-Edwards L, Khatri L, Ziff EB. Synaptic anchorage of AMPA receptors by cadherins through neural plakophilin-related arm protein AMPA receptor-binding protein complexes. J Neurosci. 2007;27:8505–8516. doi: 10.1523/JNEUROSCI.1395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, Dell'Acqua ML. cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J Neurosci. 2006;26:2391–2402. doi: 10.1523/JNEUROSCI.3092-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Colledge M, Crozier RA, Chen WS, Scott JD, Bear MF. Role for A kinase-anchoring proteins (AKAPS) in glutamate receptor trafficking and long term synaptic depression. J Biol Chem. 2005;280:16962–16968. doi: 10.1074/jbc.M409693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, Nicoll RA. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci. 2003;23:5503–5506. doi: 10.1523/JNEUROSCI.23-13-05503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Sekino Y, Tanaka S, Mizui T, Kishi S, Shirao T. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. J Neurosci. 2003;23:6586–6595. doi: 10.1523/JNEUROSCI.23-16-06586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD. Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J Neurosci. 2002;22:3044–3051. doi: 10.1523/JNEUROSCI.22-08-03044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry-Lorenzo RT, Roadcap DW, Otsuka T, Blanpied TA, Zamorano PL, Garner CC, Shenolikar S, Ehlers MD. Neurabin/protein phosphatase-1 complex regulates dendritic spine morphogenesis and maturation. Mol Biol Cell. 2005;16:2349–2362. doi: 10.1091/mbc.E04-12-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M. Cadherin regulates dendritic spine morphogenesis. Neuron. 2002;35:77–89. doi: 10.1016/s0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, Scott JD. Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci U S A. 2008;105:12557–12562. doi: 10.1073/pnas.0805922105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronin LL, Cherubini E. “Deaf, mute and whispering” silent synapses: their role in synaptic plasticity. J Physiol. 2004;557:3–12. doi: 10.1113/jphysiol.2003.058966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Photowala H, Cahill ME, Srivastava DP, Woolfrey KM, Shum CY, Huganir RL, Penzes P. Coordination of synaptic adhesion with dendritic spine remodeling by AF-6 and kalirin-7. J Neurosci. 2008;28:6079–6091. doi: 10.1523/JNEUROSCI.1170-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Schlüter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Malenka RC. Multiple functions for the cadherin/catenin complex during neuronal development. Neuropharmacology. 2004;47:779–786. doi: 10.1016/j.neuropharm.2004.07.031. [DOI] [PubMed] [Google Scholar]
- Yudowski GA, Puthenveedu MA, Leonoudakis D, Panicker S, Thorn KS, Beattie EC, von Zastrow M. Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J Neurosci. 2007;27:11112–11121. doi: 10.1523/JNEUROSCI.2465-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci. 2004;5:24–34. doi: 10.1038/nrn1300. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Zito K, Knott G, Shepherd GM, Shenolikar S, Svoboda K. Induction of spine growth and synapse formation by regulation of the spine actin cytoskeleton. Neuron. 2004;44:321–334. doi: 10.1016/j.neuron.2004.09.022. [DOI] [PubMed] [Google Scholar]