

Abstract
Background
Stearoyl-CoA Desaturase 1 (SCD1) is a well-known enhancer of the metabolic syndrome. The purpose of the present study was to investigate the role of SCD1 in lipoprotein metabolism and atherosclerosis progression.
Methods and Results
Antisense oligonucleotides were used to inhibit SCD1 in a mouse model of hyperlipidemia and atherosclerosis (LDLr-/-Apob100/100). In agreement with previous reports, inhibition of SCD1 protected against diet-induced obesity, insulin resistance, and hepatic steatosis. However, unexpectedly SCD1 inhibition strongly promoted aortic atherosclerosis, which could not be reversed by dietary oleate. Further analyses revealed that SCD1 inhibition promoted accumulation of saturated fatty acids in plasma and tissues, reduced plasma triglyceride, yet had little impact on LDL cholesterol. Since dietary SFAs have been shown to promote inflammation through toll-like receptor 4 (TLR4), we examined macrophage TLR4 function. Interestingly, SCD1 inhibition resulted in alterations in macrophage membrane lipid composition and marked hypersensitivity to TLR4 agonists.
Conclusions
This study demonstrates that atherosclerosis can occur independently of obesity and insulin resistance, and argues against SCD1 inhibition as a safe therapeutic target for the metabolic syndrome.
Keywords: SCD1, atherosclerosis, inflammation, saturated fatty acids
Introduction
The metabolic syndrome has become a leading health concern in developed countries. This syndrome is a collection of metabolic abnormalities including abdominal obesity, hypertension, insulin resistance, hypertriglyceridemia, and low high-density lipoprotein cholesterol (HDLc) levels.1-3 Recently, the metabolic syndrome has been shown to be a predictor of atherosclerotic cardiovascular disease (CVD) in humans.1-3 Therefore, potential therapeutic targets for the metabolic syndrome are actively being pursued to combat CVD, and stearoyl-CoA desaturase 1 (SCD1) has come to the forefront in this pursuit.4-6 By catalyzing the conversion of long-chain saturated fatty acids (SFA) to monounsaturated fatty acids (MUFA), SCD1 promotes multiple aspects of the metabolic syndrome. In fact, mice lacking SCD1 are largely protected against diet-induced and genetically-induced obesity7-11, hepatic steatosis11-14, hypertriglyceridemia15-16, and insulin resistance7,9,17,18.
Since mice lacking SCD1 have severely impaired hepatic neutral lipid biosynthesis19, we hypothesized that SCD1 inhibition would diminish the hepatic production of MUFA-rich cholesteryl esters (CE) and triglycerides (TG), thereby protecting against hyperlipidemia and atherosclerosis. To test this idea we utilized ASO-mediated knockdown of SCD1 in a well-characterized mouse model of hyperlipidemia and atherosclerosis. The results from this study demonstrate that the metabolic syndrome can be dissociated from atherosclerosis in mice, and warn that SFA accumulation seen with SCD1 inhibition can promote inflammation and atherosclerosis.
Methods
A detailed description of the material and methods is provided in the online data supplement. The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Experimental Design
Male apolipoprotein B100-only, low density lipoprotein receptor (LDLr) deficient (LDLr-/-Apob100/100) mice were used in this study. These mice were chosen based on previous reports documenting their “human-like” lipoprotein profile,20 atherosclerosis susceptibility,20 and responsiveness to dietary fatty acids.21 All mice were on a mixed background (∼75% C57BL/6 and ∼25% 129Sv/Jae). At 6 weeks of age, the mice were switched from a diet of rodent chow to one of two synthetic diets containing 12% of energy as saturated fatty acid (SFA)-enriched fat (palm oil) or monounsaturated fatty acid (MUFA)-enriched fat (oleinate-enriched safflower oil) with 0.1% (w/w) cholesterol added. Please refer to Supplemental Table 1 (online) for complete analysis of dietary fatty acid composition. In conjunction with diet, mice were injected biweekly with either saline, 25 mg/kg of a non-targeting ASO (control ASO; 5 ′- TCCCATTTCAGGAGACCTGG -3′), or 25 mg/kg of an ASO targeting the knockdown of SCD1 (SCD1 ASO; 5′-GCTCTAATCACCTCAGAACT -3′). These phosphorothioate modified ASO compounds were generously provided by ISIS Pharmaceuticals, Inc. (Carlsbad, CA). Body weight was measured weekly, and food intake was measured at four weeks and eight weeks of diet/ASO treatment. All experimental animals were sacrificed after 20 weeks of parallel dietary and ASO treatment. All mice were maintained in a pathogen-free animal facility, and experimental protocols were approved by the institutional animal care and use committee at the Wake Forest University School of Medicine.
Results
SCD1 ASO Treatment Inhibits Hepatic and Adipose SCD1 Function
After 20 weeks of ASO treatment, liver and adipose SCD1 mRNA levels were reduced by 99% and 83-94%, respectively (Figure 1A). Further, SCD1 protein expression was undetectable by Western blot in both liver and adipose tissue (Figure 1B). Hepatic SCD1 activity was reduced by > 95% after 20 weeks of treatment (Figure 1C). After only four weeks of ASO treatment, hepatic SCD1 protein and activity levels were reduced by > 90% (data not shown). Importantly, SCD1 protein expression in skeletal muscle and skin was not altered by 20 weeks of SCD1 ASO treatment (data not shown). This tissue-specific pattern of knockdown is likely due to the intrinsic pharmacokinetic properties of ASOs, and has been previously described with other ASO compounds of similar chemistry in mice.22,23
Figure 1.
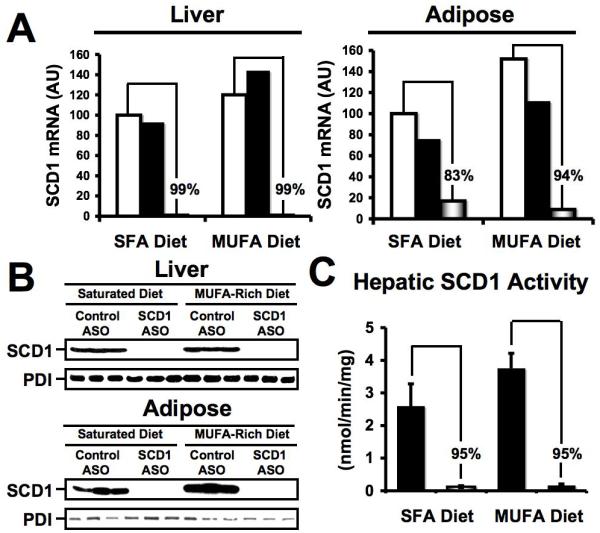
ASO-mediated knockdown of SCD1 in liver and adipose tissue. Male LDLr -/-, ApoB100/100 mice were fed diets enriched in 0.1% (w/w) cholesterol and either saturated fatty acids (SFA) or monounsaturated fatty acids (MUFA) for 20 weeks in conjunction with biweekly injections (25 mg/kg) of either saline □, a non-targeting control ASO ■, or SCD1 ASO . (A) Relative quantification of SCD1 mRNA levels in the liver or adipose tissue was conducted by real-time qPCR, and normalized to cyclophilin. Data shown in (A) represent pooled RNA samples with n=5 mice per group. (B) Western blot analysis of SCD1 and protein disulfide isomerase (PDI) protein expression in liver and adipose tissue microsomes (n=3 per group). (C) Hepatic SCD1 activity, data represent the mean ± SEM from 5 mice per group, and differences between Control ASO and SCD1 ASO group were highly significant (p<0.01).
. (A) Relative quantification of SCD1 mRNA levels in the liver or adipose tissue was conducted by real-time qPCR, and normalized to cyclophilin. Data shown in (A) represent pooled RNA samples with n=5 mice per group. (B) Western blot analysis of SCD1 and protein disulfide isomerase (PDI) protein expression in liver and adipose tissue microsomes (n=3 per group). (C) Hepatic SCD1 activity, data represent the mean ± SEM from 5 mice per group, and differences between Control ASO and SCD1 ASO group were highly significant (p<0.01).
SCD1 Inhibition Prevents Diet-Induced Obesity and Insulin Resistance in Hyperlipidemic Mice
In agreement with previous reports,7-11 SCD1 inhibition prevented diet-induced obesity in LDLr-/-Apob100/100 mice. (Figure 2A, 2B, and 2C). Epididymal fat pad mass was reduced by ∼85% on the saturated diet and ∼80% on the MUFA-rich diet, when compared to control ASO treated mice (Figure 2C), which could not be explained by reductions in food intake (Figure 2D), and may result from SCD1’s previously described role in energy expenditure.7,8,24 After only 4 weeks of treatment, fasting insulin levels were significantly lower in SCD1 ASO treated mice (Figure 2E). In parallel, SCD1 inhibition significantly improved glucose tolerance (Figure 2F) and insulin tolerance (Figure 2G). Importantly, the effects of SCD1 inhibition on adiposity and insulin resistance could not be reversed by dietary MUFA supplementation (Figure 2). Collectively, these data support the notion that ASO-mediated inhibition of SCD1 is efficacious in the prevention of diet-induced obesity and insulin resistance.10,17
Figure 2.

SCD1 inhibition prevents diet-induced obesity and insulin resistance in LDLr-/- Apob100/100 mice. Starting at six weeks of age, mice were fed diets enriched in 0.1% (w/w) cholesterol and either saturated fatty acids (SFA) or monounsaturated fatty acids (MUFA) for a period up to 20 weeks in conjunction with biweekly injections (25 mg/kg) of either saline (□ in bar graphs,  in line graphs), a non-targeting control ASO (■ in bar graphs,
in line graphs), a non-targeting control ASO (■ in bar graphs,  in line graphs), or SCD1 ASO (
in line graphs), or SCD1 ASO ( in bar graphs,
in bar graphs,  in line graphs). (A) Photographs, (B) body weights, and (C) epididymal fat pad mass of mice following 20 weeks of diet and ASO treatment. Data in panels (B) and (C) represent the mean ± SEM from 8-15 mice per group, and values in panel (B) not sharing a common superscript differ significantly (p<0.05). (D) Food intake was measured after 8 weeks of diet and ASO treatment, and no significant differences were detected. (E) Fasting plasma insulin levels were measured after 4 weeks of diet and ASO treatment. Data in panel E represent the mean ± SEM from 5 mice per group, and values not sharing a common superscript differ significantly (p<0.05). (F) Glucose tolerance tests (GTT) and (G) insulin tolerance tests (ITT) were performed following 16 weeks of diet and ASO treatment. Data shown in panels (F) and (G) represent the mean ± SEM from 5 mice per group, * = significantly different than the Control ASO group within each diet group (p<0.05).
in line graphs). (A) Photographs, (B) body weights, and (C) epididymal fat pad mass of mice following 20 weeks of diet and ASO treatment. Data in panels (B) and (C) represent the mean ± SEM from 8-15 mice per group, and values in panel (B) not sharing a common superscript differ significantly (p<0.05). (D) Food intake was measured after 8 weeks of diet and ASO treatment, and no significant differences were detected. (E) Fasting plasma insulin levels were measured after 4 weeks of diet and ASO treatment. Data in panel E represent the mean ± SEM from 5 mice per group, and values not sharing a common superscript differ significantly (p<0.05). (F) Glucose tolerance tests (GTT) and (G) insulin tolerance tests (ITT) were performed following 16 weeks of diet and ASO treatment. Data shown in panels (F) and (G) represent the mean ± SEM from 5 mice per group, * = significantly different than the Control ASO group within each diet group (p<0.05).
SCD1 Inhibition Promotes Atherosclerosis
En face morphometric analysis showed that SCD1 ASO treated mice had 2.7-fold (SFA diet) and 2.6-fold (MUFA diet) increases in total aortic lesion area (Figure 3B), when compared to control ASO treated mice. Interestingly, this SCD1 ASO-driven augmentation of atherosclerotic lesion area seemed to be regional in nature (Figure 3A and 3C). When control and SCD1 ASO groups were compared there were no significant differences in en face lesion area in the aortic arch (Figure 3C). However, there were modest increases in the thoracic aorta lesion area, and highly significant increases in the abdominal aorta lesion area, when SCD1 was inhibited (Figure 3C). In fact, SCD1 inhibition caused a striking 5-fold (MUFA diet) to 7-fold (SFA diet) increase in abdominal aortic lesion area, where greater than 70% of the abdominal aorta was covered with lesion in SCD1 inhibited mice (Figure 3A and 3C). Biochemical analysis of the complete set (n=8-15 per group) of whole aortae from this study revealed that SCD1 inhibition resulted in significant increases in both free and esterified cholesterol, compared to either saline or control ASO treated mice (Figure 3D and 3E). Furthermore, SCD1 inhibition resulted in enrichment of SFA and depletion of MUFA in aortic CE and TG (Figure 3F and 3G). Although less dramatic than the effects seen in CE (Figure 3F) and TG (Figure 3G), aortic PL was likewise significantly depleted of MUFA (Figure 3H), and desaturation indices (16:1/16:0 and 18:1/18:0) were significantly reduced with SCD1 inhibition (data not shown). Importantly, dietary MUFA did not prevent SCD1 ASO-mediated promotion of aortic atherosclerosis (Figure 3). In agreement with en face (Figure 3A, 3B, and 3C) and biochemical analyses (Figure 3D and 3E), histological evaluation of cross sections from the proximal aorta revealed that SCD1 inhibition promoted the accumulation of cholesterol clefts and necrotic core formation (Supplemental Figure 1). Similar histological lesion characteristics were seen in thoracic and abdominal aortic sections (data not shown). Collectively, these data provide evidence that SCD1 inhibition promotes SFA- and cholesterol-rich atherosclerotic lesion formation in LDLr-/-Apob100/100 mice.
Figure 3.

SCD1 inhibition promotes atherosclerosis in LDLr-/-Apob100/100 mice. Starting at six weeks of age, mice were fed diets enriched in 0.1% (w/w) cholesterol and either saturated fatty acids (SFA) or monounsaturated fatty acids (MUFA) for 20 weeks in conjunction with biweekly injections (25 mg/kg) of either saline □, a non-targeting control ASO ■, or SCD1 ASO . (A) Representative photographs after en face preparation of aortae. (B) En face morphometric analysis of total aortic lesion area. (C) En face morphometric analysis of regional (aortic arch, thoracic aorta, and abdominal aorta) differences in atherosclerosis. Data shown in panels (B) and (C) represent the mean ± SEM from 6 mice per group. GLC analysis of aortic cholesteryl ester (D) and free cholesterol (E) was determined after morphometric analysis. Data shown in panels (D) and (E) represents the mean ± SEM from 8-15 mice per group. Fatty acid (FA) composition (% of total FA that was SFA or MUFA) of aortic cholesteryl esters (F), triglycerides (G), and phospholipids (H) was determined from whole aortic lipid extracts. Data shown in panels (F), (G), and (H) represents the mean ± SEM from 5 mice per group. Values not sharing a common superscript differ significantly (p<0.05).
. (A) Representative photographs after en face preparation of aortae. (B) En face morphometric analysis of total aortic lesion area. (C) En face morphometric analysis of regional (aortic arch, thoracic aorta, and abdominal aorta) differences in atherosclerosis. Data shown in panels (B) and (C) represent the mean ± SEM from 6 mice per group. GLC analysis of aortic cholesteryl ester (D) and free cholesterol (E) was determined after morphometric analysis. Data shown in panels (D) and (E) represents the mean ± SEM from 8-15 mice per group. Fatty acid (FA) composition (% of total FA that was SFA or MUFA) of aortic cholesteryl esters (F), triglycerides (G), and phospholipids (H) was determined from whole aortic lipid extracts. Data shown in panels (F), (G), and (H) represents the mean ± SEM from 5 mice per group. Values not sharing a common superscript differ significantly (p<0.05).
SCD1 Inhibition Promotes SFA Enrichment of Plasma Lipoproteins
In agreement with previous reports,1-3 our results showed that SCD1 inhibition prevented diet-induced hypertriglyceridemia (Figure 4A). In contrast, total plasma cholesterol (TPC) was only modestly (1861 mg/dl in control ASO group vs. 1241 mg/dl in SCD1 ASO group) reduced after 20 week of feeding the SFA diet, but was not significantly altered under any other conditions (Figure 4B). When lipoprotein cholesterol distribution was analyzed, we discovered that SCD1 inhibition decreased VLDL cholesterol, had no effect on LDL cholesterol levels, and significantly reduced HDL cholesterol (Figures 4C and 4D). These SCD1 ASO-driven reductions in VLDL and HDL cholesterol levels were accompanied by reductions in plasma apoE and apoAI, while plasma apoB and LCAT were not altered by SCD1 inhibition (Figure 4G). Furthermore, VLDL particles were significantly smaller in SCD1 ASO treated mice (Figure 4F), possibly due to depletion of TG-rich core (Figure 4A). However, LDL and HDL particle size was not altered by SCD1 ASO treatment (Figure 4F). Finally, SCD1 inhibition resulted in reductions of MUFA with highly significant enrichments of SFA in LDL-CE, and similar but less impressive FA shifts in HDL-CE (Figure 4E). Collectively, SCD1 inhibition resulted in dramatic alterations in plasma lipoprotein metabolism including diminished plasma triglyceride, VLDLc, HDLc, VLDL size, apoE, and apoAI levels and striking enrichment of plasma lipoproteins with SFA. Importantly, none of the SCD1 ASO-driven alterations in lipoprotein metabolism were prevented by dietary MUFA (Figures 4A, 4B, and 4E & data not shown).
Figure 4.

Effects of SCD1 inhibition on plasma lipids in LDLr -/-, apoB100-only mice. Starting at six weeks of age, mice were fed diets enriched in 0.1% (w/w) cholesterol and either saturated fatty acids or monounsaturated fatty acids (MUFA) for a period up to 20 weeks in conjunction with biweekly injections (25 mg/kg) of either saline (□ in bar graphs,  in line graphs), a non-targeting control ASO (■ in bar graphs,
in line graphs), a non-targeting control ASO (■ in bar graphs,  in line graphs), or SCD1 ASO (
in line graphs), or SCD1 ASO ( in bar graphs,
in bar graphs,  in line graphs). Plasma samples were collected at baseline (6 weeks of age), and after 4, 8, or 20 weeks of diet and ASO treatment. Plasma triglycerides (A) and total plasma cholesterol (B) were measured enzymatically. Data shown in panels (A) and (B) represents the mean ± SEM from 5-8 mice per group, ** = significantly different than the Control ASO group within each diet group (p<0.01). Panel (C) represents the lipoprotein cholesterol distribution of pooled plasma samples (n=5 mice per pool) from mice fed a saturated diet and treated with ASO for 20 weeks. Panel (D) represents cholesterol levels in very-low-density lipoproteins (VLDLc), low-density lipoproteins (LDLc), and high-density lipoproteins (HDLc) in mice fed a saturated diet and treated with ASO for 20 weeks. Data shown in panel (D) represents the mean ± SEM from 6 mice per group, and values not sharing a common superscript differ significantly (p<0.05). (E) Fatty acid (FA) composition (% of total FA as SFA or MUFA) of LDL and HDL cholesteryl esters (LDL-CE and HDL-CE). Data shown in panel (E) represents the mean ± SEM (n=5 per group), and values not sharing a common superscript differ significantly (p<0.05). (F) Lipoprotein size was determined by dynamic light scattering, and is represented as the mean ± SEM from 5 mice fed a saturated diet, and values not sharing a common superscript differ significantly (p<0.05). (G) Western blot analysis of whole plasma from mice fed a saturated diet for 20 weeks; antibodies used were targeting apolipoproteins B (apoB), E (apoE), AI (apoAI), and lecithin:cholesterol acyltransferase (LCAT).
in line graphs). Plasma samples were collected at baseline (6 weeks of age), and after 4, 8, or 20 weeks of diet and ASO treatment. Plasma triglycerides (A) and total plasma cholesterol (B) were measured enzymatically. Data shown in panels (A) and (B) represents the mean ± SEM from 5-8 mice per group, ** = significantly different than the Control ASO group within each diet group (p<0.01). Panel (C) represents the lipoprotein cholesterol distribution of pooled plasma samples (n=5 mice per pool) from mice fed a saturated diet and treated with ASO for 20 weeks. Panel (D) represents cholesterol levels in very-low-density lipoproteins (VLDLc), low-density lipoproteins (LDLc), and high-density lipoproteins (HDLc) in mice fed a saturated diet and treated with ASO for 20 weeks. Data shown in panel (D) represents the mean ± SEM from 6 mice per group, and values not sharing a common superscript differ significantly (p<0.05). (E) Fatty acid (FA) composition (% of total FA as SFA or MUFA) of LDL and HDL cholesteryl esters (LDL-CE and HDL-CE). Data shown in panel (E) represents the mean ± SEM (n=5 per group), and values not sharing a common superscript differ significantly (p<0.05). (F) Lipoprotein size was determined by dynamic light scattering, and is represented as the mean ± SEM from 5 mice fed a saturated diet, and values not sharing a common superscript differ significantly (p<0.05). (G) Western blot analysis of whole plasma from mice fed a saturated diet for 20 weeks; antibodies used were targeting apolipoproteins B (apoB), E (apoE), AI (apoAI), and lecithin:cholesterol acyltransferase (LCAT).
SCD1 inhibition prevents diet-induced steatosis
It has been well documented that mice lacking SCD1 are protected against hepatic steatosis under a variety of conditions.1,4-7 In addition to confirming these reports, we set out to characterize hepatic cholesterol metabolism in SCD1 ASO treated mice, since cholesterol-rich atherogenic apoB-containing lipoproteins are believed to originate from the liver.25 As expected, SCD1 inhibition resulted in striking reductions in hepatic steatosis (Supplemental Figure 2A), manifested as a 93% reduction of hepatic TG concentration and 81% reduction of hepatic CE concentration (Supplemental Figure 2B), when compared to control ASO treated mice. This reduction in hepatic TG mass may be partially a result of a reduction in SREBP1c protein expression (Supplemental Figure 2F), with parallel downregulation of SREBP1c target genes (Supplemental Figure 2E) including fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC), and mitochondrial glycerol-3-phosphate acyltransferase (mGPAT). Quite unexpectedly, hepatic free cholesterol concentration was significantly increased by 1.5-fold (SFA diet) or 2.4-fold (MUFA diet, data not shown) when SCD1 was inhibited (Supplemental Figure 2B). The hepatic free cholesterol increase seen with SCD1 ASO treatment was accompanied by compensatory downregulation of HMG-CoA synthase, upregulation of Cyp7α1, but normal ATP-binding cassette protein G5 (ABCG5) mRNA expression (Supplemental Figure 2E). Protein expression of ABCG5 also was not altered by SCD1 ASO treatment (Supplemental Figure 2F). Interestingly, ACAT2 protein expression was slightly increased with SCD1 ASO treatment (Supplemental Figure 2F), without corresponding changes in mRNA expression (data not shown), indicating post-transcriptional regulation. During isolated liver perfusion it was found that SCD1 inhibition did not significantly alter the secretion rate of TG, CE, FC, or PL (Supplemental Figure 2C). There was a trend towards a decrease in TG secretion rate with SCD1 ASO treatment, but this was not statistically significant (Supplemental Figure 2C). A closer look at the fatty acid composition of hepatic CE, TG, and PL revealed that SCD1 inhibition promoted the enrichment of SFA into hepatic CE and TG, but not PL (Supplemental Figure 2D). None of the SCD1 ASO-driven alterations in hepatic lipid metabolism were prevented by dietary MUFA (data not shown). Collectively, these data support previous observations7,8,10,12,14,17 showing that SCD1 inhibition is efficacious in the prevention hepatic steatosis, but surprisingly augmented free cholesterol concentrations were also present.
SCD1 Inhibition Exacerbates TLR4-Driven Proinflammatory Response in Macrophages
Macrophages can play a pivotal role in the pathogenesis of atherosclerosis through multiple mechanisms, including their well-known role in producing atherogenic cytokine signals.26 It has been demonstrated that SFAs promote inflammatory cytokine secretion in macrophages through a toll-like receptor 4 (TLR4)-dependent mechanism.27-30 Since SCD1 inhibition resulted in striking SFA enrichment of low density lipoprotein associated cholesteryl esters (LDL-CE) (Figure 4E), we hypothesized that macrophage lipids may likewise become enriched in SFA, thereby enhancing TLR4-dependent proinflammatory cytokine secretion. We found that in vivo SCD1 ASO treatment for six weeks reduced SCD1 mRNA and protein expression by >90% and >50%, respectively, in isolated macrophages (Figure 5A). Also, macrophages isolated from SCD1 ASO-treated mice had a significantly decreased 16:1/16:0 ratio and an increased proportion of linoleic acid (18:2, n-6) in isolated phospholipids (Figure 5B). Similar effects were observed in neutral lipids, with SCD1 inhibition resulting in significant decreases in 16:1/16:0 and 18:1/18:0 ratios (data not shown). Interestingly, when macrophages isolated from SCD1 ASO-treated mice were challenged with a TLR4 agonist (10 ng/ml Kdo2-Lipid A), marked hypersensitivity was apparent (Figure 5C and 5D). In support of this, SCD1 inhibition resulted in augmented TLR4-driven proinflammatory gene expression (Figure 5C) and cytokine secretion (Figure 5D) in isolated macrophages, although basal expression (ie the absence of TLR4 ligand) of inflammatory cytokines was not significantly different between macrophages isolated from control and SCD1 ASO treated mice (data not shown). When we examined the expression of key proteins involved in TLR4-dependent signal transduction31,32 we found a slight reduction in TLR4 expression, no change in MyD88, and a slight reduction in CD14 in SCD1 inhibited macrophages (Figure 5F). Furthermore, canonical TLR4-MyD88-dependent activation of mitogen-activated protein kinases (MAPKs), phosphorylation of inhibitor kappa kinase alpha/beta (IKK-α/β), and downstream degradation of inhibitory kappaB kinase alpha (IκBα) was not different between control and SCD1 ASO treated macrophages (Figure 5E). However, the TLR4-MyD88-independent driven tyrosine phosphorylation of STAT1, a well-known interferon beta (IFNβ)-dependent event,33 was markedly elevated in SCD1 inhibited macrophages (Figure 5E).
Figure 5.

SCD1 inhibition exacerbates TLR4-driven proinflammatory response in macrophages. Starting at six weeks of age, mice were fed a diet enriched in 0.1% (w/w) cholesterol and saturated fatty acids (SFA) for 6 weeks in conjunction with biweekly injections (25 mg/kg) of either a non-targeting control ASO ■ or SCD1 ASO  . Following six weeks of treatment, freshly isolated thioglycollate-ellicited macrophages were pooled (n=5-7 mice per pool) and cultured as described in materials and methods. A) SCD1 mRNA and protein expression in freshly isolated (2h culture) macrophages. GAPDH was used to normalize mRNA levels and β-actin was used as a loading control for Western blotting. B) Fatty acid (FA) composition (16:1 to 16:0 ratio and % or total FA that were 18:2, n-6) of freshly isolated (2h culture) macrophage phospholipids. C) Proinflammatory gene expression: Freshly isolated macrophages were treated with vehicle or 10ng/ml Kdo2-Lipid A (TLR4 agonist) for 6 hours, and the mRNA levels of interleukins -1β and -6 (IL-1β and IL-6), monocyte chemotactic protein 1 (MCP-1), inducible nitric oxide synthase (iNOS), C-X-C motif ligand 10 (IP-10), and interferon-induced with tetratricopeptide repeats 1 (Garg-16) were measured by qPCR, and normalized to GAPDH. Data shown in panel C are expressed as the fold-change above vehicle treated mRNA levels (Kdo2-Lipid A treated / vehicle treated). (D) Cytokine secretion: Freshly isolated macrophages were treated with 10ng/ml Kdo2-Lipid A (TLR4 agonist) for 8 hours, and conditioned media were collected for detection of multiple cytokines using an antibody array as described in methods. E) TLR4-driven signal transduction. Freshly isolated macrophages were treated with 10ng/ml Kdo2-Lipid A (TLR4 agonist) for a period of 30 or 120 minutes (0′, 30′, 120′), and TLR-4-dependent signaling was measured by Western blotting as described in methods. (F) Western blot analysis of TLR4, MyD88, and CD14 protein expression in freshly isolated macrophages (2h culture); β-actin was used as a loading control.
. Following six weeks of treatment, freshly isolated thioglycollate-ellicited macrophages were pooled (n=5-7 mice per pool) and cultured as described in materials and methods. A) SCD1 mRNA and protein expression in freshly isolated (2h culture) macrophages. GAPDH was used to normalize mRNA levels and β-actin was used as a loading control for Western blotting. B) Fatty acid (FA) composition (16:1 to 16:0 ratio and % or total FA that were 18:2, n-6) of freshly isolated (2h culture) macrophage phospholipids. C) Proinflammatory gene expression: Freshly isolated macrophages were treated with vehicle or 10ng/ml Kdo2-Lipid A (TLR4 agonist) for 6 hours, and the mRNA levels of interleukins -1β and -6 (IL-1β and IL-6), monocyte chemotactic protein 1 (MCP-1), inducible nitric oxide synthase (iNOS), C-X-C motif ligand 10 (IP-10), and interferon-induced with tetratricopeptide repeats 1 (Garg-16) were measured by qPCR, and normalized to GAPDH. Data shown in panel C are expressed as the fold-change above vehicle treated mRNA levels (Kdo2-Lipid A treated / vehicle treated). (D) Cytokine secretion: Freshly isolated macrophages were treated with 10ng/ml Kdo2-Lipid A (TLR4 agonist) for 8 hours, and conditioned media were collected for detection of multiple cytokines using an antibody array as described in methods. E) TLR4-driven signal transduction. Freshly isolated macrophages were treated with 10ng/ml Kdo2-Lipid A (TLR4 agonist) for a period of 30 or 120 minutes (0′, 30′, 120′), and TLR-4-dependent signaling was measured by Western blotting as described in methods. (F) Western blot analysis of TLR4, MyD88, and CD14 protein expression in freshly isolated macrophages (2h culture); β-actin was used as a loading control.
Discussion
Although presence of the metabolic syndrome seems to be associated with atherosclerotic CVD outcomes in humans,1-3 dissociation of the two has been previously reported.34 Results from this study provides evidence that the metabolic syndrome can be completely dissociated from atherosclerosis in mice. That is, in the extreme case of leanness and insulin sensitivity induced by SCD1 ASO treatment, atherosclerosis independently progressed. Results from this study support the notion7-18 that SCD1 inhibitors may be efficacious in preventing many aspects of the metabolic syndrome (diet-induced obesity, hepatic steatosis, insulin resistance, hypertriglyceridemia), but this may be at the expense of the artery wall. To help explain this unexpected finding, we propose a working model in which SCD1 inhibitors promote atherosclerosis (Figure 6). Briefly, we believe that inhibition of SCD1 in the liver results in secretion of VLDL particles that are highly enriched in SFA-rich CE, giving rise to SFA-CE rich LDL particles. These SFA-CE rich LDLs deliver SFA to macrophages, which also have diminished SCD1 expression, resulting in accumulation of SFA in macrophages, enhanced TLR4-driven tyrosine phosphorylation of STAT1, and ultimately enhanced inflammatory cytokine secretion. This proinflammatory phenotype thereby promotes atherosclerosis in a hyperlipidemic setting.
Figure 6.

Proposed mechanism by which SCD1 inhibition promotes atherosclerosis. Briefly, Inhibition of SCD1 in the liver results in secretion of VLDL particles that are highly enriched in saturated fatty acid (SFA)-rich cholesteryl esters (CE) and likely other SFA metabolites, which are metabolized to SFA-CE rich LDL particles. These SFA-CE rich LDL deliver abundant SFA to macrophages, which also have diminished SCD1 expression, resulting in alterations in membrane lipid composition, enhanced TLR4-driven tyrosine phosphorylation of STAT1, and ultimately enhanced inflammatory cytokine secretion. This proinflammatory phenotype ultimately promotes atherosclerosis.
In support of this model, a role for SCD1 in protecting against another inflammation-driven disease [dextran sulfate sodium (DSS)-induced colitis] was recently reported.35 In this report, Chen et al. elegantly demonstrated that mice lacking SCD1 had elevated DSS- and bacterial-driven inflammatory gene expression and exaggerated colitis, findings analogous to our results. This study35, as well as ours, supports the long-standing notion that SFAs are potent proinflammatory molecules.27-30 Hence, one of the key roles of SCD1 may be to suppress inflammation by preventing excessive accumulation of SFA themselves and downstream metabolites such as stearoyl-lysophosphatidylcholine35 and ceramide36. Importantly, this study now joins several recent reports that have demonstrated unexpected harmful consequences of inhibiting SCD1.18,35,37
The molecular mechanism(s) by which SCD1 inhibition promotes atherosclerosis (Figure 3), inflammatory colitis,35 frank diabetes,18 and cholestasis37 has not been clearly elucidated. More work is needed to address these unexpected outcomes if SCD1 inhibitors are to be pursued as CVD therapeutics in humans. In addition to SCD1 inhibitors, TLR4 antagonists have been suggested as potential CVD therapeutics, yet whether TLR4 plays a role in atherosclerosis in humans has been a matter of intense debate.38-40 Indeed, more work is needed to investigate whether TLR4 is necessary for SFA-dependent induction of diseases such as atherosclerosis and the other diverse pathologies associated with SCD1 inhibition.18,35,37,Figure 3 Performing SCD1 inhibition studies in TLR4 deficient mice will no doubt provide useful insight into the necessity of TLR4 in promoting both endogenous and dietary SFA-driven atherosclerosis and other inflammatory diseases.
One potential unifying mechanism driving the multiple pathologies seen under conditions of SCD1 deficiency may involve the previous documented function of SCD1 in modulating the formation of cholesterol- and SFA-rich membrane microdomains, better known as “lipid rafts”. It has been previously shown that overexpression of SCD1 in macrophages results in decreased abundance of liquid-ordered domains or lipid rafts.41 This finding correlates well with the recent report that mice lacking SCD1 in a leptin deficient background have massive accumulation of free cholesterol and SFA in pancreatic beta cells.18 Our study further supports this idea, given that SCD1 inhibition resulted in accumulation of free cholesterol and SFA in the liver (Suplemental Figure 2B and 2D), aorta (Figure 3E, 3F, 3G, and 3H), and isolated macrophages (data not shown). Collectively, these data suggest that SCD1 may play a crucial role in limiting accumulation of lipids (cholesterol and SFA) known to segregate into membrane liquid-ordered domains, which could potentially alter membrane-associated signal transduction.
In summary, this study demonstrates that inhibition of SCD1 protects against development of the metabolic syndrome, but may promote atherosclerosis. These results do not support the idea that obesity and insulin resistance are causatively linked to atherosclerosis, and argue against SCD1 inhibition as a safe therapeutic target for treatment of CVD.
Supplementary Material
Acknowledgments
We thank Rosanne Crooke, Mark Graham, and Richard Lee (ISIS Pharmaceuticals, Inc. Carlsbad, CA USA) for providing ASOs used in this study. We thank James Ntambi, Alan Tall, Helen Hobbs, Joachim Herz, and Jay Horton for providing antibodies used in this study.
Sources of Funding
This work was supported by grants from the National Institutes of Health (NIH-P01-HL49373 to L.L.R. and J.S.P.), the American Heart Association (AHA postdoctoral fellowship # 0625400U to J.M.B), and the Howard Hughes Medical Institute (Gilliam Fellowship to T.N.).
Footnotes
Disclosures
None.
Clinical Perspective
The metabolic syndrome has become a leading health concern in developed countries. Importantly, presence of the metabolic syndrome has been shown to be a predictor of atherosclerotic cardiovascular disease extent in humans. Simply by catalyzing the conversion of long-chain saturated fatty acids (SFA) to monounsaturated fatty acids, stearoyl-CoA desaturase 1 (SCD1) has been shown to promote multiple aspects of the metabolic syndrome. Therefore, inhibition of SCD1 is currently regarded as a promising therapeutic strategy, yet little information exists regarding whether SCD1 inhibition could also protect against atherosclerosis. To examine this possibility we inhibited SCD1 in a hyperlipidemic mouse model of atherosclerosis. In agreement with previous reports, inhibition of SCD1 protected against diet-induced obesity, insulin resistance, hypertriglyceridemia, and hepatic steatosis. However, quite unexpectedly, SCD1 inhibition strongly promoted aortic atherosclerosis. Since dietary SFAs have been shown to promote inflammation through toll-like receptor 4 (TLR4), we examined macrophage TLR4 function. Interestingly, SCD1 inhibition resulted in marked hypersensitivity to TLR4 agonists in macrophages. This study is the first to report the consequences of inhibiting SCD1 on atherosclerosis. Although the presence of the metabolic syndrome may be associated with atherosclerosis in humans, this study provides evidence that the metabolic syndrome can be completely dissociated from atherosclerosis in mice. Taken together, these results suggest that the link between obesity and systemic insulin resistance and atherosclerosis should be approached with caution, and that SCD1 inhibition may not necessarily be a treatment for atherosclerosis and its complications.
References
- 1.Grundy SM. Metabolic syndrome pandemic. Arterioscler. Thromb. Vasc. Biol. 2008;28:629–636. doi: 10.1161/ATVBAHA.107.151092. [DOI] [PubMed] [Google Scholar]
- 2.Kahn R. Metabolic syndrome: is it a syndrome? Does it matter? Circulation. 2007;115:1806–1810. doi: 10.1161/CIRCULATIONAHA.106.658336. [DOI] [PubMed] [Google Scholar]
- 3.Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC, Jr, Spertus JA, Costa F. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–2752. doi: 10.1161/CIRCULATIONAHA.105.169404. [DOI] [PubMed] [Google Scholar]
- 4.Cohen P, Ntambi JM, Friedman JM. Stearoyl-CoA desaturase-1 and the metabolic syndrome. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2003;3:271–280. doi: 10.2174/1568008033340117. [DOI] [PubMed] [Google Scholar]
- 5.Dobrzyn A, Ntambi JM. Stearoyl-CoA desaturase as a new drug target for obesity treatment. Obes. Rev. 2005;6:169–174. doi: 10.1111/j.1467-789X.2005.00177.x. [DOI] [PubMed] [Google Scholar]
- 6.Miyazaki M, Ntambi JM. Role of stearoyl-coenzyme A desaturase in lipid metabolism. Prostaglandins Leukot. Essent. Fatty Acids. 2003;68:113–121. doi: 10.1016/s0952-3278(02)00261-2. [DOI] [PubMed] [Google Scholar]
- 7.Ntambi JM, Miyazaki M, Stoehr JP, Lan H, Kendziorski CM, Yandell BS, Song Y, Cohen P, Friedman JM, Attie AD. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. USA. 2002;99:11482–11486. doi: 10.1073/pnas.132384699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen P, Miyazaki M, Socci ND, Hagge-Greenberg A, Liedtke W, Soukas AA, Sharma R, Hudgins LC, Ntambi JM, Friedman JM. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science. 2002;297:240–243. doi: 10.1126/science.1071527. [DOI] [PubMed] [Google Scholar]
- 9.Macdonald ML, Singaraja RR, Bissada N, Ruddle P, Watts R, Karasinska JM, Gibson WT, Fievet C, Vance JE, Staels B, Hayden MR. Absence of stearoyl-CoA desaturase-1 ameloriates features of the metabolic syndrome in LDLR-deficient mice. J. Lipid Res. 2008;49:217–229. doi: 10.1194/jlr.M700478-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang G, Li Z, Liu F, Ellsworth K, Dallas-Yang Q, Wu M, Ronan J, Esau C, Murphy C, Szalkowski D, Bergeron R, Doebber T, Zhang BB. Prevention of obesity in mice by antisense oligonucleotide inhibitors of stearoyl-CoA desaturase-1. J. Clin. Invest. 2005;115:1030–1038. doi: 10.1172/JCI23962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyazaki M, Dobrzyn A, Sampath H, Lee SH, Man WC, Chu K, Peters JM, Gonzalez FJ, Ntambi JM. Reduced adiposity and liver steatosis by stearoyl-CoA desaturase deficiency are independent of peroxisome proliferators-activated receptor-alpha. J. Biol. Chem. 2004;279:35017–35024. doi: 10.1074/jbc.M405327200. [DOI] [PubMed] [Google Scholar]
- 12.Asilmaz E, Cohen P, Miyazaki M, Dobrzyn P, Ueki K, Fayzikhodjaeva G, Soukas AA, Kahn CR, Ntambi JM, Socci ND, Friedman JM. Site and mechanism of leptin action in a rodent form of congenital lipodystrophy. J. Clin. Invest. 2004;113:414–424. doi: 10.1172/JCI19511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyazaki M, Dobrzyn A, Man WC, Chu K, Sampath H, Kim HJ, Ntambi JM. Stearoyl-CoA desaturase 1 gene expression is necessary for fructose-mediated induction of lipogenic gene expression by sterol regulatory element-binding protein-1c-dependent and -independent mechanisms. J. Biol. Chem. 2004;279:25164–25171. doi: 10.1074/jbc.M402781200. [DOI] [PubMed] [Google Scholar]
- 14.Miyazaki M, Flowers MT, Sampath H, Chu K, Otzelberger C, Liu X, Ntambi JM. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab. 2007;6:484–496. doi: 10.1016/j.cmet.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 15.Attie AD, Krauss RM, Gray-Keller MP, Brownlie A, Miyazaki M, Kastelein JJ, Lusis AJ, Stalenhoef AF, Stoehr JP, Hayden MR, Ntambi JM. Relationship between stearoyl-CoA desaturase activity and plasma triglycerides in human and mouse hypertriglyeridemia. J. Lipid Res. 2002;43:1899–1907. doi: 10.1194/jlr.m200189-jlr200. [DOI] [PubMed] [Google Scholar]
- 16.Chu K, Miyazaki M, Man WC, Ntambi JM. Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol. Cell. Biol. 2006;26:6786–6798. doi: 10.1128/MCB.00077-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gutierrez-Juarez R, Pocai A, Mulas C, Ono H, Bhanot S, Monia BP, Rosetti L. Critical role of stearoyl-CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance. J. Clin. Invest. 2006;116:1686–1695. doi: 10.1172/JCI26991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flowers JB, Rabglia ME, Schueler KL, Flowers MT, Lan H, Keller MP, Ntambi JM, Attie AD. Loss of stearoyl-CoA desaturase-1 improves insulin sensitivity in lean mice but worsens diabetes in leptin-deficient obese mice. Diabetes. 2007;56:1228–1239. doi: 10.2337/db06-1142. [DOI] [PubMed] [Google Scholar]
- 19.Miyazaki M, Kim YC, Gray-Keller MP, Attie AD, Ntambi JM. The biosynthesis of hepatic cholesterol esters and triglycerides is impaired in mice with a disruption of the gene for stearoyl-CoA desaturase 1. J. Biol. Chem. 2000;275:30132–30128. doi: 10.1074/jbc.M005488200. [DOI] [PubMed] [Google Scholar]
- 20.Veniant MM, Sullivan MA, Kim SK, Ambroziak P, Chu A, Wilson MD, Hellerstein MK, Rudel LL, Walzem RL, Young SG. Defining the atherogenicity of large and small lipoproteins containing apolipoprotein B100. J. Clin. Invest. 2000;106:1501–1510. doi: 10.1172/JCI10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bell TA, III, Kelley K, Wilson MD, Sawyer JK, Rudel LL. Dietary fat-induced alterations in atherosclerosis are abolished by ACAT2-deficiency in ApoB100 only, LDLr-/- mice. Arterioscler. Thromb. Vasc. Biol. 2007;27:1396–1402. doi: 10.1161/ATVBAHA.107.142802. [DOI] [PubMed] [Google Scholar]
- 22.Levin AA, Yu RZ, Geary RS. Antisense Drug Technology. 2nd edition CRC Press; Boca Raton, FL: 2008. Basic principles of the pharmacokinetics of antisense oligonucleotide drugs. [Google Scholar]
- 23.Choi CS, Savage DB, Kulkarni A, Yu XX, Liu ZX, Morino K, Kim S, Distefano A, Samuel VT, Neschen S, Zhang D, Wang A, Zhang XM, Kahn M, Cline GW, Pandey SK, Geisler JG, Bhanot S, Monia BP, Shulman GI. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J. Biol. Chem. 2007;282:22678–22688. doi: 10.1074/jbc.M704213200. [DOI] [PubMed] [Google Scholar]
- 24.Lee SH, Dobrzyn A, Dobrzyn P, Rahman SM, Miyazaki M, Ntambi JM. Lack of stearoyl-CoA desaturase 1 upregulates basal thermogenesis but causes hypothermia in a cold environment. J. Lipid Res. 2004;45:1674–1682. doi: 10.1194/jlr.M400039-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Rudel LL, Haines J, Sawyer JK, Shah R, Wilson MS, Carr TP. Hepatic origin of cholesteryl oleate in coronary artery atherosclerosis in African green monkeys. Enrichment by dietary monounsaturated fat. J. Clin. Invest. 1997;100:74–83. doi: 10.1172/JCI119524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hansson GK, Libby P, Schonbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ. Res. 2002;91:281–291. doi: 10.1161/01.res.0000029784.15893.10. [DOI] [PubMed] [Google Scholar]
- 27.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- 29.Suganami T, Tanimoto-Koyama K, Nishida J, Itoh M, Yuan X, Mizuarai S, Kotani H, Yamaoka S, Miyake K, Aoe S, Kamei Y, Ogawa Y. Role of the Toll-like receptor4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler. Thromb. Vasc. Biol. 2007;27:84–91. doi: 10.1161/01.ATV.0000251608.09329.9a. [DOI] [PubMed] [Google Scholar]
- 30.Renier G, Skamene E, DeSanctis J, Radzioch D. Dietary n-3 polyunsaturated fatty acids prevent the development of atherosclerotic lesions in mice. Modulation of macrophage secretory activities. Arterioscler. Thromb. 1993;13:1515–1524. doi: 10.1161/01.atv.13.10.1515. [DOI] [PubMed] [Google Scholar]
- 31.Takeda K, Akira S. TLR signaling pathways. Semin. Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 32.Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, Freudenberg M, Beutler B. CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 33.Thomas KE, Galligan CL, Newman RD, Fish EN, Vogel SN. Contribution of interferon-beta to the murine macrophage response to the toll-like receptor 4 agonist, lipopolysaccharide. J. Biol. Chem. 2006;281:31119–31130. doi: 10.1074/jbc.M604958200. [DOI] [PubMed] [Google Scholar]
- 34.Semenkovich CF. Insulin resistance and atherosclerosis. J. Clin. Invest. 2006;116:1813–1822. doi: 10.1172/JCI29024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C, Shah YM, Morimura K, Krausz KW, Miyazaki M, Richardson TA, Morgan ET, Ntambi JM, Idel JR, Gonzalez FJ. Metabolomics reveals that hepatic stearoyl-CoA desaturase 1 downregulation exacerbates inflammation and acute colitis. Cell Metab. 2008;7:135–147. doi: 10.1016/j.cmet.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 37.Flowers MT, Groen AK, Oler AT, Keller MP, Choi Y, Schueler KL, Richards OC, Lan H, Miyazaki M, Kuipers F, Kendziorski CM, Ntambi JM, Attie AD. Cholestasis and hypercholesterolemia in SCD1-deficient mice fed a low-fat, high-carbohydrate diet. J. Lipid Res. 2006;47:2668–2680. doi: 10.1194/jlr.M600203-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E, Willeit J, Schwartz DA. Toll-lik receptor 4 polymorphisms and atherogenesis. N. Engl. J. Med. 2002;347:185–192. doi: 10.1056/NEJMoa012673. [DOI] [PubMed] [Google Scholar]
- 39.Yang IA, Holloway JW, Ye S. TLR4 Asp299Gly polymorphism in not associated with coronary artery stenosis. Atherosclerosis. 2003;170:187–190. doi: 10.1016/s0021-9150(03)00286-7. [DOI] [PubMed] [Google Scholar]
- 40.Netea MG, Hijmans A, van Wissen S, Smilde TJ, Trip MD, Kullberg BJ, de Boo T, Van der Meer JW, Kastelein JJ, Stalenhoef AF. Toll-like receptor-4 Asp299Gly polymorphism does not influence progression of atherosclerosis in patients with familial hypercholesterolemia. Eur. J. Clin. Invest. 2004;34:94–99. doi: 10.1111/j.1365-2362.2004.01303.x. [DOI] [PubMed] [Google Scholar]
- 41.Sun Y, Hao M, Luo Y, Liang CP, Silver DL, Cheng C, Maxfield FR, Tall AR. Stearoyl-CoA desaturase inhibits ATP-binding cassette transporter A1-mediated cholesterol efflux and modulates membrane domain structure. J. Biol. Chem. doi: 10.1074/jbc.M208687200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.