

Abstract
BACKGROUND:
Several investigations have implicated cytokines such as tumour necrosis factor-alpha (TNF-α), interleukin (IL)-1, IL-6, IL-8 and transforming growth factor-beta in the pathophysiology of cellular dysfunction in ischemia-reperfusion (I/R). Although an increase in the production of these cytokines has been detected after myocardial infarction and cardiopulmonary bypass surgery, their exact role and mechanisms for inducing cardiac dysfunction are poorly understood.
OBSERVATIONS:
TNF-α, transforming growth factor-beta, IL-1, IL-6 and IL-8 have frequently been studied in different cardiovascular diseases, including I/R injury in the heart. Low concentrations of TNF-α appear to exert cardioprotective effects, whereas high concentrations have been shown to produce deleterious actions in the heart. Some efforts have been made to explore the molecular mechanisms of cytokine actions; however, such information is insufficient to develop therapeutic strategies to combat their deleterious effects during the development of I/R injury in the heart.
CONCLUSIONS:
In addition to a time-dependent response, the conflicting effects of cytokines seem to depend on their concentrations used in different experimental studies. It is also likely that both the beneficial and pathophysiological actions of cytokines occur concomitantly. On the basis of the existing literature, it is suggested that different ways need to be found to modify the synthesis as well as the cardiodepressant actions of cytokines to improve the therapy of ischemic heart disease.
Keywords: Cardiac dysfunction, Cytokines, Interleukins, Ischemic heart disease, TNF-α
Ischemia-reperfusion (I/R) injury is one of the most common cardiovascular problems and is associated with various clinical conditions such as arteriosclerosis, coronary spasm and thrombosis (1). Several investigators (2,3) have shown that accumulation of protons, cessation of oxidative metabolism and damage of electron transport are the major characteristics of myocardial ischemic injury on stoppage of blood flow. Although reinstitution of flow by procedures such as angioplasty, thrombolysis or coronary bypass surgery is essential for salvaging ischemic myocardium, re-establishment of blood flow beyond a certain period of ischemic insult has been observed to produce adverse effects commonly known as reperfusion injury or I/R injury (1,4). It has been shown that I/R injury is associated with reperfusion arrhythmias, myocardial stunning, microvascular damage and accelerated necrosis (4). Multiple factors that are involved in myocardial cell damage and cardiac dysfunction due to I/R injury show the occurrence of intracellular Ca2+ overload, production of oxygen-derived free radicals and alterations in different enzyme activities (5,6).
Recent studies have shown that myocardial ischemic insult promotes the formation of cytokines, a group of low molecular weight polypeptides that are autocrine contributors to cardiac dysfunction and cardiomyocyte necrosis, as well as apoptosis in I/R injury. These mediators mainly include tumour necrosis factor-alpha (TNF-α), transforming growth factor-beta (TGF-β) and interleukins (ILs) such as IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8 and IL-12 (7–11). The specific targets of such mediators appear to be the endothelium and neutrophils (these deleterious mediators act on neutrophils for adherence to the vascular endothelium and, thus, induce the obstruction of capillary beds to cause a no-reflow phenomenon during reperfusion) (12,13). In fact, the accumulation of these cytokines, especially TNF-α and IL-1, within the ischemic zone damages the tissue and releases oxygen free radicals that produce myocardial cell damage and induce cardiac dysfunction (12,13). In addition, it has been reported that cytokines have numerous basic functions including activation of leukocytes, promotion of inflammation, control of cell division, induction of certain genes to produce a multitude of proteins for cellular/humoral immunity and initiation of other cytokine synthesis (14). In view of the fact that TNF-α, TGF-β, IL-1, IL-6 and IL-8 have been frequently studied in ischemic heart disease (IHD) (15,16), the present article is intended to review the role of these cytokines in I/R injury and to examine whether the modification of their synthesis is associated with alterations in cellular function.
ROLE OF TNF-α IN I/R INJURY
In the cytokine family, TNF-α is considered to be the most important mediator of cardiovascular disease. This substance was discovered by the surgeon William Coley (17), who reported that TNF-α was primarily produced by lymphocytes and macrophages, and mediates endotoxin-induced tumour necrosis; other cells such as cardiomyocytes, resident cardiac macrophages and vascular smooth muscle cells have also been shown to produce TNF-α (18). TNF-α is synthesized as a 26 kDa propeptide (pro-TNF-α) in the cytosol, which is then cleaved to a 17 kDa active form by TNF-α-converting enzyme (TACE); this cleavage occurs as pro-TNF-α passes through the cell membrane. The activated form of TNF-α binds to the receptors on the cell membrane surface and triggers alterations in cytosolic protein synthesis and activation of different kinases (18,19). Similar to other protein synthesis pathways, both transcription and translational processes are involved in the regulation of TNF-α synthesis. At the transcriptional level, nuclear factor kappa B (NFκB) is the major redox-sensitive transcriptional factor associated with TNF-α production. It has been shown that inhibition of NFκB signalling completely blocks lipopolysaccharide (LPS)-induced TNF-α production (20). At the translational level, TACE plays an important role in converting pro-TNF-α to TNF-α and, thus, inhibition of TACE activity is effective in controlling the synthesis of TNF-α (21). Although LPS has been viewed as an important trigger for the production of TNF-α, several studies (22–25) have shown that TNF-α is formed and released from the rat and human myocardium after I/R injury. The pharmacological inhibition of TNF-α synthesis significantly improves the recovery of myocardial dysfunction induced by I/R (26–33). Although LPS-induced increases in TNF-α expression have been extensively studied, the mechanisms of I/R-induced TNF-α synthesis and myocardial alterations due to TNF-α during I/R in the myocardium are not fully understood.
It has been previously shown that TNF-α directly decreases contractile function in hamster, rat, dog and human myocardium (31,32,34–36). The acute negative inotropic effect of TNF-α appears to be due to alterations in Ca2+ handling, which are manifested by attenuated Ca2+-induced Ca2+ release from the sarcoplasmic reticulum and myofilament Ca2+ sensitivity (Figure 1). The initial contractile depression induced by TNF-α is mediated by activation of sphingomyelinase (37), which hydrolyzes the phospholipid sphingomyelin to ceramide (38). In the presence of acidic or neutral ceramidase, ceramide is deacylated to sphingosine, an endogenous second messenger (39) that causes blockade of ryanodine receptors in the sarcoplasmic reticulum and, thus, decreases Ca2+-induced Ca2+ release and myocardial contractility (40). Delayed contractile depression by prolonged TNF-α exposure is mediated by the induction of inducible nitric oxide synthase (iNOS), with subsequent production of nitric oxide (NO). NO prevents Ca2+ influx via cyclic GMP-dependent inhibition of the L-type Ca2+ channels in sarcolemma (SL), depresses myofilament sensitivity to Ca2+ and, subsequently, attenuates myocardial contractility (34,41) (Figure 1). TNF-α-mediated delayed contractile depression has also been shown to be associated with desensitization of β-adrenoceptor mechanisms in the SL membrane (30). Although different investigators have revealed the involvement of both NO-dependent and NO-independent functional uncoupling of β-adrenoceptor to adenylyl cyclase (42), the exact mechanisms are not completely understood. Similarly, the cellular events in TNF-α-mediated depressed contractility after binding to the SL membrane receptors remain to be elucidated.
Figure 1).
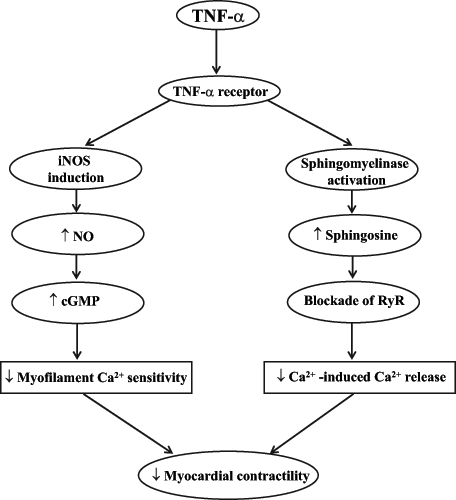
Effect of tumour necrosis factor-alpha (TNF-α) on myocardial contractility. ↓ Decrease; ↑ Increase; cGMP Cyclic GMP; iNOS Inducible nitric oxide synthase; NO Nitric oxide; RyR Ryanodine receptors
Different signal transduction pathways include an increase in phosphatidylcholine (PC)-specific phospholipase C (PLC) and phospholipase D (PLD) activities subsequent to the binding of TNF-α to the TNF-α receptors (TNF-Rs) (43,44). Activated PLC and PLD have been shown to hydrolyze PC to diacylglycerol (DAG) and phosphatidic acid (PA), respectively (45). DAG activates protein kinase C, which is involved in multiple signalling pathways that result in apoptosis, phosphorylation of troponin T and troponin I, and an increase in NFκB activity (46,47). Production of PA has also been associated with the pathogenesis of TNF-α-induced heart injury (10); moreover, PA has been shown to cause Ca2+ overload and activation of extracellular-signal-regulated protein kinase, which stimulates the activation of NFκB and, thus, plays an important role in the development of the positive feedback loop of TNF-α and NFκB (48). Furthermore, TNF-α bound to TNF-Rs triggers the activation of phospholipase A2 and generates arachidonic acid and prostaglandins; this mechanism appears to explain the proinflammatory activities of TNF-α (49). It is important to point out that DAG, PA and arachidonic acid have also been shown to activate sphingomyelinase, the key enzyme of the sphingomyelin pathway (50), thus further aggravating TNF-α-induced contractile dysfunction.
Another mechanism of cardiac depression induced by TNF-α is associated with the induction of apoptosis in cardiomyocytes (Figure 2). It has been reported (21) that apoptosis is mainly induced via TNF-α binding to either TNF-R1 or Fas. TNF-R1 and Fas are linked with cytoplasmic proteins that are referred to as the TNF-R1-associated death domain and Fas-associated death domain, respectively. Binding of TNF-α to TNF-R1 or Fas causes conformational changes in the TNF-R1-associated and Fas-associated death domains, triggering their binding with receptor-interacting protein-2, which has a kinase domain (51). McCarthy et al (52) have shown an association of initiator caspases 1, 2 and 9 with receptor-interacting protein-2. Proteolytic cleavage of initiator caspases leads to the activation of downstream effector caspases 3, 6 and 7. Activation of effector caspases promotes the activation of endonucleases, chromatin condensation and DNA fragmentation leading to apoptosis (53). On the other hand, the binding of TNF-α to TNF-R2, which is linked with TNF-R-associated factors, causes activation of NFκB (21). The association of TNF-R1 to TNF receptor-associated factors with subsequent activation of NFκB has been previously reported (54). Activation of NFκB has been implicated in the induction of genes involved in cell proliferation, growth, survival and death (55). The proposed mechanism is outlined in Figure 3. Nonetheless, the regulatory balance between cell survival and apoptosis by NFκB activation remains to be characterized in I/R heart.
Figure 2).
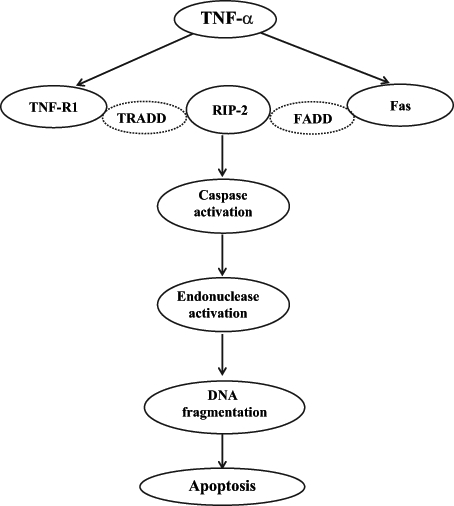
Proposed outline of the pathway of tumour necrosis factor-alpha (TNF-α)-mediated apoptosis. FADD Fas-associated death domain; RIP-2 Receptor-interacting protein-2; TNF-R1 TNF-α receptor-1; TRADD TNF-R1-associated death domain
Figure 3).
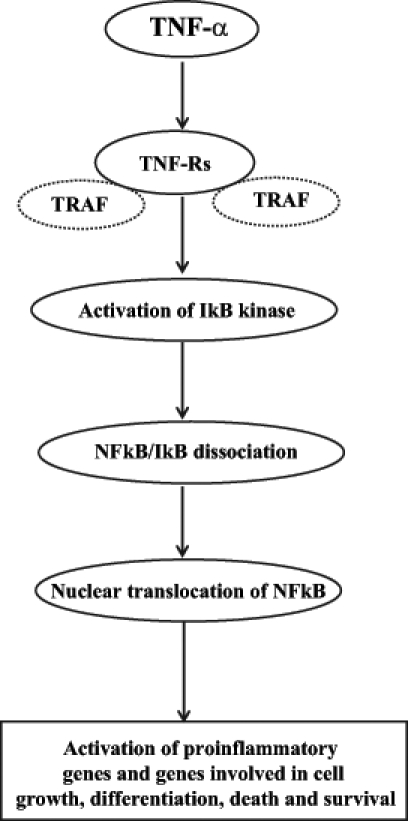
Mechanism of tumour necrosis factor-alpha (TNF-α)-mediated activation of nuclear factor kappa B (NFκB). IκB Inhibitory kappa B; TNF-Rs TNF-α receptors; TRAF TNF receptor-associated factors
TNF-α-mediated apoptosis appears to be mediated by sphingosine and NO (56,57). Additionally, TNF-α has been indicated to be an initiator of a cytokine cascade, which results in the production of IL-6, IL-1 and IL-8, and ultimately, a worsening of the deleterious alterations induced by I/R (22,58,59). These studies seem to suggest that anti-TNF-α therapy may be valuable in I/R injury. However, it has been indicated that high circulating levels of TNF-α could be an adaptive response, which acts by promoting the shedding of TNF-Rs and reducing the number of active receptors (60). The shedding of TNF-Rs causes the formation of soluble forms of TNF-Rs (sTNF-Rs) (61). Different investigators have implicated the activation of metalloproteinase in the release of sTNF-Rs (62,63). On one hand, sTNF-Rs have been shown to exert protective effects such as inhibiting TNF-α bioactivity, serving as a TNF-α antagonist, blocking the effects of high concentrations of TNF-α and maintaining the basal TNF-α level (64). On the other hand, sTNF-Rs exert adverse effects by acting as carriers that transport TNF-α to other body compartments, by slowing the release of TNF-α and by prolonging the half-life of TNF-α by stabilizing its bioactivity (61,64). Thus, it appears that both the deleterious and beneficial effects of TNF-α may be occurring concomitantly.
A large body of evidence has accumulated to provide some information on interventions for blocking TNF-α synthesis in the ischemic heart (Table 1). p38 mitogen-activated protein kinase (MAPK) and NFκB inhibitors appear to depress the synthesis of this proinflammatory factor (21,65). Adenosine and noradrenaline, released during transient ischemia, have also been shown to reduce cardiac TNF-α production in humans (21,31,66). Maekawa et al (67) have reported reduced infarct size, decreased occurrence of arrhythmia and improved cardiac function upon subjecting TNF-α knockout mice to I/R injury compared with wild-type mice. Furthermore, TNF-α antibody, IL-1 receptor antagonist (68) and sTNF-R were also found to attenuate the deleterious effects of TNF-α in I/R heart injury in rats (33). Other interventions, including agonists of glycoprotein 130 receptor subunits (69–72), an inhibitor of TACE (73,74), an inhibitor of serine protease, aprotonin (75) and heat shock proteins (HSPs) such as HSP70 and HSP72 (76–78), have also been used in clinical and experimental trials.
TABLE 1.
Modification of tumour necrosis factor-alpha (TNF-α) synthesis by different pharmacological interventions in ischemic-reperfused heart
| Target | Agent | Effect | References |
|---|---|---|---|
| Neutralization of TNF-α | Soluble TNF-α receptor | ↓ TNF-α bioactivity | 33 |
| IL-receptor antagonist | ↓ TNF-α synthesis | 68,70 | |
| NFκB antagonism | Antioxidant (vitamin E, N-acetylcysteine) | ↓ NFκB activation | 94–96 |
| Pentoxifylline/heparin | ↓ NFκB translocation | 98–100 | |
| 20S proteasome inhibitor (PS519) | ↓ NFκB activation | 97 | |
| HSP70 | ↓ NFκB translocation | 78 | |
| p38 MAPK inhibition | SB 203580 | ↓ TNF-α transcription | 65 |
| Metalloproteinase inhibition (TACE inhibitor) | GI 129471 | ↓ pro-TNF-α conversion to TNF-α | 73–75 |
| Aprotonin | |||
| Ischemic preconditioning | Adenosine | ↓ TNF-α production, ↓ iNOS expression, | 31,66 |
| Noradrenaline | ↓ TNF-α bioactivity | ||
| Induction of HSPs | HSP70, HSP72 | Binding of HSPs to cytosolic TNF-α | 76–78 |
| gp130 subunit-linked agonism | IL-6 (IL-11, CT-1, leukemia inhibitory factor, oncostatin M receptor) | ↓ TNF-α production | 51,71,72 |
↓ Decrease; CT Cardiotropin; gp130 Glycoprotein 130; HSP Heat shock protein; IL Interleukin; iNOS Inducible nitric oxide synthase; MAPK Mitogen-activated protein kinase; NFκB Nuclear factor kappa B; pro-TNF-α TNF-α propeptide; TACE TNF-α-converting enzyme
In contrast to reports showing adverse effects of TNF-α, different investigators have observed that TNF-α may have protective effects during I/R (65,67,69,79–81). Lecour et al (81) have shown that TNF-α-evoked preconditioning is an effective intervention for the protection of I/R injury. Nelson et al (69) have indicated that pretreatment with TNF-α 24 h before the I/R period results in improved cardiac contractile function in rabbits. Several studies (79,80) have also suggested that TNF-α knockout mice may display larger infarct size after undergoing coronary ligation compared with normal mice. Such results indicate that the beneficial effects of TNF-α may be due to varying time periods, during which the heart is exposed to different concentrations of TNF-α. Although it has been indicated that the adverse and beneficial effects of TNF-α are probably dependent on the absolute levels of TNF-α during the I/R period (18), the effects of TNF-α and the mechanism of the beneficial effects remain a matter of debate. It appears that low concentrations of TNF-α exert beneficial effects, whereas high concentrations produce deleterious actions in the isolated heart (35). Similarly, in isolated cardiomyocytes, low concentrations of TNF-α cause an increase in the intracellular Ca2+ concentration and cardiac contraction, whereas high concentrations attenuate the electrically stimulated Ca2+ transient and cardiac contraction (82). Hence, to understand the pathogenesis of different heart diseases, further research is needed to elucidate the role of TNF-α in both cardioprotection and cardiodepression.
ROLE OF NFκB IN CYTOKINE PRODUCTION
NFκB is a redox-sensitive transcription factor that plays a key role in the production of most cytokines. It has been shown that NFκB exists in an inactive state in the cytoplasm of unstimulated cells because it is bound to inhibitory kappa B (IκB). The NFκB family is comprised of different subunits such as p50, p52, p65 (Rel), c-Rel, p52 and Rel B. Multiple subfamilies of IκB have also been shown to exist, including IκB-α, -β, -gamma (p105), -delta (p100), -epsilon and Bcl-3 (83). The most common active form of NFκB is the p50/p65 dimer, which is associated with the inhibitory protein IκB-α. The crucial step in the activation of NFκB is the phosphorylation of IκB by a multimeric complex referred to as IκB kinase. Anoxia, reactive oxygen species, LPS, IL-1 and TNF-α are considered to be the major stimuli that activate IκB kinase, leading to the dissociation of IκB from NFκB subunits (84). It has been reported (83,84) that multiple regulatory steps are involved in the activation of NFκB, which include nuclear translocation, phosphorylation of Rel family protein, interaction with the basal transcription complex and redox regulation. Blocking of any of the phases is likely to prevent the activation of NFκB, resulting in changes in NFκB-regulated gene expression for a broad range of physiological and pathophysiological processes. Various genes regulated by NFκB activation include cyclooxygenase-2, inhibitors of apoptotic factors, manganese superoxide dismutase, ILs (IL-1, IL-6 and IL-8), TNF-α, Fas ligands and cell adhesion molecules (83–85). In addition, the action of TNF-α on its receptors causes the activation of NFκB, which further stimulates production of TNF-α and, thus, develops a positive feedback loop (51) (Figure 4).
Figure 4).
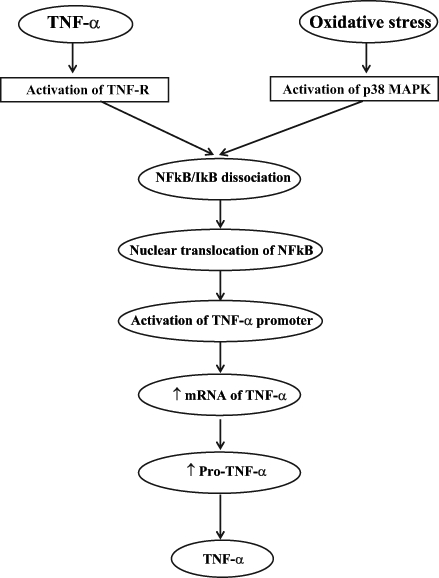
Proposed outline of the pathway of tumour necrosis factor-alpha (TNF-α) synthesis. ↑ Increase; IκB Inhibitory kappa B; MAPK Mitogen-activated protein kinase; mRNA Messenger RNA; NFκB Nuclear factor kappa B; Pro-TNF-α TNF-α propeptide; TNF-R TNF-α receptor
Recent evidence has indicated that NFκB is activated in the ischemic myocardium upon the initiation of reperfusion (86–88). Intracellular adhesion molecule-1 protein expression and iNOS are increased due to NFκB activation following I/R injury in canine heart (89). Additionally, locally produced cytokines in the myocardium have been reported to stimulate NFκB activation via PC-specific PLC and PLD pathways (48); this positive feedback loop further augments the local pathogenesis responses. Another important deleterious effect of NFκB is the promotion of apoptosis, which possibly induces irreversible myocardial damage or amplifies infarct size in myocardial infarction (90). Although Ca2+ is an important second messenger in the activation of NFκB in the kidneys and lymphocytes (91–93), oxidative stress is still considered an essential trigger for the activation of NFκB following I/R injury (83). An I/R-induced increase in oxidative stress causes the activation of p38 MAPK, which appears to be involved in NFκB activation followed by TNF-α production (21) (Figure 4). Several antioxidants such as N-acetylcysteine, α-lipoic acid and vitamin E inhibit the activation of NFκB (94–96). Cargnoni et al (94) have shown that N-acetylcysteine prevents the activation of NFκB by obstructing alterations in intracellular thiol, reduced glutathione and oxidized glutathione levels. In addition, the 20S proteasome inhibitor PS-519 was found to depress the activation of NFκB due to I/R injury in the myocardium (97). In cultured vascular smooth muscle cells, HSP70, pentoxifylline, a known phosphodiesterase inhibitor, and nonanticoagulant heparin were reported to inhibit NFκB activation (77,98–100). It is important to point out that the adverse effects of NFκB during I/R injury have been determined indirectly by functional studies of NFκB-regulated genes. Therefore, further details of this pathway are needed to understand the relationship between the activation of NFκB and I/R injury.
In contrast to the negative effect of NFκB in I/R injury, some studies have reported a protective effect of NFκB activation in hearts undergoing I/R. It has been suggested that NFκB may be a key mediator of the beneficial effect of preconditioning against I/R injury (101,102), ie, ischemic preconditioning protects the heart by activating NFκB (102). It has also been suggested that NFκB stimulates the production of cytoprotective genes (eg, HSPs) (103) and NO (104). These cytoprotective genes may inhibit NFκB activation induced by the overwhelming oxidative stress during I/R and, in turn, can lead to an inhibitory effect on the production of inflammatory genes (105). Furthermore, Bach et al (106) have also suggested that NFκB mediates numerous gene expressions of proteins that inhibit cell death; in particular, these proteins include the Bcl family, zinc finger protein, endogenous antioxidants, manganese superoxide dismutase and hemeoxygenase-1. In fact, Bcl-2 has been shown to cause activation of NFκB, with a subsequent reduction in apoptosis (107). Additionally, functional NFκB signalling seems to be crucial for suppressing TNF-α-mediated apoptosis in ventricular cardiomyocytes (108). Furthermore, recent studies (109) have shown that NFκB activation prevents hypoxia-induced cell death by preserving mitochondrial function. Thus, the role of NFκB in I/R injury is controversial and the pathways mediating protective and detrimental effects still need to be elucidated. It is also important to point out that the adverse effects of NFκB during I/R injury have been indirectly suggested by functional studies of NFκB-regulated genes. Therefore, further details of this pathway are needed to understand the relationship between the activation of NFκB and I/R injury.
ROLE OF TGF-β IN I/R INJURY
TGF is a family of peptides that exists in many mammalian tissues. Recently, TGF-β has received considerable attention for its multiple functions in controlling cell growth and responding to extracellular environmental changes (110). The most common form of TGF is TGF-β1, which modulates various biological functions and has been identified as a powerful cardioprotective agent (111–113). Lefer et al (114) have shown that the administration of TGF-β1 reduces infarct size in a feline model of myocardial I/R, and the protective effect seems to be due to inhibition of endothelial cell-neutrophil interactions, as well as anti-inflammatory actions that result from decreased TNF-α production. Baxter et al (115) have also observed a significant limitation of infarct size in rat heart and a reduction in apoptosis in ventricular myocytes due to TGF-β1 administration during the early reperfusion period. It has been noted that the p42/p44 MAPK (extracellular-signal-regulated protein kinase) signalling pathway may be involved in this cardioprotective effect; its involvement is suspected because PD98059, an inhibitor of p42/p44 MAPK, abolishes the attenuated infarct size and cardiomyocyte apoptosis after TGF-β1 treatment. In addition, the induction of the antiapoptotic protein Bcl-2 has been implicated in the protective effect of TGF-β1 during I/R injury in rat cardiac allografts (116). Furthermore, Mehta et al (117) have shown the participation of NO in TGF-β1 signalling; the hypoxia-reoxygenation-induced decrease in active TGF-β1 release was shown to be augmented by 3-morpholino-sydnonimine and nitroglycerine, known NO donors. Chen et al (118) have shown that the hypoxia-reoxygenation-mediated upregulation of iNOS expression, decrease in endothelial NOS and increase in Akt/PKB phosphorylation were attenuated by TGF-β1 treatment, indicating the involvement of these mediators in TGF-β1-mediated cardioprotection. Moreover, the expression of matrix metalloproteinases was also inhibited by TGF-β1, resulting in a significant improvement in cardiac function in I/R hearts (11). Therefore, TGF-β1 appears to promote a cardioprotective effect through a wide variety of intracellular signal pathways and, indeed, is a promising new approach to attenuate I/R injury.
ROLE OF IL-1 IN I/R INJURY
IL-1 is as an important mediator of inflammatory reactions. There are two forms of IL-1, namely, IL-α and IL-β. Because IL-β is easily detected in the blood, it has been the main focus in experimental research. Several studies (8,119,120) have shown the negative inotropic effect of IL-1 in both intact heart and isolated cardiomyocytes. IL-1 has been found to induce the expression of iNOS at both the messenger RNA (mRNA) and protein levels, and is considered to be involved in the p38 and p42/p44 MAPK signalling pathways (7). It has been observed that IL-β and TNF-α have similar pathways, causing negative inotropic effects in the myocardium (121). Furthermore, a decrease in the expression of Ca2+ regulatory genes has been shown to be involved in the deleterious effect of IL-1; this mechanism may be implicated in arrhythmogenesis after I/R injury (122–126). In addition, it has been observed that IL-1, like other cytokines, has the effect of inducing apoptosis in neonatal cardiac myocytes (127). Although most of the data suggest IL-1 has a primarily deleterious role in the heart, some investigations (125,128,129) have indicated a cardioprotective effect of IL-1.
ROLE OF IL-6 IN I/R INJURY
IL-6 was originally identified as a T cell-derived cytokine, but it has now been documented as a multifunctional cytokine produced by different cells types. The IL-6 family is comprised of IL-11, leukemia inhibitory factor, oncostatin M and cardiotropin-1 (CT-1). All cytokines of this family regulate intracellular signalling upon binding to receptors with a glyco-protein 130 subunit (121,130). Elevated levels of IL-6 have been detected in patients with myocardial infarction, especially during the period of reperfusion, indicating a role for IL-6 in the pathogenesis of IHD (131). Other experimental studies (132–134) have also reported an elevation of IL-6 mRNA and protein content in canine myocardium subjected to I/R injury. An increasing number of investigations (34) have shown that IL-6 serves as a direct cardiodepressant; it has been reported to inhibit myocardial contractility in hamster myocardium. It has also been shown that IL-6 reduces the peak systolic Ca2+ transient and contractility by increasing the production of NO and a subsequent cyclic GMP-mediated decrease in L-type Ca2+ channel current (135,136). Furthermore, it has been suggested that IL-6 induces the expression of iNOS in isolated cardiomyocytes, which subsequently causes a sustained depression of myocardial contractility (34,135,136). The induction of IL-6 has also been involved in the expression of intracellular adhesion molecule-1, which leads to inflammatory injury in canine ischemic heart (133,137). Conversely, pretreatment with CT-1, a member of the IL-6 family, has been shown to protect cultured cardiomyocytes against simulated ischemia/hypoxia, which may be mediated by enhancing the protein levels of HSP70 and HSP90 (138). In addition, Latchman (139) has reported that CT-1 protects the cultured cardiac cells and/or isolated rat heart from I/R injury by regulating the p42/p44 MAPK pathway. Furthermore, Craig et al (140) have shown the antiapoptotic effect of IL-6 in isolated cardiomyocytes. Therefore, further studies are needed to differentiate IL-6-mediated cell survival and cell death pathways that may be stimulated concomitantly by this cytokine.
ROLE OF IL-8 IN I/R INJURY
Like other cytokines, IL-8 is also expressed at the mRNA level in myocardium subjected to I/R injury (141). In addition, various reports (142–144) have suggested that IL-8 is important in the development of myocardial injury in human myocardial infarction. Cells such as neutrophils, monocytes, T lymphocytes and endothelial cells have also been shown to produce IL-8 (145). IL-8 is considered to be a potent participant in granule enzymatic release and oxidative burst in neutrophils, which in turn lead to further damage in the ischemic heart (121). Boyle et al (146) have shown that a depression in IL-8 level protects the heart from I/R injury in rabbits; however, the role of IL-8 in the interaction of neutrophils and endothelial cells has been controversial. Some results have shown inhibitory effects (9,147,148), whereas others have shown stimulatory effects on the migration of neutrophils through the swollen endothelium, tyrosine kinases Src and focal adhesion kinase activity (145,149–152). Although the effect of IL-8 on neutrophils is conflicting, it has been reported (121) that IL-8 is not involved in the regulation of myocardial contractility after I/R. Hence, IL-8 may be a minor factor in mediating the inflammatory effect in hearts subjected to I/R.
CONCLUSIONS
Different cytokines produced during I/R act as intracellular communication molecules within a complex network of interrelated and interacting signals, and participate in the alteration of myocardial function. Recent studies (153,154) have shown the involvement of an ischemic stimulus in cytokine production during postmyocardial infarction remodelling. As shown in Table 2, most cytokines cause deleterious activities; TNF-α, IL-1 and IL-6 cause a negative inotropic effect and induce apoptosis in myocardium subjected to I/R injury, whereas IL-8 has a mild inflammatory effect in the ischemic heart. However, some cytokines play cardioprotective roles in I/R injuries; TGF-β has been shown to promote beneficial effects by reducing cell apoptosis, decreasing infarct size and depressing TNF-α synthesis by activating multiple protein kinases and MAPK. Furthermore, CT-1, a subfamily of IL-6, has been reported to present a cardioprotective effect in I/R injury and has become an important target in cardiovascular research. From the forgoing discussion, it is also evident that cytokines such as TNF-α, IL-1 and IL-8 show conflicting results because both adverse effects and cardioprotective actions in I/R heart have been reported. It appears that the deleterious actions of these cytokines, especially TNF-α, occur with high concentrations and/or prolonged exposure, whereas the beneficial effects occur as initial responses or at low cytokine concentrations. Although both the adverse and cardioprotective effects of cytokines appear to be occurring concomitantly, the predominance of deleterious actions may play a critical role in the development of heart disease. Because interventions such as pentoxifylline have been reported to reduce the level of TNF-α and improve cardiac function in Ca2+ overloaded (Ca2+ paradox) hearts and in I/R hearts (155,156), it is suggested that pharmacological manipulation of cytokine levels may prove helpful in designing improved therapy for IHD.
TABLE 2.
Effects of cytokines on ischemia-reperfusion-induced cardiac injury
| Cytokine | Receptors | Second messengers/pathways | Effect | References |
|---|---|---|---|---|
| TNF-α | TNF-α receptors | NO, sphingosine, MAPK, PKC, NFκB | Apoptosis, negative inotropic effect, initiator of synthesis of other cytokines, and inflammatory effect | 27,32,34,39, 41–44,56,64 |
| TGF-β | TGF-β1 receptors | PKC, Bcl-2, p42/p44 MAPK | ↓ Apoptosis, ↓ infarct size, ↓ endothelial cell/neutrophil interaction, anti-inflammatory, ↑ endothelium-dependent relaxation, ↓ TNF-α production | 111,114–118 |
| IL-1 | IL-1β receptors | NO, MAPK, NFκB | Apoptosis, negative inotropic effect, ↓ Ca2+-regulated gene expression, ↑ arrhythmogenesis | 121–127 |
| IL-6 | gp130 receptors | MAPK, NO | Apoptosis, ↓ contractility, ↑ ICAM-1 production | 121,130–134 |
| IL-8 | IL-8 receptors | Tyrosine kinases (Src, focal adhesion kinase) | Inflammatory effect, neutrophil migration, ↑ granule enzymatic release, ↑ oxidative burst in neutrophil | 142,145,149–152 |
↓ Decrease; ↑ Increase; gp130 Glycoprotein 130; ICAM-1 Intracellular adhesion molecule-1; IL Interleukin; MAPK Mitogen-activated protein kinase; NFκB Nuclear factor kappa B; NO Nitric oxide; PKC Protein kinase C; TGF-β Transforming growth factor-beta; TNF-α Tumour necrosis factor-alpha
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research, as well as a CHFNET/IHRT program grant from the Institute of Circulatory and Respiratory Health and the Heart and Stroke Foundation of Canada. Harjot K Saini is a predoctoral fellow of the Heart and Stroke Foundation of Canada. Dr Lorrie A Kirshenbaum holds the Canadian Research Chair in Molecular Cardiology. Dr Peter P Liu holds the Heart and Stroke/Polo Chair Professor of Medicine and Physiology at the University Health Network, University of Toronto.
REFERENCES
- 1.Braunwald E, Kloner RA. Myocardial reperfusion: A double-edged sword? J Clin Invest. 1985;76:1713–9. doi: 10.1172/JCI112160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierce GN, Czubryt MP. The contribution of ionic imbalance to ischemia/reperfusion-induced injury. J Mol Cell Cardiol. 1995;27:53–63. doi: 10.1016/s0022-2828(08)80007-7. [DOI] [PubMed] [Google Scholar]
- 3.Piper HM, Garcia-Dorado D, Ovize M. A fresh look at reperfusion injury. Cardiovasc Res. 1998;38:291–300. doi: 10.1016/s0008-6363(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 4.Opie LH. Reperfusion injury and its pharmacologic modification. Circulation. 1989;80:1049–62. doi: 10.1161/01.cir.80.4.1049. [DOI] [PubMed] [Google Scholar]
- 5.Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev. 1999;79:609–34. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- 6.Dhalla NS, Golfman L, Takeda S, Takeda N, Nagano M. Evidence for the role of oxidative stress in acute ischemic heart disease: A brief review. Can J Cardiol. 1999;15:587–93. [PubMed] [Google Scholar]
- 7.Guillen I, Blanes M, Gomez-Lechon MJ, Castell JV. Cytokine signaling during myocardial infarction: Sequential appearance of IL-1 beta and IL-6. Am J Physiol. 1995;269:R229–35. doi: 10.1152/ajpregu.1995.269.2.R229. [DOI] [PubMed] [Google Scholar]
- 8.Weisensee D, Bereiter-Hahn J, Schoeppe W, Low-Friedrich I. Effects of cytokines on the contractility of cultured cardiac myocytes. Int J Immunopharmacol. 1993;15:581–7. doi: 10.1016/0192-0561(93)90075-a. [DOI] [PubMed] [Google Scholar]
- 9.Hebert CA, Luscinskas FW, Kiely JM, et al. Endothelial and leukocyte forms of IL-8. Conversion by thrombin and interactions with neutrophils. J Immunol. 1990;145:3033–40. [PubMed] [Google Scholar]
- 10.Bursten SL. Interaction of lipopolysaccharide with a mammalian lyso-phosphatidate acyltransferase (LPAAT) transfected into E coli, and effect of lisofylline on LPAAT transfected into mammalian cells. Prog Clin Biol Res. 1998;397:345–56. [PubMed] [Google Scholar]
- 11.Chen H, Li D, Saldeen T, Mehta JL. TGF-beta 1 attenuates myocardial ischemia-reperfusion injury via inhibition of upregulation of MMP-1. Am J Physiol Heart Circ Physiol. 2003;284:H1612–7. doi: 10.1152/ajpheart.00992.2002. [DOI] [PubMed] [Google Scholar]
- 12.Bellisarii FL, Gallina S, De Caterina R. Tumor necrosis factor-alpha and cardiovascular diseases. Ital Heart J. 2001;2:408–17. [PubMed] [Google Scholar]
- 13.Dhote-Burger P, Vuilleminot A, Lecompte T, et al. Neutrophil degranulation related to the reperfusion of ischemic human heart during cardiopulmonary bypass. J Cardiovasc Pharmacol. 1995;25(Suppl 2):S124–9. doi: 10.1097/00005344-199500252-00026. [DOI] [PubMed] [Google Scholar]
- 14.Cavaillon JM. Cytokines and macrophages. Biomed Pharmacother. 1994;48:445–53. doi: 10.1016/0753-3322(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 15.Herskowitz A, Choi S, Ansari AA, Wesselingh S. Cytokine mRNA expression in postischemic/reperfused myocardium. Am J Pathol. 1995;146:419–28. [PMC free article] [PubMed] [Google Scholar]
- 16.Marx N, Neumann FJ, Ott I, et al. Induction of cytokine expression in leukocytes in acute myocardial infarction. J Am Coll Cardiol. 1997;30:165–70. doi: 10.1016/s0735-1097(97)00116-2. [DOI] [PubMed] [Google Scholar]
- 17.Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res. 1991;262:3–11. [PubMed] [Google Scholar]
- 18.Sack M. Tumor necrosis factor-alpha in cardiovascular biology and the potential role for anti-tumor necrosis factor-alpha therapy in heart disease. Pharmacol Ther. 2002;94:123–35. doi: 10.1016/s0163-7258(02)00176-6. [DOI] [PubMed] [Google Scholar]
- 19.Torre-Amione G, Kapadia S, Lee J, Bies RD, Lebovitz R, Mann DL. Expression and functional significance of tumor necrosis factor receptors in human myocardium. Circulation. 1995;92:1487–93. doi: 10.1161/01.cir.92.6.1487. [DOI] [PubMed] [Google Scholar]
- 20.Celec P. Nuclear factor kappa B – molecular biomedicine: The next generation. Biomed Pharmacother. 2004;58:365–71. doi: 10.1016/j.biopha.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 21.Meldrum DR. Tumor necrosis factor in the heart. Am J Physiol. 1998;274:R577–95. doi: 10.1152/ajpregu.1998.274.3.R577. [DOI] [PubMed] [Google Scholar]
- 22.Meldrum DR, Cleveland JC, Jr, Cain BS, Meng X, Harken AH. Increased myocardial tumor necrosis factor-alpha in a crystalloid-perfused model of cardiac ischemia-reperfusion injury. Ann Thorac Surg. 1998;65:439–43. doi: 10.1016/s0003-4975(97)01297-6. [DOI] [PubMed] [Google Scholar]
- 23.Shames BD, Barton HH, Reznikov LL, et al. Ischemia alone is sufficient to induce TNF-alpha mRNA and peptide in the myocardium. Shock. 2002;17:114–9. doi: 10.1097/00024382-200202000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Meldrum DR, Meng X, Dinarello CA, et al. Human myocardial tissue TNFalpha expression following acute global ischemia in vivo. J Mol Cell Cardiol. 1998;30:1683–9. doi: 10.1006/jmcc.1998.0776. [DOI] [PubMed] [Google Scholar]
- 25.Gurevitch J, Frolkis I, Yuhas Y, et al. Tumor necrosis factor-alpha is released from the isolated heart undergoing ischemia and reperfusion. J Am Coll Cardiol. 1996;28:247–52. doi: 10.1016/0735-1097(96)00105-2. [DOI] [PubMed] [Google Scholar]
- 26.Cain BS, Harken AH, Meldrum DR. Therapeutic strategies to reduce TNF-alpha mediated cardiac contractile depression following ischemia and reperfusion. J Mol Cell Cardiol. 1999;31:931–47. doi: 10.1006/jmcc.1999.0924. [DOI] [PubMed] [Google Scholar]
- 27.Bergman MR, Holycross BJ.Pharmacological modulation of myocardial tumor necrosis factor alpha production by phosphodiesterase inhibitors J Pharmacol Exp Ther 1996279247–54.(Erratum in 1997;280:520). [PubMed] [Google Scholar]
- 28.Wagner DR, McTiernan C, Sanders VJ, Feldman AM. Adenosine inhibits lipopolysaccharide-induced secretion of tumor necrosis factor-alpha in the failing human heart. Circulation. 1998;97:521–4. doi: 10.1161/01.cir.97.6.521. [DOI] [PubMed] [Google Scholar]
- 29.Peng J, Xiao J, Ye F, Deng HW, Li YJ. Inhibition of cardiac tumor necrosis factor-alpha production by calcitonin gene-related peptide-mediated ischemic preconditioning in isolated rat hearts. Eur J Pharmacol. 2000;407:303–8. doi: 10.1016/s0014-2999(00)00702-0. [DOI] [PubMed] [Google Scholar]
- 30.Meldrum DR, Dinarello CA, Shames BD, et al. Ischemic preconditioning decreases postischemic myocardial tumor necrosis factor-alpha production. Potential ultimate effector mechanism of preconditioning. Circulation. 1998;98:II214–8. [PubMed] [Google Scholar]
- 31.Cain BS, Meldrum DR, Dinarello CA, Meng X, Banerjee A, Harken AH. Adenosine reduces cardiac TNF-alpha production and human myocardial injury following ischemia-reperfusion. J Surg Res. 1998;76:117–23. doi: 10.1006/jsre.1998.5304. [DOI] [PubMed] [Google Scholar]
- 32.Cain BS, Meldrum DR, Meng X, et al. p38 MAPK inhibition decreases TNF-alpha production and enhances postischemic human myocardial function. J Surg Res. 1999;83:7–12. doi: 10.1006/jsre.1998.5548. [DOI] [PubMed] [Google Scholar]
- 33.Gurevitch J, Frolkis I, Yuhas Y, et al. Anti-tumor necrosis factor-alpha improves myocardial recovery after ischemia and reperfusion. J Am Coll Cardiol. 1997;30:1554–61. doi: 10.1016/s0735-1097(97)00328-8. [DOI] [PubMed] [Google Scholar]
- 34.Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–9. doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- 35.Rathi SS, Xu YJ, Dhalla NS. Mechanism of cardioprotective action of TNF-α in the isolated rat heart. Exp Clin Cardiol. 2002;7:146–50. [PMC free article] [PubMed] [Google Scholar]
- 36.Thielmann M, Dorge H, Martin C, et al. Myocardial dysfunction with coronary microembolization: Signal transduction through a sequence of nitric oxide, tumor necrosis factor-alpha, and sphingosine. Circ Res. 2002;90:807–13. doi: 10.1161/01.res.0000014451.75415.36. [DOI] [PubMed] [Google Scholar]
- 37.Cailleret M, Amadou A, Andrieu-Abadie N, et al. N-acetylcysteine prevents the deleterious effect of tumor necrosis factor-(alpha) on calcium transients and contraction in adult rat cardiomyocytes. Circulation. 2004;109:406–11. doi: 10.1161/01.CIR.0000109499.00587.FF. [DOI] [PubMed] [Google Scholar]
- 38.Oral H, Dorn GW, II, Mann DL. Sphingosine mediates the immediate negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian cardiac myocyte. J Biol Chem. 1997;272:4836–42. doi: 10.1074/jbc.272.8.4836. [DOI] [PubMed] [Google Scholar]
- 39.Mathias S, Dressler KA, Kolesnick RN. Characterization of a ceramide-activated protein kinase: Stimulation by tumor necrosis factor alpha. Proc Natl Acad Sci USA. 1991;88:10009–13. doi: 10.1073/pnas.88.22.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dettbarn CA, Betto R, Salviati G, Palade P, Jenkins GM, Sabbadini RA. Modulation of cardiac sarcoplasmic reticulum ryanodine receptor by sphingosine. J Mol Cell Cardiol. 1994;26:229–42. doi: 10.1006/jmcc.1994.1026. [DOI] [PubMed] [Google Scholar]
- 41.Goldhaber JI, Kim KH, Natterson PD, Lawrence T, Yang P, Weiss JN. Effects of TNF-alpha on [Ca2+]i and contractility in isolated adult rabbit ventricular myocytes. Am J Physiol. 1996;271:H1449–55. doi: 10.1152/ajpheart.1996.271.4.H1449. [DOI] [PubMed] [Google Scholar]
- 42.Prabhu SD. Cytokine-induced modulation of cardiac function. Circ Res. 2004;95:1140–53. doi: 10.1161/01.RES.0000150734.79804.92. [DOI] [PubMed] [Google Scholar]
- 43.Schutze S, Machleidt T, Kronke M. Mechanisms of tumor necrosis factor action. Semin Oncol. 1992;19(Suppl 4):16–24. [PubMed] [Google Scholar]
- 44.Park H, Park SG, Kim J, Ko YG, Kim S. Signaling pathways for TNF production induced by human aminoacyl-tRNA synthetase-associating factor, p43. Cytokine. 2002;20:148–53. doi: 10.1006/cyto.2002.1992. [DOI] [PubMed] [Google Scholar]
- 45.Dhalla NS, Xu YJ, Sheu SS, Tappia PS, Panagia V. Phosphatidic acid: A potential signal transducer for cardiac hypertrophy. J Mol Cell Cardiol. 1997;29:2865–71. doi: 10.1006/jmcc.1997.0522. [DOI] [PubMed] [Google Scholar]
- 46.Cain BS, Meldrum DR, Harken AH. Protein kinase C in normal and pathologic myocardial states. J Surg Res. 1999;81:249–59. doi: 10.1006/jsre.1998.5508. [DOI] [PubMed] [Google Scholar]
- 47.Schutze S, Potthoff K, Machleidt T, Berkovic D, Wiegmann K, Kronke M. TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell. 1992;71:765–76. doi: 10.1016/0092-8674(92)90553-o. [DOI] [PubMed] [Google Scholar]
- 48.Plo I, Lautier D, Levade T, et al. Phosphatidylcholine-specific phospholipase C and phospholipase D are respectively implicated in mitogen-activated protein kinase and nuclear factor kappaB activation in tumour-necrosis-factor-alpha-treated immature acute-myeloid-leukaemia cells. Biochem J. 2000;351:459–67. [PMC free article] [PubMed] [Google Scholar]
- 49.Liu SJ, McHowat J. Stimulation of different phospholipase A2 isoforms by TNF-alpha and IL-1beta in adult rat ventricular myocytes. Am J Physiol. 1998;275:H1462–72. doi: 10.1152/ajpheart.1998.275.4.H1462. [DOI] [PubMed] [Google Scholar]
- 50.Alessenko AV. Functions of sphingosine in cell proliferation and death. Biochemistry. 1998;63:62–8. [PubMed] [Google Scholar]
- 51.Cleveland JL, Ihle JN. Contenders in FasL/TNF death signaling. Cell. 1995;81:479–82. doi: 10.1016/0092-8674(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 52.McCarthy JV, Ni J, Dixit VM. RIP2 is a novel NF-kappaB-activating and cell death-inducing kinase. J Biol Chem. 1998;273:16968–75. doi: 10.1074/jbc.273.27.16968. [DOI] [PubMed] [Google Scholar]
- 53.van den Hoff MJ, van den Eijnde SM, Viragh S, Moorman AF. Programmed cell death in the developing heart. Cardiovasc Res. 2000;45:603–20. doi: 10.1016/s0008-6363(99)00401-0. [DOI] [PubMed] [Google Scholar]
- 54.Natoli G, Costanzo A, Guido F, Moretti F, Levrero M. Apoptotic, non-apoptotic, and anti-apoptotic pathways of tumor necrosis factor signalling. Biochem Pharmacol. 1998;56:915–20. doi: 10.1016/s0006-2952(98)00154-3. [DOI] [PubMed] [Google Scholar]
- 55.Perkins ND. Achieving transcriptional specificity with NF-kappa B. Int J Biochem Cell Biol. 1997;29:1433–48. doi: 10.1016/s1357-2725(97)00088-5. [DOI] [PubMed] [Google Scholar]
- 56.Krown KA, Page MT, Nguyen C, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. 1996;98:2854–65. doi: 10.1172/JCI119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song W, Lu X, Feng Q. Tumor necrosis factor-alpha induces apoptosis via inducible nitric oxide synthase in neonatal mouse cardiomyocytes. Cardiovasc Res. 2000;45:595–602. doi: 10.1016/s0008-6363(99)00395-8. [DOI] [PubMed] [Google Scholar]
- 58.Wan S, DeSmet JM, Barvais L, Goldstein M, Vincent JL, LeClerc JL. Myocardium is a major source of proinflammatory cytokines in patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1996;112:806–11. doi: 10.1016/S0022-5223(96)70068-5. [DOI] [PubMed] [Google Scholar]
- 59.Hennein HA, Ebba H, Rodriguez JL, et al. Relationship of the proinflammatory cytokines to myocardial ischemia and dysfunction after uncomplicated coronary revascularization. J Thorac Cardiovasc Surg. 1994;108:626–35. [PubMed] [Google Scholar]
- 60.Lantz M, Malik S, Slevin ML, Olsson I. Infusion of tumor necrosis factor (TNF) causes an increase in circulating TNF-binding protein in humans. Cytokine. 1990;2:402–6. doi: 10.1016/1043-4666(90)90048-x. [DOI] [PubMed] [Google Scholar]
- 61.Aderka D. The potential biological and clinical significance of the soluble tumor necrosis factor receptors. Cytokine Growth Factor Rev. 1996;7:231–40. doi: 10.1016/s1359-6101(96)00026-3. [DOI] [PubMed] [Google Scholar]
- 62.Mullberg J, Durie FH, Otten-Evans C, et al. A metalloprotease inhibitor blocks shedding of the IL-6 receptor and the p60 TNF receptor. J Immunol. 1995;155:5198–205. [PubMed] [Google Scholar]
- 63.Crowe PD, Walter BN, Mohler KM, Otten-Evans C, Black RA, Ware CF. A metalloprotease inhibitor blocks shedding of the 80-kD TNF receptor and TNF processing in T lymphocytes. J Exp Med. 1995;181:1205–10. doi: 10.1084/jem.181.3.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferrari R. The role of TNF in cardiovascular disease. Pharmacol Res. 1999;40:97–105. doi: 10.1006/phrs.1998.0463. [DOI] [PubMed] [Google Scholar]
- 65.Zhao TC, Taher MM, Valerie KC, Kukreja RC. p38 Triggers late preconditioning elicited by anisomycin in heart: Involvement of NF-kappaB and iNOS. Circ Res. 2001;89:915–22. doi: 10.1161/hh2201.099452. [DOI] [PubMed] [Google Scholar]
- 66.van der Poll T, Coyle SM, Barbosa K, Braxton CC, Lowry SF. Epinephrine inhibits tumor necrosis factor-alpha and potentiates interleukin 10 production during human endotoxemia. J Clin Invest. 1996;97:713–9. doi: 10.1172/JCI118469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maekawa N, Wada H, Kanda T, et al. Improved myocardial ischemia/reperfusion injury in mice lacking tumor necrosis factor-alpha. J Am Coll Cardiol. 2002;39:1229–35. doi: 10.1016/s0735-1097(02)01738-2. [DOI] [PubMed] [Google Scholar]
- 68.Latini R, Bianchi M, Correale E, et al. Cytokines in acute myocardial infarction: Selective increase in circulating tumor necrosis factor, its soluble receptor, and interleukin-1 receptor antagonist. J Cardiovasc Pharmacol. 1994;23:1–6. [PubMed] [Google Scholar]
- 69.Nelson SK, Wong GH, McCord JM. Leukemia inhibitory factor and tumor necrosis factor induce manganese superoxide dismutase and protect rabbit hearts from reperfusion injury. J Mol Cell Cardiol. 1995;27:223–9. doi: 10.1016/s0022-2828(08)80021-1. [DOI] [PubMed] [Google Scholar]
- 70.Gershenwald JE, Fong YM, Fahey TJ, III, et al. Interleukin 1 receptor blockade attenuates the host inflammatory response. Proc Natl Acad Sci USA. 1990;87:4966–70. doi: 10.1073/pnas.87.13.4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alexander HR, Wong GG, Doherty GM, Venzon DJ, Fraker DL, Norton JA. Differentiation factor/leukemia inhibitory factor protection against lethal endotoxemia in mice: Synergistic effect with interleukin 1 and tumor necrosis factor. J Exp Med. 1992;175:1139–42. doi: 10.1084/jem.175.4.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benigni F, Villa P, Demitri MT, et al. Ciliary neurotrophic factor inhibits brain and peripheral tumor necrosis factor production and, when coadministered with its soluble receptor, protects mice from lipopolysaccharide toxicity. Mol Med. 1995;1:568–75. [PMC free article] [PubMed] [Google Scholar]
- 73.McGeehan GM, Becherer JD, Bast RC, Jr, et al. Regulation of tumour necrosis factor-alpha processing by a metalloproteinase inhibitor. Nature. 1994;370:558–61. doi: 10.1038/370558a0. [DOI] [PubMed] [Google Scholar]
- 74.Mohler KM, Sleath PR, Fitzner JN, et al. Protection against a lethal dose of endotoxin by an inhibitor of tumour necrosis factor processing. Nature. 1994;370:218–20. doi: 10.1038/370218a0. [DOI] [PubMed] [Google Scholar]
- 75.Hill GE, Springall DR, Robbins RA. Aprotinin is associated with a decrease in nitric oxide production during cardiopulmonary bypass. Surgery. 1997;121:449–55. doi: 10.1016/s0039-6060(97)90316-0. [DOI] [PubMed] [Google Scholar]
- 76.Nakano M, Mann DL, Knowlton AA. Blocking the endogenous increase in HSP 72 increases susceptibility to hypoxia and reoxygenation in isolated adult feline cardiocytes. Circulation. 1997;95:1523–31. doi: 10.1161/01.cir.95.6.1523. [DOI] [PubMed] [Google Scholar]
- 77.Feinstein DL, Galea E, Aquino DA, Li GC, Xu H, Reis DJ. Heat shock protein 70 suppresses astroglial-inducible nitric-oxide synthase expression by decreasing NFkappaB activation. J Biol Chem. 1996;271:17724–32. doi: 10.1074/jbc.271.30.17724. [DOI] [PubMed] [Google Scholar]
- 78.Plumier JC, Ross BM, Currie RW, et al. Transgenic mice expressing the human heat shock protein 70 have improved post-ischemic myocardial recovery. J Clin Invest. 1995;95:1854–60. doi: 10.1172/JCI117865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sumeray MS, Rees DD, Yellon DM. Infarct size and nitric oxide synthase in murine myocardium. J Mol Cell Cardiol. 2000;32:35–42. doi: 10.1006/jmcc.1999.1050. [DOI] [PubMed] [Google Scholar]
- 80.Smith RM, Suleman N, McCarthy J, Sack MN. Classic ischemic but not pharmacologic preconditioning is abrogated following genetic ablation of the TNFalpha gene. Cardiovasc Res. 2002;55:553–60. doi: 10.1016/s0008-6363(02)00283-3. [DOI] [PubMed] [Google Scholar]
- 81.Lecour S, Smith RM, Woodward B, Opie LH, Rochette L, Sack MN. Identification of a novel role for sphingolipid signaling in TNF alpha and ischemic preconditioning mediated cardioprotection. J Mol Cell Cardiol. 2002;34:509–18. doi: 10.1006/jmcc.2002.1533. [DOI] [PubMed] [Google Scholar]
- 82.Amadou A, Nawrocki A, Best-Belpomme M, Pavoine C, Pecker F. Arachidonic acid mediates dual effect of TNF-alpha on Ca2+ transients and contraction of adult rat cardiomyocytes. Am J Physiol Cell Physiol. 2002;282:C1339–47. doi: 10.1152/ajpcell.00471.2001. [DOI] [PubMed] [Google Scholar]
- 83.Bowie A, O’Neill LA. Oxidative stress and nuclear factor-kappaB activation: A reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol. 2000;59:13–23. doi: 10.1016/s0006-2952(99)00296-8. [DOI] [PubMed] [Google Scholar]
- 84.Valen G, Yan ZQ, Hansson GK. Nuclear factor kappa-B and the heart. J Am Coll Cardiol. 2001;38:307–14. doi: 10.1016/s0735-1097(01)01377-8. [DOI] [PubMed] [Google Scholar]
- 85.Mann DL. Stress activated cytokines and the heart. Cytokine Growth Factor Rev. 1996;7:341–54. doi: 10.1016/s1359-6101(96)00043-3. [DOI] [PubMed] [Google Scholar]
- 86.Li C, Browder W, Kao RL. Early activation of transcription factor NF-kappaB during ischemia in perfused rat heart. Am J Physiol. 1999;276:H543–52. doi: 10.1152/ajpheart.1999.276.2.H543. [DOI] [PubMed] [Google Scholar]
- 87.Chandrasekar B, Freeman GL. Induction of nuclear factor kappaB and activation protein 1 in postischemic myocardium. FEBS Lett. 1997;401:30–4. doi: 10.1016/s0014-5793(96)01426-3. [DOI] [PubMed] [Google Scholar]
- 88.Shimizu N, Yoshiyama M, Omura T, et al. Activation of mitogen-activated protein kinases and activator protein-1 in myocardial infarction in rats. Cardiovasc Res. 1998;38:116–24. doi: 10.1016/s0008-6363(97)00327-1. [DOI] [PubMed] [Google Scholar]
- 89.Fan H, Sun B, Gu Q, Lafond-Walker A, Cao S, Becker LC. Oxygen radicals trigger activation of NF-kappaB and AP-1 and upregulation of ICAM-1 in reperfused canine heart. Am J Physiol Heart Circ Physiol. 2002;282:H1778–86. doi: 10.1152/ajpheart.00796.2000. [DOI] [PubMed] [Google Scholar]
- 90.Yang J, Marden JJ, Fan C, et al. Genetic redox preconditioning differentially modulates AP-1 and NFkappaB responses following cardiac ischemia/reperfusion injury and protects against necrosis and apoptosis. Mol Ther. 2003;7:341–53. doi: 10.1016/s1525-0016(02)00061-8. [DOI] [PubMed] [Google Scholar]
- 91.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI.Differential activation of transcription factors induced by Ca2+ response amplitude and duration Nature 1997386855–8.(Erratum in 1997;388:308). [DOI] [PubMed] [Google Scholar]
- 92.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–6. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 93.Hu Q, Deshpande S, Irani K, Ziegelstein RC. [Ca2+]i oscillation frequency regulates agonist-stimulated NF-kappaB transcriptional activity. J Biol Chem. 1999;274:33995–8. doi: 10.1074/jbc.274.48.33995. [DOI] [PubMed] [Google Scholar]
- 94.Cargnoni A, Ceconi C, Gaia G, Agnoletti L, Ferrari R. Cellular thiols redox status: A switch for NF-kappaB activation during myocardial post-ischaemic reperfusion. J Mol Cell Cardiol. 2002;34:997–1005. doi: 10.1006/jmcc.2002.2046. [DOI] [PubMed] [Google Scholar]
- 95.Suzuki YJ, Packer L. Inhibition of NF-kappa B DNA binding activity by alpha-tocopheryl succinate. Biochem Mol Biol Int. 1993;31:693–700. [PubMed] [Google Scholar]
- 96.Suzuki YJ, Aggarwal BB, Packer L. Alpha-lipoic acid is a potent inhibitor of NF-kappa B activation in human T cells. Biochem Biophys Res Commun. 1992;189:1709–15. doi: 10.1016/0006-291x(92)90275-p. [DOI] [PubMed] [Google Scholar]
- 97.Pye J, Ardeshirpour F, McCain A, et al. Proteasome inhibition ablates activation of NF-kappa B in myocardial reperfusion and reduces reperfusion injury. Am J Physiol Heart Circ Physiol. 2003;284:H919–26. doi: 10.1152/ajpheart.00851.2002. [DOI] [PubMed] [Google Scholar]
- 98.Bretschneider E, Wittpoth M, Weber AA, Glusa E, Schror K. Activation of NFkappaB is essential but not sufficient to stimulate mitogenesis of vascular smooth muscle cells. Biochem Biophys Res Commun. 1997;235:365–8. doi: 10.1006/bbrc.1997.6788. [DOI] [PubMed] [Google Scholar]
- 99.Bellas RE, Lee JS, Sonenshein GE. Expression of a constitutive NF-kappa B-like activity is essential for proliferation of cultured bovine vascular smooth muscle cells. J Clin Invest. 1995;96:2521–7. doi: 10.1172/JCI118313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Thourani VH, Brar SS, Kennedy TP, et al. Nonanticoagulant heparin inhibits NF-kappaB activation and attenuates myocardial reperfusion injury. Am J Physiol Heart Circ Physiol. 2000;278:H2084–93. doi: 10.1152/ajpheart.2000.278.6.H2084. [DOI] [PubMed] [Google Scholar]
- 101.Xuan YT, Tang XL, Banerjee S, et al. Nuclear factor-kappaB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits. Circ Res. 1999;84:1095–109. doi: 10.1161/01.res.84.9.1095. [DOI] [PubMed] [Google Scholar]
- 102.Maulik N, Sato M, Price BD, Das DK. An essential role of NFkappaB in tyrosine kinase signaling of p38 MAP kinase regulation of myocardial adaptation to ischemia. FEBS Lett. 1998;429:365–9. doi: 10.1016/s0014-5793(98)00632-2. [DOI] [PubMed] [Google Scholar]
- 103.Marber MS, Latchman DS, Walker JM, Yellon DM. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–72. doi: 10.1161/01.cir.88.3.1264. [DOI] [PubMed] [Google Scholar]
- 104.Zhao T, Xi L, Chelliah J, Levasseur JE, Kukreja RC. Inducible nitric oxide synthase mediates delayed myocardial protection induced by activation of adenosine A1 receptors: Evidence from gene-knockout mice. Circulation. 2000;102:902–7. doi: 10.1161/01.cir.102.8.902. [DOI] [PubMed] [Google Scholar]
- 105.Morgan EN, Boyle EM, Jr, Yun W, et al. An essential role for NF-kappaB in the cardioadaptive response to ischemia. Ann Thorac Surg. 1999;68:377–82. doi: 10.1016/s0003-4975(99)00646-3. [DOI] [PubMed] [Google Scholar]
- 106.Bach FH, Hancock WW, Ferran C. Protective genes expressed in endothelial cells: A regulatory response to injury. Immunol Today. 1997;18:483–6. doi: 10.1016/s0167-5699(97)01129-8. [DOI] [PubMed] [Google Scholar]
- 107.Regula KM, Ens K, Kirshenbaum LA. IKK beta is required for Bcl-2-mediated NF-kappa B activation in ventricular myocytes. J Biol Chem. 2002;277:38676–82. doi: 10.1074/jbc.M206175200. [DOI] [PubMed] [Google Scholar]
- 108.Mustapha S, Kirshner A, De Moissac D, Kirshenbaum LA. A direct requirement of nuclear factor-kappa B for suppression of apoptosis in ventricular myocytes. Am J Physiol Heart Circ Physiol. 2000;279:H939–45. doi: 10.1152/ajpheart.2000.279.3.H939. [DOI] [PubMed] [Google Scholar]
- 109.Regula KM, Baetz D, Kirshenbaum LA. Nuclear factor-kappaB represses hypoxia-induced mitochondrial defects and cell death of ventricular myocytes. Circulation. 2004;110:3795–802. doi: 10.1161/01.CIR.0000150537.59754.55. [DOI] [PubMed] [Google Scholar]
- 110.Roberts AB, Sporn MB. Physiological actions and clinical applications of transforming growth factor-beta (TGF-beta) Growth Factors. 1993;8:1–9. doi: 10.3109/08977199309029129. [DOI] [PubMed] [Google Scholar]
- 111.Mehta JL, Yang BC, Strates BS, Mehta P. Role of TGF-beta1 in platelet-mediated cardioprotection during ischemia-reperfusion in isolated rat hearts. Growth Factors. 1999;16:179–90. doi: 10.3109/08977199909002128. [DOI] [PubMed] [Google Scholar]
- 112.Yellon DM, Baxter GF. Reperfusion injury revisited: Is there a role for growth factor signaling in limiting lethal reperfusion injury? Trends Cardiovasc Med. 1999;9:245–9. doi: 10.1016/s1050-1738(00)00029-3. [DOI] [PubMed] [Google Scholar]
- 113.Lefer AM, Tsao P, Aoki N, Palladino MA., Jr Mediation of cardioprotection by transforming growth factor-beta. Science. 1990;249:61–4. doi: 10.1126/science.2164258. [DOI] [PubMed] [Google Scholar]
- 114.Lefer AM, Ma XL, Weyrich AS, Scalia R. Mechanism of the cardioprotective effect of transforming growth factor beta 1 in feline myocardial ischemia and reperfusion. Proc Natl Acad Sci USA. 1993;90:1018–22. doi: 10.1073/pnas.90.3.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Baxter GF, Mocanu MM, Brar BK, Latchman DS, Yellon DM. Cardioprotective effects of transforming growth factor-beta1 during early reoxygenation or reperfusion are mediated by p42/p44 MAPK. J Cardiovasc Pharmacol. 2001;38:930–9. doi: 10.1097/00005344-200112000-00015. [DOI] [PubMed] [Google Scholar]
- 116.Grunenfelder J, Miniati DN, Murata S, Falk V, Hoyt EG, Robbins RC. Up-regulation of Bcl-2 through hyperbaric pressure transfection of TGF-beta1 ameliorates ischemia-reperfusion injury in rat cardiac allografts. J Heart Lung Transplant. 2002;21:244–50. doi: 10.1016/s1053-2498(01)00377-1. [DOI] [PubMed] [Google Scholar]
- 117.Mehta JL, Chen HJ, Li DY. Protection of myocytes from hypoxia-reoxygenation injury by nitric oxide is mediated by modulation of transforming growth factor-beta1. Circulation. 2002;105:2206–11. doi: 10.1161/01.cir.0000015602.94990.3d. [DOI] [PubMed] [Google Scholar]
- 118.Chen H, Li D, Saldeen T, Mehta JL. TGF-beta1 modulates NOS expression and phosphorylation of Akt/PKB in rat myocytes exposed to hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2001;281:H1035–9. doi: 10.1152/ajpheart.2001.281.3.H1035. [DOI] [PubMed] [Google Scholar]
- 119.Evans HG, Lewis MJ, Shah AM. Interleukin-1 beta modulates myocardial contraction via dexamethasone sensitive production of nitric oxide. Cardiovasc Res. 1993;27:1486–90. doi: 10.1093/cvr/27.8.1486. [DOI] [PubMed] [Google Scholar]
- 120.Oyama J, Shimokawa H, Momii H, et al. Role of nitric oxide and peroxynitrite in the cytokine-induced sustained myocardial dysfunction in dogs in vivo. J Clin Invest. 1998;101:2207–14. doi: 10.1172/JCI986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stangl V, Baumann G, Stangl K, Felix SB. Negative inotropic mediators released from the heart after myocardial ischaemia-reperfusion. Cardiovasc Res. 2002;53:12–30. doi: 10.1016/s0008-6363(01)00420-5. [DOI] [PubMed] [Google Scholar]
- 122.LaPointe MC, Isenovic E. Interleukin-1beta regulation of inducible nitric oxide synthase and cyclooxygenase-2 involves the p42/44 and p38 MAPK signaling pathways in cardiac myocytes. Hypertension. 1999;33:276–82. doi: 10.1161/01.hyp.33.1.276. [DOI] [PubMed] [Google Scholar]
- 123.Schulz R, Nava E, Moncada S. Induction and potential biological relevance of a Ca2+-independent nitric oxide synthase in the myocardium. Br J Pharmacol. 1992;105:575–80. doi: 10.1111/j.1476-5381.1992.tb09021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pinsky DJ, Cai B, Yang X, Rodriguez C, Sciacca RR, Cannon PJ. The lethal effects of cytokine-induced nitric oxide on cardiac myocytes are blocked by nitric oxide synthase antagonism or transforming growth factor beta. J Clin Invest. 1995;95:677–85. doi: 10.1172/JCI117713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brown JM, White CW, Terada LS, et al. Interleukin 1 pretreatment decreases ischemia/reperfusion injury. Proc Natl Acad Sci USA. 1990;87:5026–30. doi: 10.1073/pnas.87.13.5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Thaik CM, Calderone A, Takahashi N, Colucci WS. Interleukin-1 beta modulates the growth and phenotype of neonatal rat cardiac myocytes. J Clin Invest. 1995;96:1093–9. doi: 10.1172/JCI118095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.McTiernan CF, Lemster BH, Frye C, Brooks S, Combes A, Feldman AM. Interleukin-1 beta inhibits phospholamban gene expression in cultured cardiomyocytes. Circ Res. 1997;81:493–503. doi: 10.1161/01.res.81.4.493. [DOI] [PubMed] [Google Scholar]
- 128.Maulik N, Engelman RM, Wei Z, Lu D, Rousou JA, Das DK. Interleukin-1 alpha preconditioning reduces myocardial ischemia reperfusion injury. Circulation. 1993;88:II387–94. [PubMed] [Google Scholar]
- 129.Nogae C, Makino N, Hata T, et al. Interleukin 1 alpha-induced expression of manganous superoxide dismutase reduces myocardial reperfusion injury in the rat. J Mol Cell Cardiol. 1995;27:2091–9. doi: 10.1016/s0022-2828(95)91155-3. [DOI] [PubMed] [Google Scholar]
- 130.Wollert KC, Drexler H. The role of interleukin-6 in the failing heart. Heart Fail Rev. 2001;6:95–103. doi: 10.1023/a:1011401825680. [DOI] [PubMed] [Google Scholar]
- 131.Miyao Y, Yasue H, Ogawa H, et al. Elevated plasma interleukin-6 levels in patients with acute myocardial infarction. Am Heart J. 1993;126:1299–304. doi: 10.1016/0002-8703(93)90526-f. [DOI] [PubMed] [Google Scholar]
- 132.Chandrasekar B, Mitchell DH, Colston JT, Freeman GL. Regulation of CCAAT/Enhancer binding protein, interleukin-6, interleukin-6 receptor, and gp130 expression during myocardial ischemia/reperfusion. Circulation. 1999;99:427–33. doi: 10.1161/01.cir.99.3.427. [DOI] [PubMed] [Google Scholar]
- 133.Gwechenberger M, Mendoza LH, Youker KA, et al. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–51. doi: 10.1161/01.cir.99.4.546. [DOI] [PubMed] [Google Scholar]
- 134.Kukielka GL, Youker KA, Michael LH, et al. Role of early reperfusion in the induction of adhesion molecules and cytokines in previously ischemic myocardium. Mol Cell Biochem. 1995;147:5–12. doi: 10.1007/BF00944777. [DOI] [PubMed] [Google Scholar]
- 135.Finkel MS, Hoffman RA, Shen L, Oddis CV, Simmons RL, Hattler BG. Interleukin-6 (IL-6) as a mediator of stunned myocardium. Am J Cardiol. 1993;71:1231–2. doi: 10.1016/0002-9149(93)90654-u. [DOI] [PubMed] [Google Scholar]
- 136.Kinugawa K, Takahashi T, Kohmoto O, et al. Nitric oxide-mediated effects of interleukin-6 on [Ca2+]i and cell contraction in cultured chick ventricular myocytes. Circ Res. 1994;75:285–95. doi: 10.1161/01.res.75.2.285. [DOI] [PubMed] [Google Scholar]
- 137.Kukielka GL, Smith CW, Manning AM, Youker KA, Michael LH, Entman ML. Induction of interleukin-6 synthesis in the myocardium. Potential role in postreperfusion inflammatory injury. Circulation. 1995;92:1866–75. doi: 10.1161/01.cir.92.7.1866. [DOI] [PubMed] [Google Scholar]
- 138.Stephanou A, Brar B, Heads R, et al. Cardiotrophin-1 induces heat shock protein accumulation in cultured cardiac cells and protects them from stressful stimuli. J Mol Cell Cardiol. 1998;30:849–55. doi: 10.1006/jmcc.1998.0651. [DOI] [PubMed] [Google Scholar]
- 139.Latchman DS. Cardiotrophin-1 (CT-1): A novel hypertrophic and cardioprotective agent. Int J Exp Pathol. 1999;80:189–96. doi: 10.1046/j.1365-2613.1999.00114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Craig R, Larkin A, Mingo AM, et al. p38 MAPK and NF-kappa B collaborate to induce interleukin-6 gene expression and release. Evidence for a cytoprotective autocrine signaling pathway in a cardiac myocyte model system. J Biol Chem. 2000;275:23814–24. doi: 10.1074/jbc.M909695199. [DOI] [PubMed] [Google Scholar]
- 141.Kukielka GL, Smith CW, LaRosa GJ, et al. Interleukin-8 gene induction in the myocardium after ischemia and reperfusion in vivo. J Clin Invest. 1995;95:89–103. doi: 10.1172/JCI117680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dame JB, Juul SE. The distribution of receptors for the pro-inflammatory cytokines interleukin (IL)-6 and IL-8 in the developing human fetus. Early Hum Dev. 2000;58:25–39. doi: 10.1016/s0378-3782(00)00064-5. [DOI] [PubMed] [Google Scholar]
- 143.Oz MC, Liao H, Naka Y, et al. Ischemia-induced interleukin-8 release after human heart transplantation. A potential role for endothelial cells. Circulation. 1995;92:II428–32. doi: 10.1161/01.cir.92.9.428. [DOI] [PubMed] [Google Scholar]
- 144.Abe Y, Kawakami M, Kuroki M, et al. Transient rise in serum interleukin-8 concentration during acute myocardial infarction. Br Heart J. 1993;70:132–4. doi: 10.1136/hrt.70.2.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Huber AR, Kunkel SL, Todd RF, III, Weiss SJ.Regulation of transendothelial neutrophil migration by endogenous interleukin-8 Science 199125499–102.(Erratum in 1991;254:631). [DOI] [PubMed] [Google Scholar]
- 146.Boyle EM, Jr, Kovacich JC, Hebert CA, et al. Inhibition of interleukin-8 blocks myocardial ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 1998;116:114–21. doi: 10.1016/S0022-5223(98)70249-1. [DOI] [PubMed] [Google Scholar]
- 147.Moser R, Olgiati L, Patarroyo M, Fehr J. Chemotaxins inhibit neutrophil adherence to and transmigration across cytokine-activated endothelium: Correlation to the expression of L-selectin. Eur J Immunol. 1993;23:1481–7. doi: 10.1002/eji.1830230713. [DOI] [PubMed] [Google Scholar]
- 148.Luscinskas FW, Kiely JM, Ding H, et al. In vitro inhibitory effect of IL-8 and other chemoattractants on neutrophil-endothelial adhesive interactions. J Immunol. 1992;149:2163–71. [PubMed] [Google Scholar]
- 149.Lee LF, Louie MC, Desai SJ, et al. Interleukin-8 confers androgen-independent growth and migration of LNCaP: Differential effects of tyrosine kinases Src and FAK. Oncogene. 2004;23:2197–205. doi: 10.1038/sj.onc.1207344. [DOI] [PubMed] [Google Scholar]
- 150.Smart SJ, Casale TB. TNF-alpha-induced transendothelial neutrophil migration is IL-8 dependent. Am J Physiol. 1994;266:L238–45. doi: 10.1152/ajplung.1994.266.3.L238. [DOI] [PubMed] [Google Scholar]
- 151.Smith WB, Gamble JR, Clark-Lewis I, Vadas MA. Interleukin-8 induces neutrophil transendothelial migration. Immunology. 1991;72:65–72. [PMC free article] [PubMed] [Google Scholar]
- 152.Neumann FJ, Ott I, Gawaz M, et al. Cardiac release of cytokines and inflammatory responses in acute myocardial infarction. Circulation. 1995;92:748–55. doi: 10.1161/01.cir.92.4.748. [DOI] [PubMed] [Google Scholar]
- 153.Irwin MW, Mak S, Mann DL, et al. Tissue expression and immunolocalization of tumor necrosis factor-alpha in postinfarction dysfunctional myocardium. Circulation. 1999;99:1492–8. doi: 10.1161/01.cir.99.11.1492. [DOI] [PubMed] [Google Scholar]
- 154.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–53. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 155.Zhang M, Xu YJ, Saini HK, Turan B, Liu PP, Dhalla NS. TNF-alpha as a potential mediator of cardiac dysfunction due to intracellular Ca2+-overload. Biochem Biophys Res Commun. 2005;327:57–63. doi: 10.1016/j.bbrc.2004.11.131. [DOI] [PubMed] [Google Scholar]
- 156.Zhang M, Xu YJ, Saini HK, Turan B, Liu PP, Dhalla NS. Pentoxifylline attenuates cardiac dysfunction and reduces TNF-alpha level in ischemic-reperfused heart. Am J Physiol Heart Circ Physiol. 2005;289:H832–9. doi: 10.1152/ajpheart.00178.2005. [DOI] [PubMed] [Google Scholar]