

Abstract
Ischemic preconditioning results not only in a reduction in myocardial ischemic damage, but also in a marked suppression of those ventricular arrhythmias that result from a more prolonged period of ischemia and reperfusion insult. This protection is time-dependent and occurs in two distinct phases. There is an ‘early phase’ which is apparent immediately after the preconditioning stimulus but fades quickly (within 1 h to 2 h), and a ‘delayed protection phase’ in which the antiarrhythmic protection reappears 20 h to 24 h later. In both phases, the intensity of protection largely depends on the nature of the preconditioning stimulus. This can be ischemia resulting from brief coronary artery occlusions, cardiac pacing or vigorous physical exercise. Both cardiac pacing and exercise results in a marked reduction in the incidence and severity of ischemia and reperfusion-induced ventricular arrhythmias 24 h later. Although the precise mechanisms of the delayed protection that results from cardiac pacing and exercise are not yet fully understood, there is some evidence that similar endogenous protective substances (such as bradykinin, prostanoids and nitric oxide), as with ischemic preconditioning, play a pivotal trigger and mediator role in this anti-arrhythmic protection.
Keywords: Cardiac pacing, Exercise, Ischemia, Preconditioning, Ventricular arrhythmias
One of the most important consequences of the abrupt reduction in coronary blood flow that results from coronary artery occlusion, in both humans and experimental animals, is the occurrence of life-threatening ventricular arrhythmias. In the clinical situation, these are responsible for most instances of sudden cardiac death. Despite advances in drug therapy, surgical and interventional cardiology, further research is required to explore new strategies that may ultimately provide practical therapeutic interventions against these fatal ventricular arrhythmias.
In the past 17 years, a number of experiments have provided evidence that ischemic preconditioning not only protects the myocardium against ischemic damage, but also reduces those ventricular arrhythmias that result from coronary artery occlusion and reperfusion (1,2). Although this protection is extremely pronounced, there are at least two major observations that limit the clinical exploitation of this phenomenon. First, the protection which results from preconditioning is short-lived; indeed, only 1 h to 2 h after the preconditioning stimulus, the protection has almost completely disappeared. Second, coronary artery occlusion, apart from some acute surgical and/or interventional cardiological situations, cannot be applied as a preconditioning stimulus in humans. These concerns are, however, partially solved by two observations that show that the protection spontaneously reappears approximately 20 h to 24 h after the initial preconditioning stimulus (3,4), and that protection can be induced by means other than short coronary artery occlusions (ie, cardiac pacing or physical exercise) (5–7). These findings have revealed new perspectives for both experimental and clinical investigations.
The purpose of the present paper is to outline the evidence for the antiarrhythmic effects of preconditioning induced by ischemic preconditioning, cardiac pacing and physical exercise. We also provide evidence that various preconditioning procedures apply similar trigger mechanisms to induce early and delayed protection against arrhythmias in a canine model of myocardial ischemia and reperfusion. These findings were presented at the North Atlantic Treaty Organisation Advanced Research Workshop, held in Antalya, Turkey from February 2 to 7, 2005.
THE ANTIARRHYTHMIC EFFECT OF ISCHEMIC PRECONDITIONING, CARDIAC PACING AND PHYSICAL EXERCISE
It is worthwhile to give a brief summary of the previous findings demonstrating that preconditioning protects the heart against arrhythmias. Apart from the study of Shiki and Hearse in 1987 (8) who demonstrated in anesthetized rats that brief periods of coronary artery occlusion of the same duration (5 min) reduce the incidence and severity of reperfusion-induced ventricular arrhythmias, the first evidence that preconditioning provides protection against arrhythmias occurring during a more prolonged ischemia and reperfusion insult comes from our own previous dog studies (1,2). If dogs are subjected to either one or two 5 min occlusions of the left anterior descending coronary artery, 20 min before prolonged (25 min) ischemia, the number of ventricular premature beats (VPBs) during this prolonged ischemic insult is markedly reduced compared with the controls that are simply subjected to a 25 min occlusion-reperfusion insult (Figure 1). Similarly, the more malignant ventricular arrhythmias, such as ventricular tachycardia and ventricular fibrillation (VF), are significantly reduced by preconditioning. Thus, compared with the control group, in which 52 of 110 dogs (47%) fibrillated during coronary artery occlusion and only three dogs survived the combined ischemia/reperfusion insult, in dogs subjected to preconditioning, no dog died during occlusion and 47% of the dogs survived reperfusion. This truly is a marked protection against these life-threatening ventricular arrhythmias.
Figure 1).
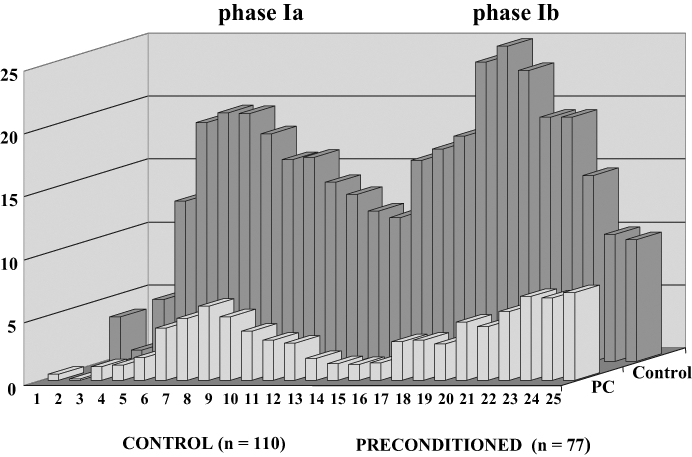
Distribution of ventricular premature beats (VPBs) at 1 min intervals over a 25 min occlusion period in 110 control dogs and in 77 preconditioned (PC) dogs which were subjected to a 25 min occlusion of the left anterior descending coronary artery. Following occlusion, the early postocclusion ventricular arrhythmias occur in two phases: there is marked ventricular ectopic activity between 3 min and 8 min of the occlusion (phase Ia), which is followed by a relatively quiet period during which the arrhythmias (VPBs) are transiently reduced. Then, after approximately 12 min to 15 min of ischemia comes a later phase (phase Ib), in which the number of VPBs is again increased and, in which, the more malignant ventricular arrhythmias, such as ventricular tachycardia or fibrillation, are common. Preconditioning, induced either by one or two 5 min periods of left anterior descending coronary artery occlusion 20 min before prolonged (25 min) ischemia, markedly reduces the number of VPBs over the entire occlusion period
In this anesthetized canine model of ischemia and reperfusion, a similar protection can be achieved if the dogs are subjected to rapid (overdrive) pacing (four times for 5 min at a rate of 220 beats/min) through the right ventricle at various time intervals before coronary artery occlusion (6). The time course of this pacing-induced antiarrhythmic protection is shown in Figure 2. It can be seen that the most marked protection (eg, against VF) occurs 5 min after the preconditioning stimulus. Increasing the time interval between the pacing stimulus and the occlusion to 15 min, 1 h or 6 h results in loss of the protection. However, the antiarrhythmic protection achieved by cardiac pacing reappears approximately 24 h later. At this time, the incidence of VF both during occlusion and reperfusion is again significantly reduced. This delayed protection against VF is not observed when the time interval between the pacing stimulus and the occlusion was increased to 48 h or 72 h (Figure 2). These results indicate that the protection against arrhythmias that resulted from cardiac pacing lasts for a somewhat shorter time than that with preconditioning induced by brief coronary artery occlusions. We do not know how cardiac pacing preconditions the heart against the consequences of a subsequent coronary artery occlusion. One explanation may be that these short periods of cardiac overdrive pacing stimuli result in an imbalance between O2 supply and demand, leading to a transient myocardial ischemia. This, together with the increase in left ventricular diastolic pressure during the pacing procedure, may induce transient ischemic injury, especially in the subendocardial region of the left ventricular wall (6). Nevertheless, we think that cardiac pacing results in less severe ischemia and, thus, a shorter protective response compared with the more ‘severe ischemia’ following complete coronary artery occlusion, indicating that the intensity of the preconditioning stimulus largely determines the degree and duration of the protection (9). This hypothesis is supported by experiments demonstrating that if the preconditioning pacing stimulus is repeated at a time when the antiarrhythmic protection from the previous pacing stimulus has already waned (ie, 48 h after the first pacing), then the protection against ischemia and reperfusion-induced arrhythmias can be prolonged (10). Thus, at this time, 48 h and 72 h after the second preconditioning pacing stimulus, no VF occurs during coronary artery occlusion and, for example, 72 h after the second pacing stimulus, nearly 80% of the dogs survive the combined ischemia-reperfusion insult (Figure 3). This prolonged protection achieved by repeated pacing is no longer apparent if the time interval between the second pacing stimulus and the occlusion is increased to 96 h (Figure 3).
Figure 2).
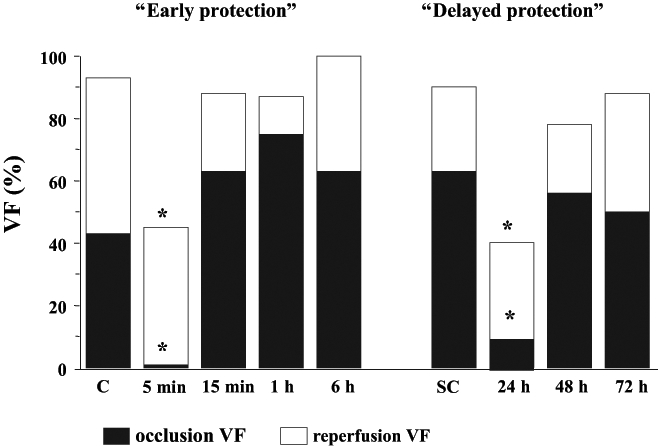
The incidence of ventricular fibrillation (VF) during a 25 min occlusion and then reperfusion of the left anterior descending coronary artery in control dogs (C) or in sham-operated controls (SC) and in dogs subjected to preconditioning by cardiac pacing (for four 5 min periods at a rate of 220 beats/min) at different times before the coronary occlusion. The incidence of VF both during occlusion and reperfusion is markedly reduced 5 min and 24 h after cessation of pacing. *P<0.05 versus control
Figure 3).
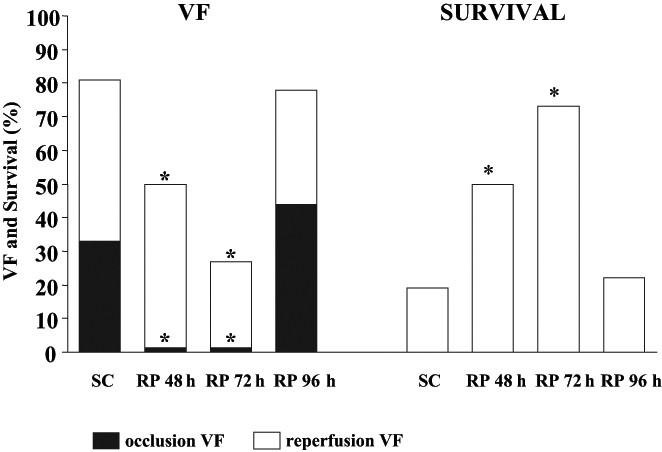
The incidence of ventricular fibrillation (VF) and survival from the combined ischemia-reperfusion insult in dogs subjected to repeated cardiac pacing (RP) (48 h after the initial pacing stimulus) and then subjected to coronary artery occlusion either 48 h, 72 h or 96 h after the end of the second pacing stimulus. The results show that repeated pacing prolongs the protection for at least 72 h against occlusion (filled histograms) and reperfusion-induced (open histograms) VF. *P<0.05 versus controls. SC Sham-operated controls
Interestingly, a similar protection against ischemia and reperfusion-induced ventricular arrhythmias could be achieved if the dogs are subjected to a single episode of treadmill exercise 24 h before coronary artery occlusion. In these experiments, dogs with an indwelling arterial catheter (to measure changes in arterial blood pressure during exercise) were subjected to a 21 min period of treadmill exercise. The slope and speed of the treadmill was increased every 3 min, reaching the maximum load (speed 13 km/h and slope 13.5%) during the final 3 min period. Such an exercise protocol results in an almost immediate marked increase in arterial blood pressure and heart rate (to approximately 200 beats/min), which are maintained during the entire running period. Twenty-four hours or 48 h later, these dogs were anesthetized, thoracotomized and subjected to a 25 min occlusion of the left anterior descending coronary artery. At the end of this period, the ischemic myocardium was rapidly reperfused. Figure 4 shows that 24 h after exercise, arrhythmia severity was much less than in the controls. Thus, in the exercised dogs, the number of VPBs and the incidence and number of episodes of ventricular tachycardia during coronary artery occlusion are significantly reduced compared with the nonexercised controls. Furthermore, no dog in the exercise group died during ischemia, compared with a 40% incidence of VF in the controls and more than two-thirds of the exercised dogs survived the combined ischemia-reperfusion insult, in contrast to only 10% of the controls (Figure 4). This marked antifibrillatory effect of a single period of exercise disappears when the time between exercise and coronary artery occlusion is increased to 48 h (Figure 5). However, as with cardiac pacing, if the exercise protocol is repeated 48 h after the first exercise, the protection against VF is again observed. Thus, at this time, no dog fibrillated during coronary artery occlusion and all the dogs subjected to repeat pacing survived the combined ischemia-reperfusion insult (Figure 5).
Figure 4).
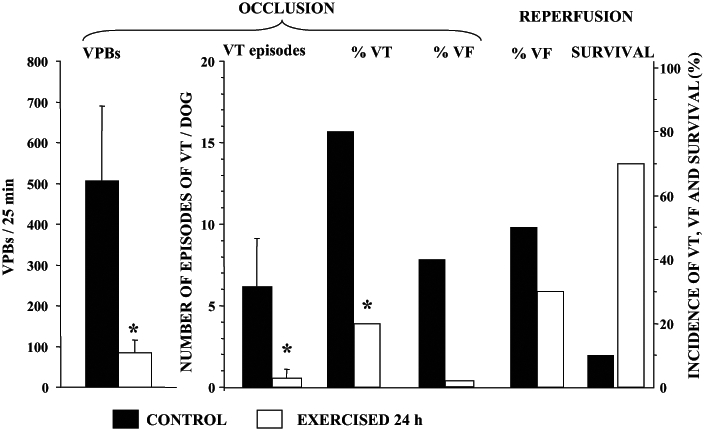
The severity of ventricular arrhythmias in control dogs (filled histograms) and in dogs subjected to treadmill exercise 24 h previously (open histograms). In the exercised dogs, the number of ventricular premature beats (VBPs) and both the incidence and the number of episodes of ventricular tachycardia (VT) are markedly reduced. Compared with the controls, in which 40% of the animals fibrillated during occlusion, no dog in the exercised group died during the same occlusion period. Furthermore, greater than two-thirds of the dogs subjected to exercise survived the combined ischemia-reperfusion insult, in contrast to only 10% of the controls. VF Ventricular fibrillation
Figure 5).
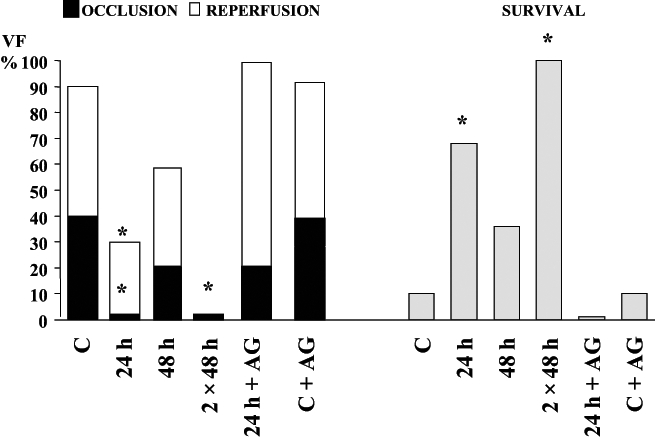
The incidence of ventricular fibrillation (VF) during coronary artery occlusion (filled histograms) and following reperfusion (open histograms) in control dogs (C), in dogs exercised 24 h and 48 h previously as well as in dogs in which the exercise protocol was repeated (two times 48 h). The effect of aminoguanidine (AG) in exercised dogs (24 h plus AG) and in nonexercised dogs (C plus AG) is also illustrated. Also shown in the right hand panels (shaded histograms) is survival from the combined ischemia-reperfusion insult. *P<0.05 versus controls
Thus, we conclude from these results that the anti-arrhythmic effect of preconditioning, no matter how induced, is extremely pronounced (it is as marked as any pharmacological intervention); however, the protection is unfortunately rather transient (it lasts for only minutes or at most 1 h to 2 h, reappearing 24 h later). This delayed protection, however, disappears 48 h or 72 h after preconditioning but, if at this time, the preconditioning stimulus (cardiac pacing or exercise) is repeated, the protection can be extended over a longer period.
EVIDENCE FOR A ROLE OF ENDOGENOUS PROTECTIVE SUBSTANCES IN THE PROTECTION AGAINST ARRHYTHMIAS INDUCED BY PRECONDITIONING, CARDIAC PACING AND EXERCISE
Whereas the general features of both the early and delayed preconditioning-induced cardioprotection have been well described, at present, little is known regarding the precise mechanisms involved in this protection. It seems most likely that several neuroendocrine and paracrine mediators are released in response to the transient ischemic injury which occurs during the preconditioning stimulus. These substances may either be protective or potentially injurious (11,12). A strong case can be made for the hypothesis that, after preconditioning, there is an increased liberation of protective substances, such as adenosine, bradykinin, nitric oxide (NO) and certain prostanoids either from ischemic cardiac myocytes and/or endothelial cells. These may compensate for the harmful consequences of a subsequent ischemic stress. Furthermore, if preconditioning is really a mechanism through which the heart is able to protect itself against ischemic stress, than it would be reasonable that more than one protective mediator is released. These mediators, acting on different receptors, may induce protection in different ways or through a final common pathway.
We were the first to propose that bradykinin, NO and prostanoids are involved both in the early and delayed protection induced by short coronary artery occlusions, cardiac pacing and physical exercise. The proposal that bradykinin may be involved in the antiarrhythmic effects of preconditioning comes first from studies which demonstrated that bradykinin has profound antiarrhythmic effects during myocardial ischemia (13) and that icatibant (HOE 140), an antagonist of bradykinin at bradykinin B2 receptors, attenuates the anti-arrhythmic protection induced either by coronary artery occlusion or cardiac pacing (14,15). For example, in dogs preconditioned by cardiac pacing, icatibant completely abolishes the early protection, markedly attenuates the delayed protection against VF and reduces the increased survival that resulted from cardiac pacing either 5 min or 24 h before coronary artery occlusion (Figure 6). These results clearly indicate that bradykinin, released early during ischemia (preconditioning), plays an important role both in early and delayed antiarrhythmic protection. We also showed that bradykinin has both a direct effect (ie, acts as a cardioprotective mediator) and acts as a trigger in preconditioning by releasing other cardioprotective mediators, such as NO and prostanoids (16).
Figure 6).

The role of bradykinin in the early and delayed antiarrhythmic-effect preconditioning induced by cardiac pacing. Compared with the controls (filled histograms) cardiac pacing, commenced either 5 min (‘classical’) or 24 h (second window of protection [‘SWOP’]) before coronary artery occlusion, markedly reduces the incidence of ventricular fibrillation and increases survival (open histograms). Icatibant given either before (hatched histograms) or after pacing, but before occlusion (dotted histograms), abolishes the early protection and markedly attenuates delayed protection against arrhythmias. *P<0.05 versus controls
As we were the first to propose, NO is one of the key mediators in both early and delayed preconditioning. The first piece of evidence that NO modulates arrhythmia severity comes from canine studies in which we demonstrated that the protective effects of preconditioning were markedly attenuated or completely abolished if the generation of NO was prevented by NG-nitro-L-arginine methyl ester (L-NAME) (17) and if the effect of NO was prevented by the inhibition of guanylyl cyclase with methylene blue (18).
Similarly, the evidence that NO plays a role in delayed cardioprotection was proposed by us in 1994 (19). In these experiments, we used dexamethasone to show that this drug prevented the delayed antiarrhythmic protection that results from cardiac pacing 24 h before ischemia (19). That particular study provided the first evidence that the delayed protection may be due to the induction of NO synthase (NOS) and/or cyclooxygenase-2 (COX-2), which are both inhibited by dexamethasone. Because of these early experiments, several more selective and specific inhibitors of the inducible NOS (iNOS) enzyme have been developed. These, in our canine ischemia/reperfusion model, also attenuated the pacing-induced antiarrhythmic protection. For example, aminoguanidine (20) and aminoethyl-methyl-isothiourea (AEST), a more selective inhibitor of iNOS (10), reversed the antiarrhythmic protection achieved by single and repeated pacing.
Similarly, there is also evidence that NO plays a role in exercise-induced delayed antiarrhythmic protection because administration of aminoguanidine to dogs subjected to a single period of exercise 24 h before coronary artery occlusion prevented protection against arrhythmias (Figure 5) (7). Also, L-NAME, a specific but nonselective NOS inhibitor, administered before exercise, and AEST, a specific and selective iNOS inhibitor, administered after exercise (but before coronary artery occlusion) prevented the antiarrhythmic protection that resulted from exercise 24 h previously. Figure 7 shows that compared with controls, exercise markedly reduces the number of VPBs, the incidences of occlusion and reperfusion-induced VF, and increases survival. This protective effect is markedly attenuated by the administration of L-NAME and AEST.
Figure 7).
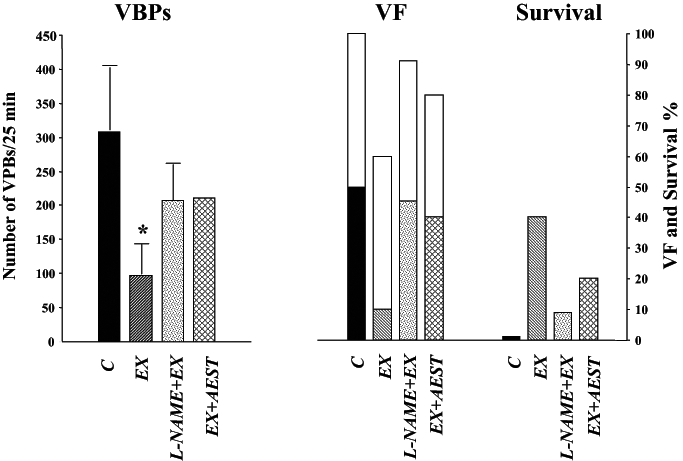
Arrhythmia severity during coronary artery occlusion and subsequent reperfusion. Shown are the incidence of ventricular fibrillation (VF), survival from the combined ischemia-reperfusion insult and the total number of ventricular premature beats (VPBs) during the 25 min occlusion. Arrhythmia severity is reduced by exercise (EX) 24 h previously and this is reversed by nitric oxide inhibition either before the exercise stimulus (NG-nitro-L-arginine methyl ester [L-NAME]) or on the following day and before coronary occlusion (aminoethyl-methyl-isothiourea [AEST]). *P<0.05 versus control (C)
To date, we do not know whether this exercise-induced NO is derived (as with ischemic preconditioning) from coronary vascular endothelial cells. However, there is evidence that chronic exercise increases coronary vascular NO production and endothelial NOS gene expression in the canine model (21). As we have previously proposed (9,22,23), one mechanism of this protection, which certainly occurs following preconditioning, cardiac pacing and exercise, might be the bradykinin-mediated increased generation and subsequent release of NO from the endothelium, and possibly also from cardiac myocytes (Figure 8). This release of NO is the means by which the endothelium ‘talks’ to cardiac myocytes and modifies myocardial function (22). One mechanism for this protection might be the elevation of cyclic GMP (cGMP) as a result of the activation of soluble guanylyl cyclase by NO. This would depress myocardial contractility and reduce energy demand, perhaps by limiting myocardial cyclic AMP (cAMP) levels by stimulation of the cGMP-dependent phosphodiesterase enzyme. cGMP may also inhibit L-type type Ca2+ channels in myocardial cells through activation of cGMP-dependent protein kinase. More recent evidence suggests that NO inhibits nor-adrenaline release from the nerve endings (24) and enhances, via a cGMP-dependent presynaptic pathway, the effects of vagal nerve stimulation (25). These mechanisms (ie, increased vagal tone, inhibition of cardiac sympathetic transmission and the modulation of the cAMP/cGMP balance in cardiac myocytes) may explain the antiarrhythmic effect of preconditioning, cardiac pacing and exercise during myocardial ischemia. Furthermore, because in our study the administration of L-NAME before exercise abolished the delayed anti-arrhythmic protection (Figure 7), we suggest that it is the generation of NO from the constitutive (endothelial NOS and/or neuronal NOS) NOS enzyme during the single exercise period that induces further NO formation from the induced enzyme 24 h later.
Figure 8).
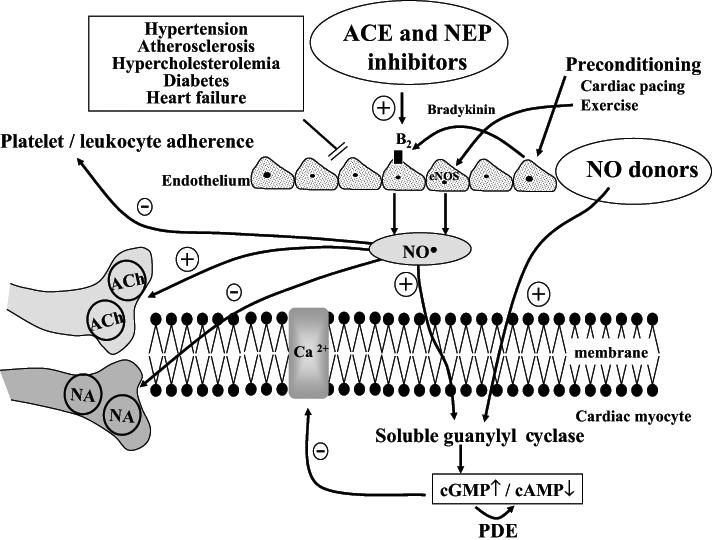
Hypothesis for the role of nitric oxide (NO) and other endothelium-derived endogenous protective mediators in the anti-arrhythmic effects of preconditioning. Ischemic preconditioning, cardiac pacing and exercise result in either a direct or a bradykinin-mediated increase in generation and subsequent release of NO from endothelial cells, and possibly also from cardiac myocytes. NO diffuses into the cardiac myocytes, stimulating soluble guanylyl cyclase and raising cyclic GMP (cGMP) levels. This both reduces cyclic AMP (cAMP) levels and Ca2+ entry through the L-type Ca2+ channels, as well as directly suppressing myocyte O2 consumption. NO can also inhibit noradrena-line (NA) release from sympathetic nerve endings and enhance, acting via a cGMP-dependent pathway, the effect of vagal nerve stimulation on presynaptic muscarinic receptors. It is hypothesized that under conditions where endothelial dysfunction is evident (hypertension, atherosclerosis, hypercholesterolemia, diabetes, etc), the ability to generate endogenous myocardial protective substances such as NO may become impaired. Thus, drugs that are able to replace the loss of these substaces (eg, NO donors), or drugs that enhance the production of these substances (eg, inhibition of kinin metabolism with angiotensin converting enzyme [ACE] and neutral endopeptidase [NEP] inhibitors which increase NO production) would be cardioprotective. Similarly, regular physical exercise, perhaps by increasing endothelial NO synthase (eNOS) gene expression, leads to increased NO production, reduced platelet and leukocyte adherence to the vascular endothelium, inhibition of cardiac sympathetic transmission and modulation of the cAMP/cGMP balance in cardiac myocytes. ACh Acetylcholine; B2 Bradykinin B2 receptor; PDE Phosphodiesterase
There are, of course, a number of other possible mediators of delayed cardioprotection that are released during preconditioning. These include catecholamines, which are able to induce late protection (for example, against ischemia and reperfusion arrhythmias [26,27]) through alpha-adrenoceptor-mediated protein kinase C activation (28,29), and which then induce iNOS gene expression (30). Exercise also releases opioid peptides which, acting on delta-opioid receptors, induce late preconditioning by a mechanism which involves COX-2 activation and the subsequent formation of prostacyclin and prostaglandin E2 (31). These prostanoids are also released by bradykinin and provide protection against arrhythmias resulting from myocardial ischemia. Because both NO and prostanoids derived from iNOS and COX-2, respectively, seem to be involved in the late protection afforded by various preconditioning procedures, it is tempting to suggest that it is the early generation of NO, together with prostacyclin (resulting from the endothelial interaction of bradykinin with bradykinin B2 receptors), that triggers delayed myocardial protection induced by preconditioning and exercise.
Finally, what might the implications of these findings be? Presumably, under those conditions where endothelial dysfunction is evident (hypertension, atherosclerosis, hypercholesterolemia, diabetes, etc), the ability to generate endogenous myocardial protective substances such as NO may be impaired (32). This would lead to the attenuation of a major pathway for protection and may explain, in part, the increased susceptibility of such patients to the arrhythmic consequences of acute ischemia. Thus, one might expect that those drugs which are able to replace the loss of these substaces resulting from such acute and chronic injury (eg, NO donors [33,34]), or drugs that enhance the production of these substances (eg, inhibition of kinin metabolism with angiotensin converting enzyme and neutral endopeptidase inhibitors with the subsequent increase in NO production [35]) would be cardioprotective (Figure 8). Similarly, regular physical exercise, perhaps by increasing endothelial NOS gene expression (36), leads to increased NO production, reduced platelet and leukocyte adherence to the vascular endothelium, inhibition of cardiac sympathetic transmission and modulation of the cAMP/cGMP balance in cardiac myocytes. Furthermore, if NO is an important signalling mechanism in preconditioning induced by cardiac pacing and exercise, then exercise by increasing NO availability, even in individuals with elevated cardiovascular risk or established disease, may represent an important means by which benefit is provided in the setting of secondary prevention.
Acknowledgments
This work was supported by the Hungarian Scientific Research Foundation (OTKA; Project number T037520 and T046243), the Health Scientific Committee of the Hungarian Ministry of Health (ETT; 25/2003) and the Hungarian Ministry of Education (FKFP 0064/2001). Professor Parratt was also the holder of a Leverhulme Trust Emeritus Fellowship and of an Albert Szent-Györgyi Fellowship, awarded by the Hungarian Government.
REFERENCES
- 1.Végh Á, Szekeres L, Parratt JR. Protective effects of preconditioning of the ischaemic myocardium involve cyclo-oxygenase products. Cardiovasc Res. 1990;24:1020–3. doi: 10.1093/cvr/24.12.1020. [DOI] [PubMed] [Google Scholar]
- 2.Végh Á, Komori S, Szekeres L, Parratt JR. Antiarrhythmic effects of preconditioning in anaesthetised dogs and rats. Cardiovasc Res. 1992;26:487–95. doi: 10.1093/cvr/26.5.487. [DOI] [PubMed] [Google Scholar]
- 3.Kuzuya T, Hoshida S, Yamashita N, et al. Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia. Circ Res. 1993;72:1293–9. doi: 10.1161/01.res.72.6.1293. [DOI] [PubMed] [Google Scholar]
- 4.Marber MS, Latchman DS, Walker JM, Yellon DM. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–72. doi: 10.1161/01.cir.88.3.1264. [DOI] [PubMed] [Google Scholar]
- 5.Végh Á, Szekeres L, Parratt JR. Transient ischaemia induced by rapid cardiac pacing results in myocardial preconditioning. Cardiovasc Res. 1991;25:1051–3. doi: 10.1093/cvr/25.12.1051. [DOI] [PubMed] [Google Scholar]
- 6.Kaszala K, Végh Á, Papp JG, Parratt JR. Time course of the protection against ischaemia and reperfusion-induced ventricular arrhythmias resulting from brief periods of cardiac pacing. J Mol Cell Cardiol. 1996;28:2085–95. doi: 10.1006/jmcc.1996.0201. [DOI] [PubMed] [Google Scholar]
- 7.Babai L, Szigeti Z, Parratt JR, Végh Á. Delayed cardioprotective effects of exercise in dogs are aminoguanidine sensitive: Possible involvement of nitric oxide. Clin Sci. 2002;102:435–45. [PubMed] [Google Scholar]
- 8.Shiki K, Hearse DJ. Preconditioning of ischemic myocardium: Reperfusion-induced arrhythmias. Am J Physiol. 1987;253:H1470–6. doi: 10.1152/ajpheart.1987.253.6.H1470. [DOI] [PubMed] [Google Scholar]
- 9.Parratt JR, Végh Á, Kaszala K, Papp JG. Suppression of life-threatening ventricular arrhythmias by brief periods of ischaemia and by cardiac pacing with particular reference to delayed myocardial protection. In: Marber M, Yellon DM, editors. Ischaemia, Preconditioning and Adaptation. Oxford; BIOS Scientific Publishers: 1996. pp. 85–113. [Google Scholar]
- 10.Kis A, Végh Á, Papp JG, Parratt JR. Repeated cardiac pacing extends the time during which canine hearts are protected against ischaemia-induced arrhythmias: Role of nitric oxide. J Mol Cell Cardiol. 1999;31:1229–41. doi: 10.1006/jmcc.1999.0955. [DOI] [PubMed] [Google Scholar]
- 11.Parratt J. Endogenous myocardial protective (antiarrhythmic) substances. Cardiovasc Res. 1993;27:693–702. doi: 10.1093/cvr/27.5.693. [DOI] [PubMed] [Google Scholar]
- 12.Végh Á, Parratt JR. Ischaemic preconditioning markedly reduces the severity of ischaemia and reperfusion-induced arrhythmias; role of endogenous myocardial protective substances. In: Wainwright CL, Parratt JR, editors. Myocardial Preconditioning. Berlin: Springer; 1996. pp. 35–55. [Google Scholar]
- 13.Végh Á, Szekeres L, Parratt JR. Local intracoronary infusions of bradykinin profoundly reduce the severity of ischaemia induced arrhythmias in anaesthetized dogs. Br J Pharmacol. 1991;104:294–5. doi: 10.1111/j.1476-5381.1991.tb12424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vegh A, Papp JG, Parratt J. Attenuation of the antiarrhythmic effects of ischaemic preconditioning by blockade of bradykinin B2 receptors. Br J Pharmacol. 1994;113:1167–72. doi: 10.1111/j.1476-5381.1994.tb17120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaszala K, Végh Á, Papp JG, Parratt JR. Modification by bradykinin B2 receptor blockade of the protection by pacing against iachaemia-induced arrhythmias. Eur J Pharmacol. 1997;328:51–60. doi: 10.1016/s0014-2999(97)83027-0. [DOI] [PubMed] [Google Scholar]
- 16.Végh Á, Papp JG, Szekeres L, Parratt JR. Prevention by an inhibitor of the L-arginine nitric oxide pathway of the antiarrhythmic effects of bradykinin in anaesthetized dogs. Br J Pharmacol. 1993;110:18–9. doi: 10.1111/j.1476-5381.1993.tb13764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Végh Á, Szekeres L, Parratt J. Preconditioning of the ischaemic myocardium; involvement of the L-arginine nitric oxide pathway. Br J Pharmacol. 1992;107:648–52. doi: 10.1111/j.1476-5381.1992.tb14501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Végh Á, Papp JG, Szekeres L, Parratt J. The local intracoronary administration of methylene blue prevents the pronounced antiarrhythmic effect of ischaemic preconditioning. Br J Pharmacol. 1992;107:910–1. doi: 10.1111/j.1476-5381.1992.tb13384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Végh Á, Papp JG, Parratt JR. Prevention by dexamethasone of the marked antiarrhythmic effects of preconditioning induced 20 h after rapid cardiac pacing. Br J Pharmacol. 1994;113:1081–2. doi: 10.1111/j.1476-5381.1994.tb17104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kis A, Végh Á, Papp J, Parratt J. Pacing-induced delayed protection against arrhythmias is attenuated by aminoguanidine, an inhibitor of nitric oxide synthase. Br J Pharmacol. 1999;127:1545–50. doi: 10.1038/sj.bjp.0702695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sessa WC, Pritchard K, Seyedi N, Wang J, Hintze TH. Chronic exercise in dogs increases coronary vascular nitric oxide production and endothelial cell nitric oxide synthase gene expression. Circ Res. 1994;74:349–53. doi: 10.1161/01.res.74.2.349. [DOI] [PubMed] [Google Scholar]
- 22.Végh Á, Kis A, Papp JG, Parratt JR. Early and delayed protection against ventricular arrhythmias induced by preconditioning. In: Mochizuki S, Takeda N, Nagano M, Dhalla NS, editors. The Ischaemic Heart. Kluwer Academic Publisher; 1998. pp. 279–305. [Google Scholar]
- 23.Parratt JR, Végh Á. Delayed protection against ventricular arrhythmias by cardiac pacing. Heart. 1997;78:423–5. doi: 10.1136/hrt.78.5.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwarz P, Diem R, Dun NJ, Forstermann U. Endogenous and exogenous nitric oxide inhibits norepinephrine release from rat heart sympathetic nerves. Circ Res. 1995;77:841–8. doi: 10.1161/01.res.77.4.841. [DOI] [PubMed] [Google Scholar]
- 25.Sears CE, Choate JK, Paterson DJ. NO-cGMP pathway accentuates the decrease in heart rate caused by cardiac vagal nerve stimulation. J Appl Physiol. 1999;86:510–6. doi: 10.1152/jappl.1999.86.2.510. [DOI] [PubMed] [Google Scholar]
- 26.Ravingerova T, Song W, Pancza D, Dzurba A, Ziegelhoeffer A, Parratt J. Pretreatment with catecholamines can suppress severe ventricular arrhythmias in rats: Relevance to ischemic preconditioning. J Exp Clin Cardiol. 1997;2:19–24. [Google Scholar]
- 27.Végh Á, Parratt JR. Noradrenaline, infused locally, reduces arrhythmia severity during coronary artery occlusion in anaesthetised dogs. Cardiovasc Res. 2002;55:53–63. doi: 10.1016/s0008-6363(02)00342-5. [DOI] [PubMed] [Google Scholar]
- 28.Banerjee A, Locke-Winter C, Rogers KB, et al. Preconditioning against myocardial dysfunction after ischemia and reperfusion by an alpha 1-adrenergic mechanism. Circ Res. 1993;73:656–70. doi: 10.1161/01.res.73.4.656. [DOI] [PubMed] [Google Scholar]
- 29.Bankwala Z, Hale SL, Kloner RA. Alpha-adrenoceptor stimulation with exogenous norepinephrine or release of endogenous catecholamines mimics ischemic preconditioning. Circulation. 1994;90:1023–8. doi: 10.1161/01.cir.90.2.1023. [DOI] [PubMed] [Google Scholar]
- 30.Ikeda U, Murakami Y, Kanbe T, Shimada K. Alpha-adrenergic stimulation enhances inducible nitric oxide synthase expression in rat cardiac myocytes. J Mol Cell Cardiol. 1996;28:1539–45. doi: 10.1006/jmcc.1996.0144. [DOI] [PubMed] [Google Scholar]
- 31.Kodani E, Xuan YT, Shinmura K, Takano H, Tang XL, Bolli R. Delta-opioid receptor-induced late preconditioning is mediated by cyclooxygenase-2 in conscious rabbits. Am J Physiol Heart Circ Physiol. 2002;283:H1943–57. doi: 10.1152/ajpheart.00150.2002. [DOI] [PubMed] [Google Scholar]
- 32.Parratt JR. Possibilities for the pharmacological exploitation of ischaemic preconditioning. J Mol Cell Cardiol. 1995;27:991–1000. doi: 10.1016/0022-2828(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 33.Végh Á, Györgyi K, Papp JG, Sakai K, Parratt JR. Nicorandil suppressed ventricular arrhythmias in a canine model of myocardial ischaemia. Eur J Pharmacol. 1996;305:163–8. doi: 10.1016/0014-2999(96)00166-5. [DOI] [PubMed] [Google Scholar]
- 34.György K, Végh Á, Rastegar MA, Papp JG, Parratt JR. Isosorbide-2-mononitrate reduces the consequences of myocardial ischaemia, including arrhythmia severity: Implications for preconditioning. Cardiovasc Drugs Ther. 2000;14:481–8. doi: 10.1023/a:1007832921391. [DOI] [PubMed] [Google Scholar]
- 35.Rastegar MA, Marchini F, Morazzoni G, Végh Á, Papp JG, Parratt JR. The effects of Z13752A, a combined ACE/NEP inhibitor, on responses to coronary artery occlusion; a primary protective role for bradykinin. Br J Pharmacol. 2000;129:671–80. doi: 10.1038/sj.bjp.0703109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kingwell BA. Nitric oxide as a metabolic regulator during exercise: Effects of training in health and disease. Clin Exp Pharmacol Physiol. 2000;27:239–50. doi: 10.1046/j.1440-1681.2000.03232.x. [DOI] [PubMed] [Google Scholar]