

Abstract
In 1983, a delayed and prolonged cardioprotection induced by drugs was described. This pharmacologically induced adaptation to stress represents a new trend in cardioprotection as opposed to the classical drug treatment that was based on the presence of drug-receptor binding. Such a long-lasting, delayed adaptation can be induced by non-injurious pharmacological stimuli (eg, prostacyclin and its stable analogues, catecholamines and other substances) and manifests as a marked protection against the severe consequences of ischemia; attenuation of early morphological changes (limitation in infarct size) and reduction in ventricular arrhythmias as results of coronary artery occlusion and reperfusion or ouabain toxication. The protection is time- and dose-dependent; the maximum effects occur 24 h and 48 h after drug treatment. These effects can be prolonged for a longer period by the periodic administration of maintenance doses. Concerning the mechanism of this marked delayed protection, the findings show that these adaptive stresses stimulate the adenylate cyclase/cyclic AMP (cAMP) system and result in elevation in cardiac cAMP level. This triggers the induction of Na+/K+-ATPase and activates phosphodiesterase (PDE) isoforms, most likely PDE1 and PDE4. The increased amount of PDE isoforms and activated Na+/K+-ATPase moderates ischemic myocardial potassium loss, and reduces sodium and calcium accumulation during myocardial ischemia. This also attenuates ouabain toxicity. Induction of PDE isoforms may lead to a reduction in the accumulation of excess cAMP and contribute to a lessened response to beta-adrenergic stimuli. The antiarrhythmic effects can be explained by electrophysiological changes, such as prolongations of the effective refractory period and the action potential duration during ischemia and reperfusion. The advantages of pharmacologically induced adaptation to stress in preventive therapy are that an exact dosage can be applied, the risk of the harmful effects is minimal, the protection can be prolonged, and it can be induced under pathological conditions (eg, atherosclerosis, hypercholesterolemia). Pharmacologically induced long-term protection may represent a new approach in the therapy of cardiovascular diseases.
Keywords: Cardiac protection, Delayed adaptation to stress, Na+/K+-ATPase, Phosphodiesterase isoforms, Prostanoids
The delayed and prolonged cardiac protection evoked by drug-induced adaptation to stress was described 20 years ago (1,2) – three years before the publication of the ‘classical’ short-lived protection (3) and 10 years before the description of delayed protection, both induced by ischemic preconditioning (4). This drug-induced endogenous ‘self-defense’ of the heart is a new principle in cardioprotection, as opposed to the classical drug treatment, which was based on drug-receptor binding.
This pioneer work seems to be forgotten, and is rarely quoted. Thus, the present review is devoted to this phenomenon, emphasizing the role of prostacyclin and its stable derivatives (7-oxo-prostacyclin or carbacyclin [iloprost, Ilomedin, Schering AG, Berlin) used in our research.
Drug-induced delayed protection as the form of cardiac adaptation
Adaptation to changing conditions is a basic function of organisms that enables the individual and the species to survive. An adaptive process, evoked by a preparatory ‘preconditioning’ (possibly noninjurious) stress might generate protection against severe forms of stress. This stress might induce functional and metabolic changes in the myocardium, which protect the heart against harmful consequences of a subsequent, severe stress.
In his first experiments (1,2), the author used the dog angina model, which has previously been described (5). In this model, pacing the heart at a normal rate in the presence of critical constriction of the left anterior descending coronary artery provides a sufficient blood supply to the myocardium. However, increasing the rate to approximately twice the original rate caused myocardial ischemia to occur, as indicated by an elevation of the epicardial ST segment. In this reproducible angina model, prostacyclin (PGI2) and 7-oxo-PGI2 – both well known to have a short half-life – markedly reduced ST segment elevation. This protection was apparent when the hypotensive and platelet aggregation inhibitory direct effects of these drugs wore off (Figure 1).
Figure 1).
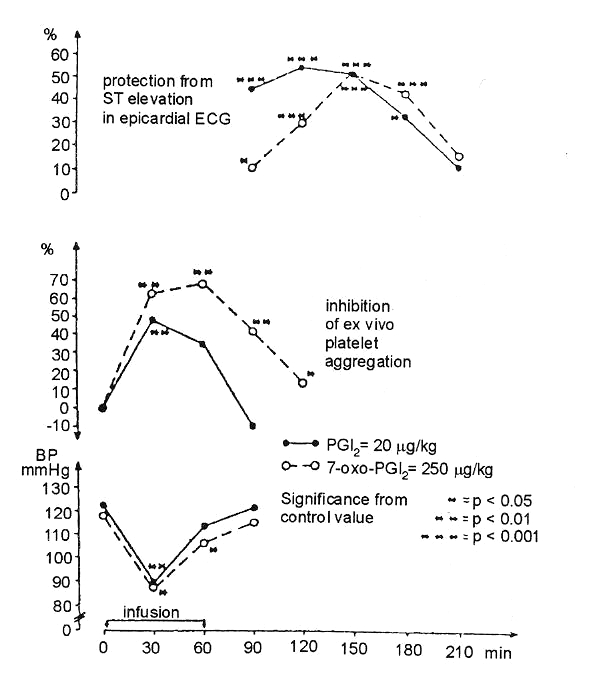
Drug-induced protection in a canine model of experimental angina. Time-dependent protective effects of the 20 μg/kg dose of prostacyclin (PGI2) (continuous line) and the 250 μg/kg dose of 7-oxo-PGI2 (broken line) on pacing-induced epicardial ST segment changes, ex vivo platelet aggregation and arterial blood pressure (BP). ECG Electrocardiogram
After this first observation, the cardioprotective effects of 7-oxo-PGI2 (and later carbacyclin [iloprost]) were thoroughly analyzed in several species (dog, rabbit, guinea pig and rat [2,6–9]) and in various experimental models. The time course and the dose dependence of the protection were also investigated. The protective effect was assessed by functional, cytoprotective and metabolic changes.
FUNCTIONAL EFFECTS
Anti-ischemic actions
As mentioned above, PGI2 and 7-oxo-PGI2 resulted in delayed protection, as shown by the reduction of ischemia-induced epicardial ST elevation in the dog angina model (2) and also by the attenuation of the vasopressin-induced T wave rise in rats (2). Similarly, in conscious, chronically instrumented rabbits, iloprost attenuated the elevations of ST segment and left ventricular end-diastolic pressure (LVEDP) that occurred during high rate pacing (200 beats/min for 5 min), performed 24 h and 48 h after treatment (10) (Figure 2). In rats, 7-oxo-PGI2 treatment 48 h before excision of the heart moderated the loss of ATP, creatine phosphate and accumulation of lactate that resulted from ischemia produced by incubation in Ringer solution for 1 min (11) (Figure 3).
Figure 2).
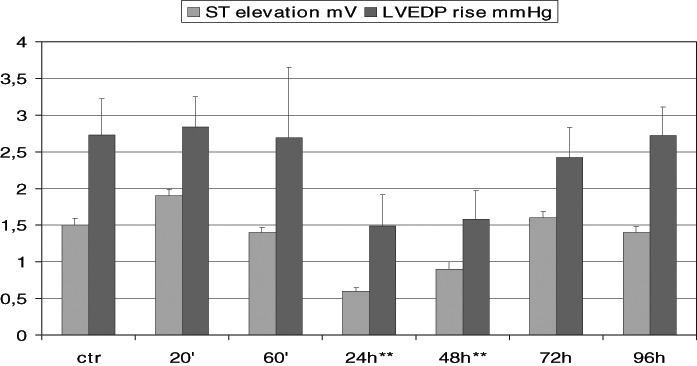
Drug-induced protection in conscious, chronically instrumented rabbits. Time-dependent changes in ST segment and left ventricular end-diastolic pressure (LVEDP) elevation resulted from high rate pacing (200 beats/min for 5 min) before (control [ctr]) and 20 min, 60 min, 24 h, 48 h, 72 h and 96 h after the administration of 10 μg/kg iloprost. **Significantly different from control values at P<0.01
Figure 3).
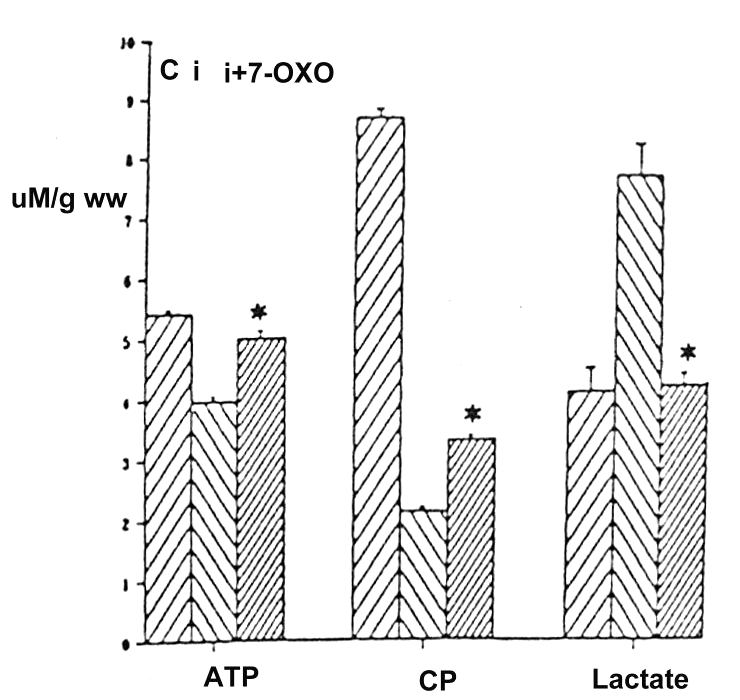
The effect of 7-oxo-prostacyclin (7-OXO) on ischemia-induced (incubation in Ringer solution for 1 min) metabolic changes in the rat heart. ATP, creatine phosphate (CP) and lactate levels (μM/g wet weight) were determined in control (C), ischemic (i) and ischemic hearts treated with 50 μg/kg 7-OXO (i + 7-OXO) 48 h before ischemia. Ischemia significantly reduced ATP and CP content and increased the lactate level in rat hearts. These changes were reversed by the prior treatment with 7-OXO
Antiarrhythmic actions
Antiarrhythmic actions were examined in anesthetized dogs subjected to occlusion of the left anterior descending coronary artery at various times after the administration of 7-oxo- PGI2. The most marked reduction in the number of ventricular ectopic beats both during occlusion and following reperfusion could be observed 48 h after 7-oxo-PGI2 treatment (12) (Figure 4). Furthermore, in a ouabain-induced arrhythmia model in guinea pigs, the arrhythmogenic doses of ouabain were increased if the guinea pigs had been treated with 7-oxo-PGI2 24 h, 48 h and 72 h previously (13,14) (Figure 5). This indicates an improved safety when cardiac glycosides were used.
Figure 4).
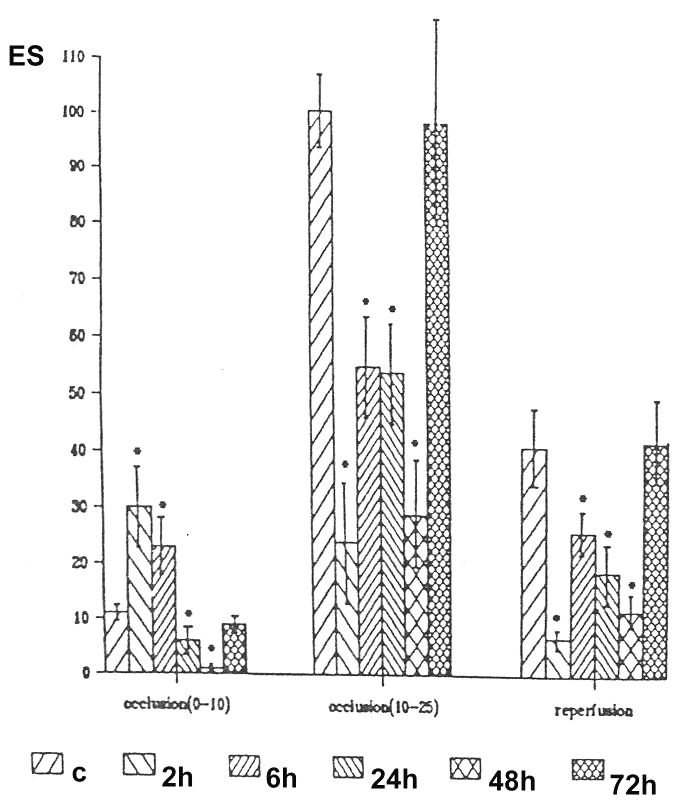
Early and delayed effects of 7-oxo-prostacyclin (7-oxo-PGI2) on ischemia and reperfusion-induced ventricular arrhythmias in anesthetized dogs. The number of extrasystoles (ES/min) during phase I (0 min to 10 min) and phase II (10 min to 25 min) of occlusion of the left anterior descending coronary artery and following reperfusion is shown in control (C) dogs and in dogs treated with 7-oxo-PGI2 2 h, 6 h, 24 h, 48 h and 72 h before the ischemia-reperfusion insult
Figure 5).
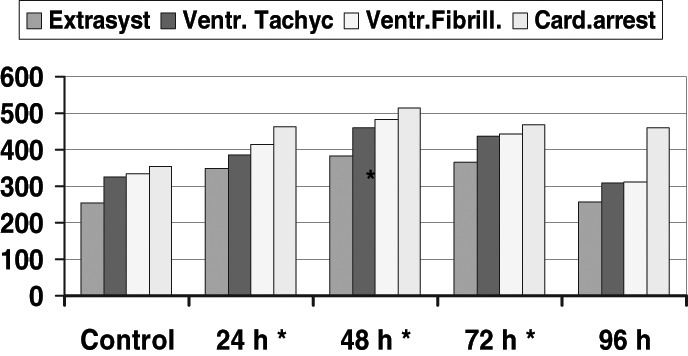
The effect of 7-oxo-prostacyclin (7-oxo-PGI2) ouabain-induced arrhyhmias in anesthetized guinea pigs. Ventricular arrhythmias (ventricular extrasystoles [Extrasyst], tachycardiac episodes [Ventr Tachyc], ventricular fibrillation [Ventr Fibrill] and cardiac arrest [Card arrest]) were evaluated in the controls and in guinea-pigs treated with 7-oxo-PGI2 24 h, 48 h, 72 h and 96 h before the induction of ouabain arrhythmias. *Significantly different from control values at P<0.01
Electrophysiological actions
Electrophysiological actions were examined in conscious rabbits. Administration of 7-oxo-PGI2 resulted in a transient reduction in the ventricular effective refractory period, which lasted for 20 min. After 20 min, the ventricular effective refractory period started to increase and a marked prolongation could be observed in those rabbits that were treated with 7-oxo-PGI2 24 h, 48 h and 72 h before (6). Similarly, QT intervals were increased when rabbits were given an intramuscular injection of 50 μg/kg 7-oxo-PGI2 at various times previously. This prolongation of the QT interval was apparent 6 h after the injection and it was still present 72 h later (6). As shown in Figure 6, atrial and ventricular fibrillation thresholds in anesthetized cats increased 24 h, 48 h and 72 h after 7-oxo-PGI2 treatment (6).
Figure 6).
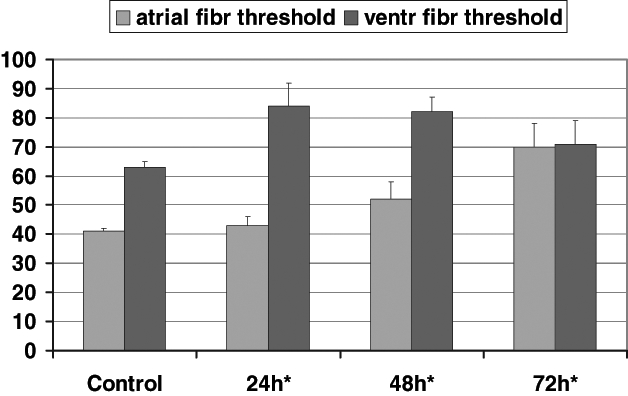
The time course of the protective effect of 7-oxo-prostacyclin (7-oxo-PGI2) treatment on changes of atrial and ventricular electrical fibrillation (atrial fibr and ventr fibr) threshold in anesthetized cats. 7-oxo-PGI2 significantly increased ventricular fibrillation threshold 24 h, 48 h and 72 h after 7-oxo-PGI2 treatment. The atrial fibrillation threshold was markedly increased 48 h and 72 h after drug administration. *Significantly different from control values at P<0.01
In a rabbit papillary muscle preparation, incubation with 7-oxo-PGI2 for 20 min, followed by washout, markedly prolonged the action potential duration and the effective refractory period. This prolongation in action potential duration and effective refractory period started 2 h after the washout and was still present 4 h later (15).
Antiadrenergic action
Antiadrenergic action was evaluated in conscious rabbits. 7-oxo-PGI2 markedly attenuated responses to beta-adrenergic stimuli. For example, the increase in heart rate following increasing doses of isoproterenol was markedly attenuated if rabbits were given 7-oxo-PGI2. This protective effect occurred 6 h after 7-oxo-PGI2 treatment and was still present 24 h and 48 h later (11).
Cytoprotective actions
In Langendorff-perfused rabbit hearts, 7-oxo-PGI2, administered 48 h before the 25 min period of global ischemia, prevented the early ultrastructural changes (7,16). Further, an evaluation of the time course effects of iloprost, given intravenously in a dose of 10 μg/kg, in rabbits showed that the most marked reduction in infarct size (necrotic area) occurred 24 h and 48 h after treatment. This protection against ischemic damage was not apparent if the time interval between the injection of iloprost and the coronary occlusion was increased to 72 h (Figure 7). Figure 7 also shows that iloprost did not cause early protection, ie, the infarct size was not reduced 2 h after treatment compared with the controls. The risk area was not affected (17).
Figure 7).
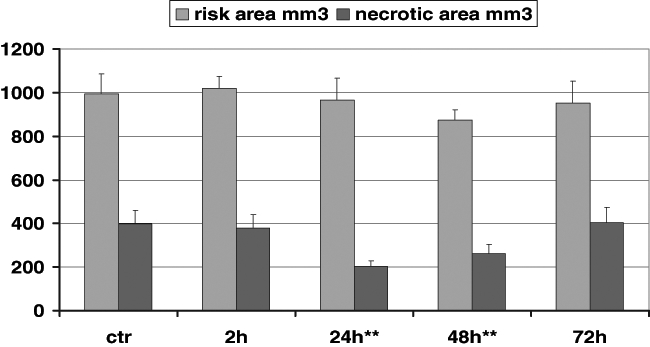
The protective effect of iloprost on infarct size in the rabbit heart. Iloprost (10 μg/kg) was administered 2 h, 24 h, 48 h and 72 h before ischemia, and the reduction in infarct size (mm3) was compared with the reduction in the untreated controls. There was no difference between the groups in the risk area. Iloprost resulted in only late protection against ischemic damage. **Significantly different from control values at P<0.01
The marked delayed protection resulting from the administration of 7-oxo-PGI2 and iloprost could be prolonged for as long as 14 days if a maintenance dose of 25 μg/kg was given every three days (8). Figure 8 shows that in dogs treated with 7-oxo-PGI2 for 14 days (50 μg/kg starting dose and then 25 μg/kg maintenance dose every three days) before coronary artery occlusion, the severity of ventricular arrhythmias (ie, the number of ventricular premature beats) and myocardial ischemia (ST segment elevation) that occurred during occlusion were markedly reduced.
Figure 8).

The effect of 7-oxo-prostacyclin (7-oxo-PGI2) in anesthetized dogs on changes of epicardial ST segment (left panel) and on the number of extrasystoles (right panel) resulting from a 25 min occlusion of the left anterior descending coronary artery (LAD coron art) performed at various times (6 h, 24 h, 48 h, 72 h and 14 days) after 7-oxo-PGI2 treatment. ctr Control
Similarly, drugs that activate the adenylate cyclase/cyclic AMP (cAMP) pathway, such as isoprenaline, induce late protection. Isoprenaline, given in fractions to avoid overdose (2 μg/kg in 5 min intervals), reduced epicardial ST segment elevation and increased LVEDP, which resulted from adrenergic loading induced by both isoprenaline (2 μg/kg) and phenylephrine (16 μg/kg) (17).
Recapitulating the effects of the stable PGI2 analogues and isoprenaline, we propose that they might induce dose- and time-dependent delayed protection (maximal protection occurs 24 h and 48 h after administration of 50 μg/kg 7-oxo- PGI2 or 10 μg/kg iloprost or 10 × 2 μg/kg isoprenaline) against the consequences of a severe stress. In our case, this protection manifested in anti-ischemic actions, such as reduction in coronary artery occlusion-induced ischemic ST segment changes in anesthetized dogs, reduction in elevations of the ST segment and LVEDP induced by cardiac pacing in conscious rabbits, and attenuation of the ischemic breakdown of ATP and creatine phosphate and increase of lactate in rat hearts. The protection also manifested in antiarrhythmic actions; these drugs reduced both the early and the late post-occlusion and reperfusion arrhythmias in anesthetized dogs, and in cytoprotective actions, by limiting early structural changes due to ischemia in isolated guinea pig hearts, limiting the ischemia-induced loss of intracellular K+ and Na+ and the accumulation of Ca2+ in isolated guinea pig hearts, and limiting infarct size in rabbits.
Molecular basis of delayed cardiac protection
The delayed protective action of PGI2 analogues and of isoprenaline is not due to a long-standing drug-receptor binding. It appears late, when the direct effects of these drugs with a short ‘half-life’ are over. Time is needed to develop changes leading to prolonged protection. These changes are also dose-dependent and appear even after elimination of the agent. The protection can be observed in isolated hearts, ie, in the absence of extra-cardiac factors (nervous and hormonal influences or blood constituents), and is not dependent on the species used.
The protective effect of PGI2 analogues is a ubiquitous metabolic effect. Further, the drug-induced delayed and prolonged protection differs from what could be evoked by physical stresses (eg, brief periods of rapid pacing, coronary artery occlusion or heat stress), ie, there is no early (‘first window’) phase of protection because only a delayed, late-appearing protection can be induced by drugs.
On the other hand, both the drug and the other stresses induced the release of endogenous substances that resulted in delayed protection. These substances include catecholamines and prostanoids, which activate the adenylate cyclase/cAMP pathway, and nitric oxide, which preferentially activates the guanylate cyclase/cyclic GMP pathway. Other mediators such as adenosine, endothelin, bradykinin and angiotensin II are also released during these stresses and involved in protection, but the present paper does not deal with them.
It is well known that catecholamines and PGI2 are released during ischemia. By stimulating the adenylate cyclase/cAMP pathway, these substances may result in excess amounts of cAMP, which then lead to severe ischemic changes. cAMP then produces an increase in the free fatty acid (FFA) level by activating triglyceride lipase. This blocks Na+/K+-ATPase and results in a loss of intracellular K+ and a rise in cytosolic Na+ and Ca2+, thus facilitating the generation of arrhythmias. The rise in myocardial oxygen consumption that results from the increased FFA oxidation contributes to ischemia.
Possible mechanism of drug-induced delayed cardioprotection
What might be the mechanism of pharmacological and other stress-induced prolonged and delayed cardiac protection? We propose that the antiadrenergic effects of drug treatment protect the heart by mitigating the harmful rise of cAMP that occurs during ischemia. This might happen either by inhibiting the formation of cAMP or by enhancing its breakdown. Inhibition of cAMP formation can be excluded because 7-oxo- PGI2 treatment failed to affect the density of beta1 adrenoceptors, the G proteins or the activity of adenylate cyclase (18). However, there is evidence for the enhanced breakdown. For example, in the rat myocardium, 48 h after 7-oxo-PGI2 treatment, the activity of cardiac phosphodiesterase (PDE) 1 and PDE4 isoforms is markedly increased. This increase is sufficient for splitting the excess of cAMP (18).
A similar increase in Na+/K+-ATPase activity occurs 24 h and 48 h after 7-oxo-PGI2 treatment. This may prevent the intracellular ionic changes, such as the loss of Ki+ and the rise in Nai+ and Cai2+ (19). Accordingly, a sufficient (but not yet harmful) amount of cAMP is needed to trigger de novo protein synthesis of some key enzymes in the nucleus, which are responsible for delayed and prolonged cardiac protection.
We suggest the following mechanism for the delayed and prolonged cardioprotection via the cAMP pathway. Increase in the activity of Na+/K+-ATPase restores ionic homeostasis, whereas the increased formation of PDE isoforms split the excess of cAMP and thereby moderate the rise of FFA and oxygen consumption during ischemia. Increased release of PGI2 may also play a role in the delayed and prolonged cardioprotection. Our evidence for this is that under both basal and ischemic conditions, PGI2 levels (but not thromboxane) increased 24 h and 48 h after 7-oxo-PGI2 treatment in guinea pig hearts, upregulation of cyclo-oxygenase-2 (COX-2) expression and COX-2-dependent synthesis of prostanoids (prostaglandin E2, PGI2) have been found 24 h after ischemic preconditioning, and COX-2-inhibitors abolished both the ischemia-induced prostanoid formation and the preconditioning-induced reduction of infarct size in rabbits (20).
Furthermore, our findings are also supported by the results that an altered gene expression of PDE1 and PDE4 cyclic nucleotide PDE isoforms was shown 48 h after 7-oxo-PGI2 treatment in the rat heart (21) and that administration of 7-oxo-PGI2 in rats markedly reduced the infarct size, the diminution of the R wave and the increase in serum malonyldialdehyde level that resulted from coronary artery occlusion 72 h later (22).
The membrane stabilizing action of 7-oxo-PGI2 was shown in rats where treatment with 24 h and 48 h before excision of the heart prevented the Ca2+ depletion-induced depression in Na+/K+-ATPase activity, the accompanying structural and functional damages and the development of Ca2+ overload, following repletion of Ca2+ (calcium paradox) (9). Thus, 7-oxo-PGI2 prevented the intracellular Na+ accumulation and the loss in cytosolic K+ that occurred during ischemia (23).
Furthermore, our studies showed that 7-oxo-PGI2 pretreatment in rats suppressed the reperfusion-induced ventricular arrhythmias when the hearts were isolated and subjected to an ischemia/reperfusion challenge (24). Also in isolated rat hearts, the administration of 7-oxo-PGI2 48 h before improved the recovery of developed pressure and coronary flow during the reperfusion-induced Ca2+ overload and provided a preserved macroergic phosphate content and cardiac ultrastructure during the Ca2+ depletion phase (25).
Possible therapeutic implications of the drug-induced delayed cardioprotection
Requirements for the use of preconditioning in therapy can be summarized as follows: the stimulus that elicits protection should be applicable in patients; the duration of the protection should be long; and, if possible, further prolongation of the protection should be achieved.
In patients, the use of ischemic preconditioning that is sufficiently intensive to evoke delayed protection is hazardous. Coronary occlusions (even brief ones) to induce protection cannot be used in patients. Similarly, immobilization, thermal stress, and the use of global ischemia by rapid pacing or tachycardia following heavy physical exercise are limited by the risk of arrhythmias in patients suffering from heart disease. Drugs that mimic the effect of ischemic preconditioning (eg, A1 receptor agonists) evoke early and delayed protection, but the duration of cardioprotection is short, just as with ischemic preconditioning. Cardioprotection induced by drugs, however, persists longer and fulfills the requirements for therapeutic application. Thus, drugs can be applied in patients by enabling an exact (if necessary, fractionated) dosage and causing minimal side effects. They can provide prolonged protection; therefore, timing of the trigger impulse is not a critical criterion. The duration of protection can be prolonged by periodically using a lower maintenance dose. The protection induced by drugs can be produced under pathological conditions, such as atherosclerosis and hypercholesterolemia (26).
We think that in the future drug-induced delayed cardiac adaptation to stress will be a valuable tool in preventing life-threatening cardiac events.
Acknowledgments
The author thanks Prof Ágnes Végh for revision of the manuscript.
Footnotes
Paper was presented at the Fourth International Symposium on Myocardial Cytoprotection, Pécs, Hungary, September 25 to 27, 2003
REFERENCES
- 1.Szekeres L, Krassói I, Udvary É. Delayed antiischemic effect of PGI2 and of a new stable PGI2 analogue 7-oxo-prostacyclin-Na in experimental model angina in dogs. J Mol Cell Cardiol. 1983;15(Suppl l):394. (Abst) [PubMed] [Google Scholar]
- 2.Szekeres L, Koltai M, Pataricza J, Takáts I, Udvary É. On the late antiischaemic action of the stable PGI2 analogue: 7-oxo-PGI2-Na and its possible mode of action. Biomed Biochim Acta. 1984;43:S135–42. [PubMed] [Google Scholar]
- 3.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–6. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 4.Kuzuya T, Hoshida S, Yamashita N, et al. Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia. Circ Res. 1993;72:1293–9. doi: 10.1161/01.res.72.6.1293. [DOI] [PubMed] [Google Scholar]
- 5.Szekeres L, Csik V, Udvary É. Nitroglycerin and dipyridamole on cardiac metabolism and dynamics in a new experimental model of angina pectoris. J Pharmacol Exp Ther. 1976;196:15–28. [PubMed] [Google Scholar]
- 6.Szekeres L, Szilvássy Z, Udvary É, Végh Á. 7-oxo-PgI2 induced late appearing and long-lasting electrophysiological changes in the heart in situ of the rabbit, guinea pig, dog and cat. J Mol Cell Cardiol. 1989;21:545–54. doi: 10.1016/0022-2828(89)90820-1. [DOI] [PubMed] [Google Scholar]
- 7.Szekeres L, Bálint ZS, Karcsú S, Tósaki Á. Delayed protection by 7-oxo-PGI2 against cardiac transmembrane ion shifts and early morphological changes due to ischemia and reperfusion. Cardioscience. 1990;1:279–86. [PubMed] [Google Scholar]
- 8.Udvary É, Végh Á, Szekeres L. 7-oxo-PGI2 induced late protective action from arrhythmias due to local myocardial ischemia. Bratisl Lek Listy. 1991;92:146–9. [PubMed] [Google Scholar]
- 9.Ravingerova T, Styk J, Tregerova V, et al. Protective effect of 7-oxo-prostacyclin on myocardial function and metabolism during postischemic reperfusion and calcium paradox. Basic Res Cardiol. 1991;86:245–53. doi: 10.1007/BF02190604. [DOI] [PubMed] [Google Scholar]
- 10.Krause EG, Szekeres L. On the mechanism and possible therapeutic application of delayed adaptation of the heart to stress situations. Mol Cell Biochem. 1995;147:115–22. doi: 10.1007/BF00944791. [DOI] [PubMed] [Google Scholar]
- 11.Szekeres L, Németh M, Szilvássy Z, Tósaki Á, Udvary É, Végh Á. On the nature and molecular basis of prostacyclin induced late cardiac changes. Biomed Biochim Acta. 1988;47:S6–11. [PubMed] [Google Scholar]
- 12.Udvary É, Szekeres L. Prostacyclin antiischaemic or cardioprotective? In: Kecskeméti V, Gyires K, Kovács G, editors. Advances in Pharmacological Research and Practice, Volume 3. Prostanoids. Budapest: Akadémiai Kiadó; 1986. pp. 333–8. [Google Scholar]
- 13.Szekeres L, Szilvássy Z, Udvary É, Végh Á. 7-oxo-PgI2 induced late appearing protection against ouabain induced cardiac arrhythmia in anaesthetised guinea pigs. Pharmacol Res Commun. 1988;20:77–8. [Google Scholar]
- 14.Szilvássy Z, Szekeres L, Udvary É, Végh Á. On the 7-oxo-PgI2 induced lasting protection against ouabain arrhythmias in anesthetized guinea pigs. Biomed Biochim Acta. 1988;47:S35–8. [PubMed] [Google Scholar]
- 15.Szekeres L, Németh M, Papp JG, Udvary É. Short incubation with 7-oxo-prostacyclin induces long lasting prolongation of repolarisation time and effective refractory period in rabbit papillary muscle preparation. Cardiovasc Res. 1990;24:37–41. doi: 10.1093/cvr/24.1.37. [DOI] [PubMed] [Google Scholar]
- 16.Szekeres L, Bálint ZS, Karcsú S, Tósaki Á, Udvary É. On the 7-oxo-PgI2 induced late appearing long-lasting cytoprotective effect. In: Schrör K, Sinzinger H, editors. Prostaglandins in Clinical Research: Cardiovascular System. New York: Alan R Liss Inc; 1989. pp. 143–7. [PubMed] [Google Scholar]
- 17.Szekeres L. Delayed adaptation to stress – A clinically useful form of cardiac protection. Exp Clin Cardiol. 2000;5:116–21. [Google Scholar]
- 18.Borchert G, Bartel S, Beyerdorfer I, Kuttner I, Szekeres L, Krause EG. Long lasting anti-adrenergic effect of 7-oxo-prostacyclin in the heart: A cycloheximide sensitive increase of phosphodiesterase isoform I and IV activities. Mol Cell Biochem. 1994;132:57–67. doi: 10.1007/BF00925675. [DOI] [PubMed] [Google Scholar]
- 19.Dzurba A, Ziegelhoeffer A, Breier A, Vrbjar N, Szekeres L. Increased activity of sarcolemmal (Na+K+)-ATPase is involved in the late cardioprotective action of 7-oxo-prostacyclin. Cardioscience. 1991;2:105–8. [PubMed] [Google Scholar]
- 20.Shinmura K, Tang XL, Wang Y, et al. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proc Natl Acad Sci. 2000;97:10197–202. doi: 10.1073/pnas.97.18.10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kostic MM, Erdogan S, Rena G, et al. Altered expression of PDE1 and PDE4 cyclic nucleotide phosphodiesterase isoforms in 7-oxo-prostacyclin-preconditioned rat heart. J Mol Cell Cardiol. 1997;29:3135–46. doi: 10.1006/jmcc.1997.0544. [DOI] [PubMed] [Google Scholar]
- 22.Ranaut K, Singh M, Chopra K, Ganguly NK. Effect of 7-oxo-PGI2 on myocardial infarct size and role of oxygen radicals in its protective effect. Arch Int Pharmacodyn Ther. 1993;324:75–86. [PubMed] [Google Scholar]
- 23.Ziegelhoffer A, Ravingerova T, Dzurba A, Tribulova N, Slezak J, Breier A. Prevention by 7-oxo-prostacyclin of the calcium paradox in rat heart: Role of the sarcolemmal (Na,K)-ATPase. Mol Cell Biochem. 1996:160–1. 257–63. doi: 10.1007/BF00240057. [DOI] [PubMed] [Google Scholar]
- 24.Ravingerova T, Tribulova N, Ziegelhoffer A, Styk J, Szekeres L. Suppression of reperfusion induced arrhythmias in the isolated rat heart: Pretreatment with 7-oxoprostacyclin in vivo. Cardiovasc Res. 1993;27:1051–5. doi: 10.1093/cvr/27.6.1051. [DOI] [PubMed] [Google Scholar]
- 25.Ravingerova T, Tribulova N, Ziegelhoffer A. 7-oxo-prostacyclin protects the rat heart against calcium paradox. Life Sci. 1993;53:1309–16. doi: 10.1016/0024-3205(93)90576-o. [DOI] [PubMed] [Google Scholar]
- 26.Szekeres L, Szilvássy Z, Ferdinándy P, Nagy I, Karcsu S, Csáti S. Delayed cardiac protection against harmful consequences of stress can be induced in experimental atherosclerosis in rabbits. J Mol CeIl Cardiol. 1997;29:1977–83. doi: 10.1006/jmcc.1997.0418. [DOI] [PubMed] [Google Scholar]