

Abstract
Oxidative stress can generate a mass of oxygen free radicals (OFR) in the cells, and these OFRs can induce several acute and chronic symptoms and diseases. If the amount of the generated OFRs overwhelms the antioxidant capacity of the cells, the pathophysiological changes may lead to the death of the cell or the development of chronic degenerative diseases.
The phenomenon of ischemic preconditioning has demonstrated the important role of these aggressive and harmful molecules in the endogenous adaptation mechanism of the cells to oxidative stress. After sublethal oxidative stress – mild ischemic insult – the resulting development of a few OFRs can stimulate the intracellular signal-transduction cascade of ischemic preconditioning and, through the induction of severe transcription factors, new antioxidant enzymes and heat shock proteins will be synthesized. These newly synthesized proteins will protect the cellagainst another, more serious oxidative insult in the future.
Keywords: Endogenous adaptation, Free radicals, Ischemic preconditioning, Signal transduction
PRECONDITIONING AND THE ANTIOXIDANT DEFENCE SYSTEM OF THE CELL
Ischemic preconditioning (IPC) is a cellular adaptive response of the heart to stress, whereby one or more brief, sublethal periods of ischemia enhance the tissue’s tolerance to a later, more sustained ischemia and reperfusion. The evoked cytoprotection is biphasic: the classic or early preconditioning (PC) appears immediately after the PC stimuli and lasts for 2 h to 3 h, while the ‘second window of protection’ (SWOP) may be exhibited 24 h to 72 h later. PC as an endogenous adaptation form of the myocardium was first described by Murry et al (1). Since then, PC has been most intensively examined in the myocardium, but recent evidence demonstrates that the same endogenous adaptation is available in several other tissues (eg, bowel mucosa, parenchimal tissues, liver, skeletal muscle) (2–6). The role of oxygen free radicals in ischemia-reperfusion syndrome, chronic degenerative diseases and carcinogenesis is unquestionable. Because delayed PC (SWOP) stimulates the endogenous antioxidant system of the cells, application of this phenomenon may provide a therapeutic line to treat these diseases. The goal of this research (the exact definition of the cellular signalling mechanism of the PC) is to develop the possibility of pharmacological PC. By using drugs – without ischemia-reperfusion cycles – the endogenous adaptation of the cells is accelerated and, therefore, the endogenous antioxidant defence system is enhanced. Drug-induced PC may provide a great opportunity in the prevention, in case of serious risk factors, or therapy of the ‘free radical diseases’.
IPC
The protection conferred by PC with ischemia (now termed classic PC) appears to be an acute and immediate response lasting not more than a few hours. The protection has been evoked by various PC protocols and tested using different endpoints such as limitation in infarct size, reduced susceptibility to arrhythmia, better recovery from contractile dysfunction and cardiac enzyme release.
In 1993, two separate studies by Marber et al (7) and Kuzuya et al (8) observed that, in addition to the initial phase, a second wave of protection appears 24 h following the PC protocol. This second wave of protection is now referred to as SWOP, late PC or delayed PC. SWOP has certain characteristics distinct from classic PC. It appears gradually, yet lasts as long as 72 h or more. Also, the protection offered is not as marked as with classic PC. A fundamental difference between classic and delayed PC may be in the means by which cardioprotection is conveyed. In the former, ATP-sensitive potassium channels are suspected to be the end-effectors; in the latter, newly synthesized cardioprotective proteins may convey protection. Several such proteins have been identified that seem to be upregulated 24 h following IPC, which corresponds with the appearance of SWOP. They include the heat shock protein (HSP) family (such as HSP72), manganese superoxide dismutase (SOD) and, more recently, nitric oxide synthase (NOS). HSP72 is a chaperone protein involved in the folding, transport and denaturation of other proteins during the cellular response to injury. Manganese SOD is a mitochondrial antioxidant capable of detoxifying accumulated superoxide anions.
Most early SWOP studies took infarct size reduction as the end point of cardioprotection, and there were little data regarding any delayed antiarrhythmic effect in the second window. In fact, a study conducted by Shiki and Hearse (9) found no protection against reperfusion-induced arrhythmias if the period between stimulus and insult was extended to 24 h. However, in 1994, Vegh et al (10) published a study that positively confirmed delayed protection against reperfusion arrhythmias in the canine heart using ventricular rapid pacing to globally precondition the heart.
The positive role of oxygen free radicals in the delayed adaptation
Oxygen free radicals are highly reactive molecules with an unpaired electron, and are associated widely with ischemic-reperfusion injury (11–13). Although better known for their toxicity, in large quantities they overwhelm the endogenous antioxidant systems or, if the antioxidant system is insufficient or damaged, they accelerate the oxidative stress (Figure 1).
Figure 1).
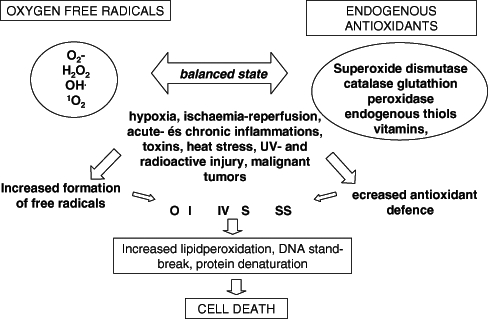
Oxidative balance of the cell: development of oxidative stress. H2O2 Hydrogen peroxide; O2− Superoxide anion; OH• Hydroxyl radical; UV Ultraviolet
It has been suggested that at low concentrations, oxygen free radicals can modulate functions within the cell. Soon after their initial findings (1), Murry et al (14) investigated a role for oxygen free radicals in classic IPC. They showed that administration of oxygen free radicals scavengers blocked the protection afforded by IPC against infarction in dogs. Similar findings achieved with the use of N-2-mercaptopropionyl glycine or SOD further substantiated these results (15). Furthermore, Osada et al (16) could abolish the antiarrhythmic effects of IPC by administration of SOD during the PC protocol. However, there have also been numerous negative studies. Iwamoto et al (17) failed to show diminished protection using either SOD or catalase in a rabbit model of 4×5 min ischemia with an intermittant 5 min reperfusion. Similarly, our published results demonstrated that administering N-2-mercaptopropionyl glycine in an in vivo model could not abolish the protective effects of three cycles of delayed PC (18–21). However, as previously mentioned, the induction of IPC seems to be very much model- and species-dependent.
Redox regulation, adaptation and survival signals
Recurrent episodes of myocardial ischemia are commonly observed in patients with coronary artery disease who suffer from frequent angina pectoris or who have had angioplasty of the left anterior descending coronary artery. Reversibly injured myocardium (by a short episode of ischemia followed by a short period of reperfusion) renders the heart more resistant to a longer ischemic-reperfusion period. Such adaptation (IPC) is mediated through the upregulation of the heart’s own cellular defense via the accumulation of intracellular mediators and reprogramming of gene expression. Recent studies suggest that nuclear factor-kappa B (NF-κB) and activation protein-1 (AP1) transcription factors have a possible role in the signal transduction pathways of this cytoprotection, resulting from ischemic adaptation. A positive role of free radicals in the activation cascade of these transcription factors has been confirmed (Figure 2).
Figure 2).
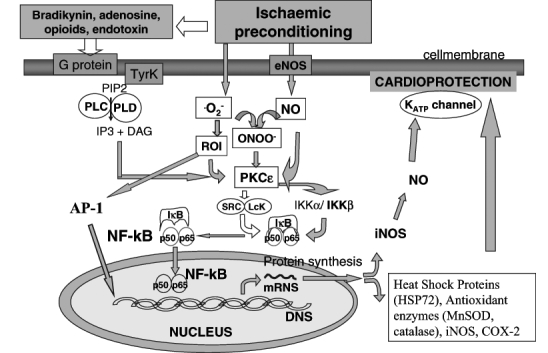
Intracellular signalling of ischemic preconditioning. DAG Diacyl glycerol; IKK Inhibitor kappa kinase; iNOS Inducible nitric oxide synthase; IP3 Inositol triphosphate; Mn-SOD Manganese super-oxide-dismutase; NF-κB Nuclear factor-kappa B; NO nitric oxide; ONOO− Peroxynitrite; PLC and PLD Phospholipase C and D; PIP2 Phosphatidyl inositol diphosphate; PKCɛ Protein kinase Cɛ; ROI Reactive oxygen intermediaries; TyrK Tyrosine kinase
A potential role for NF-κB in the endogenous adaptation of IPC has recently been suggested (22,23). Myocardial protection by NF-κB activation may be caused by induction of an NF-κB-regulated mediator, such as manganese SOD (24), inducible cyclooxygenase (25) and inducible NOS (26). The protective effect of NF-κB activation could also be caused by a downregulation of the inflammatory response during reperfusion. In human umbilical vein endothelial cells preconditioned by hydrogen peroxide, reduced upregulation of cytokines and leukocyte adhesion molecules after subsequent stimulation with tumour necrosis factor (TNF)-αwas found (27). HSPs of the 70 kDa family, which are upregulated during PC by other pathways, have been suggested as mediators of ischemic adaptation (28). HSPs modulate AP1 and NF-κB DNA binding activity (29) and may reduce NF-κB activation, thereby reducing inflammation during reperfusion. A last possible route of NF-κB-mediated cardioprotection is through the antiapoptotic effect of PC. Preconditioning reduces apoptosis during reperfusion, which may be linked to an NF-κB-dependent increase of cardiac Bcl-2 (30).
The authors’ results under publication show that there is a biphasic activation of NF-κB in the preconditioned myocardium, with increased levels at an early time point (30 min) and again at 3 h reperfusion. There are presumed to be two different pathways leading to early NF-κB activation after IPC. Through receptor-dependent triggers (adenosine A1 agonists [31], opioid δ1 agonists [32], bradykinin, prostaglandins, noradrenaline, angiotensin, endothelin), the receptor is coupled through G proteins to, among others, phospholipase C (PLC) and phospholipase D. PLC catalyzes the hydrolysis of membrane inositol-containing phospholipids into inositol trisphosphate and diacyl glycerol (DAG) (33). DAG stimulates the translocation and activation of protein kinase Cɛ (PKCɛ). The onset of the PLC reaction is typically very rapid, and DAG production is short-lived, peaking at 30 s. PKCɛ activation then triggers a complex signalling cascade that involves Src and/or Lck tyrosine kinases and probably other kinases, leading to phosphorylation of inhibitor-κB (IκB)α and to mobilization (nuclear translocation) and activation of the transcription factor NF-κB (34).
Another possible way of NF-κB activation in IPC is through the increased production of nitric oxide (NO) (most likely via endothelial cell [e]NOS) and •O2− (leading to formation of secondary reactive oxygen species) after a brief episode of myocardial ischemia/reperfuson (35). Both NO and •O2− derived reactive oxygen species could directly activate PKCɛ via nitrosylation and oxidative modification, respectively; alternatively, NO and •O2− are known to react to form peroxynitrite which, in turn, could activate PKCɛ. Thus, PKCɛ is thought to be a critical component in both pathways (36).
IPC has recently been found to activate Janus-activated kinase 1 and 2 with a subsequent tyrosine phosphorylation and activation of signal transducers and activators of transcription (STAT) 1 and STAT3, which are essential for inducible (i)NOS upregulation. Binding of NF-κB and STAT1 and STAT3 to the iNOS promoter results in transcriptional activation of the iNOS gene and leads to synthesis of new iNOS proteins (eNOS-dependent iNOS induction). iNOS-derived NO is supposed to produce a second wave of PKCɛ activation, leading to the late phase of NF-κB activation and translocation (37).
The other possible explanation of the late increase in NF-κB activation may be the feedback mechanism of NF-κB-induced proinflammatory cytokines (TNFα, interleukin [IL]-1β). After ischemia-reperfusion, even in the case of IPC, NF-κB can induce TNFαand IL-1βgene expression. Through a currently unknown mechanism, these cytokines generate a mass of reactive oxygen intermediates in the myocardium, which can, via the above mentioned pathway, newly activate NF-κB in the cytoplasm and lead to a delayed wave of nuclear translocation of this transcription factor. TNFαand IL-1βcan also start up a signaling pathway leading to IκB phosphorylation and, thus, NF-κB activation, through cell membrane receptors (38). A number of signal transduction proteins have been identified as associated with these receptors, including TNF receptor-associated factors 2 and 6 death domain-containing proteins (TNF receptor associated death domain and Fas-associated death domain), kinases associated with IL-1 receptor (IL-1 receptor-associated kinase 1 and 2, and myeloid differentiation primary response gene 88). These kinases phosphorylate members of the IκB family at specific serines within their N-termini, leading to site-specific ubiquitination and degradation of NF-κB by the 26S proteosome. This circle cascade (NF-κB→TNFα, IL-1β→NF-κB feedback) might also be an explanation of the biphase activation of NF-κB after IPC (39).
Clinical aspects of endogenous adaptation
This detailed signalling description demonstrates that free radicals have a positive role in the activation of transcription factors, which leads to clinically applicable cardioprotection. However, a number of free radicals can cause well-known free radical diseases, a small amount of which can start an adaptation response without causing free radical damage. The therapeutic goals are to induce the endogenous adaptation with a mild oxidative stress and increase the tissue tolerance against serious oxidative damage.
The first study to assess adaptation to ischemia during coronary angioplasty was reported by Deutsch et al (40), and involved 12 patients with an isolated stenosis undergoing two sequential 90 s balloon inflations. In comparison with the initial balloon occlusion, the second occlusion was characterized by less subjective anginal pain, smaller ST segment shift and lower mean pulmonary artery pressure. Collateral recruitment was excluded as a cause. Other investigators have observed similar findings in percutaneous transluminal coronary angioplasty models (41–43), thus confirming an adaptive response to repeated ischemic episodes, akin to IPC.
Intermittent ischemia achieved by aortic cross-clamping in a fibrillating heart during coronary artery bypass grafting has been used as a clinical model of IPC. In such a model, the confounding effects due to collateral flow are overcome by using global instead of regional ischemia. Yellon et al (44) examined the effect of two 3-min ischemic episodes on high energy phosphate metabolism during 10-min cross-clamping. Myocardial biopsies exhibited a significantly higher ATP content than in controls not previously exposed to brief ischemic episodes, demonstrating typical biochemical features of IPC. However, this finding was contradicted by a more recent study (45) that used warm-blood cardioplegic arrest during bypass surgery. Nevertheless, more positive evidence has been uncovered in similar investigations (46–48). Taken together, these findings suggest that IPC occurs in this human model with potentially relevant beneficial clinical effects. This is particularly attractive in the management of high-risk patients with poor left ventricular function.
Further examinations demonstrated that PC not only increases the ischemia tolerance of the cells, but it also turns up the intracellular antioxidant armament of the cells and ensures efficient protection against a further oxidative stress insult. Therefore, in normal conditions, a low level of cellular stress is useful because it activates and maintains the cellular antioxidant defence. In healthy organisms, preventive applied exogenous antioxidants (uncontrolled treatment with vitamin C and E) can be even more harmful, because they may suppress this defence mechanism. This ability of the cells to adapte to oxidative stress changes with aging; in senior populations, this adaptive response is less efficient. Furthermore, it has been demonstrated that in some chronic diseases (diabetes mellitus, asthma, essential hypertension, chronic arthritis, malignant tumours) the capacity of this endogenous protective mechanism is depleted, or the intracellular signalling pathway of the adaptation is injured. Thus, in these critical conditions, the cells can lose their own defence mechanism. With a more exact understanding of this signalling cascade, drug-induced PC and endogenous adaptation may be achieved.
Footnotes
Paper was presented at the Fourth International Symposium on Myocardial Cytoprotection, Pécs, Hungary, September 25 to 27, 2003
REFERENCES
- 1.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: A delay of lethal injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 2.Lin AM, Dung SW, Chen CF, Chen WH, Ho LT. Hypoxic preconditioning prevents cortical infarction by transient focal ischemia-reperfusion. Ann N Y Acad Sci. 2003;993:168–78. 195–6. doi: 10.1111/j.1749-6632.2003.tb07527.x. [DOI] [PubMed] [Google Scholar]
- 3.Dembinski A, Warzecha Z, Ceranowicz P, et al. Ischemic preconditioning reduces the severity of ischemia/reperfusion-induced pancreatitis. Eur J Pharmacol. 2003;473:207–16. doi: 10.1016/s0014-2999(03)01994-0. [DOI] [PubMed] [Google Scholar]
- 4.Fernandez L, Heredia N, Peralta C, et al. Role of ischemic preconditioning and the portosystemic shunt in the prevention of liver and lung damage after rat liver transplantation. Transplantation. 2003;76:282–9. doi: 10.1097/01.TP.0000067529.82245.4E. [DOI] [PubMed] [Google Scholar]
- 5.Wang SF, Li GW. Early protective effect of ischemic preconditioning on small intestinal graft in rats. World J Gastroenterol. 2003;9:1866–70. doi: 10.3748/wjg.v9.i8.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Badhwar A, Dungey AA, Harris KA, et al. Limitations of ischemic tolerance in oxidative skeletal muscle: Perfusion vs tissue protection. J Surg Res. 2003;109:62–7. doi: 10.1016/s0022-4804(02)00044-6. [DOI] [PubMed] [Google Scholar]
- 7.Marber MS, Latchman DS, Walker JM, Yellon DM. Cardiac stress protein elevation 24 hours following brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–72. doi: 10.1161/01.cir.88.3.1264. [DOI] [PubMed] [Google Scholar]
- 8.Kuzuya T, Hoshida S, Yamashita N, et al. Delayed effects of sublethal ischemia on the acqusition of tolerance to ischemia. Circ Res. 1993;72:1293–9. doi: 10.1161/01.res.72.6.1293. [DOI] [PubMed] [Google Scholar]
- 9.Shiki K, Hearse DJ. Preconditioning of ischemic myocardium: Reperfusion-induced arrhythmias. Am J Physiol. 1987;253:H1470–6. doi: 10.1152/ajpheart.1987.253.6.H1470. [DOI] [PubMed] [Google Scholar]
- 10.Vegh A, Papp JG, Parratt JR. Prevention by dexamethasone of the marked antiarrhythmic effects of preconditioning induced 20h after rapid cardiac pacing. Br J Pharmacol. 1994;113:1081–2. doi: 10.1111/j.1476-5381.1994.tb17104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Becker J, Mezger W, Courgeon AM, Best-Belpomme M. On the mechanism of action of H2O2 in the cellular stress. Free Radic Res Commun. 1991:12–13. 455–60. doi: 10.3109/10715769109145817. [DOI] [PubMed] [Google Scholar]
- 12.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci USA. 1987;84:1404–7. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kloner RA, Pryzyklenk K, Whittaker P. Deleterious effects of oxygen free radicals in ischemia/reperfusion. Circulation. 1989;80:1115–27. doi: 10.1161/01.cir.80.5.1115. [DOI] [PubMed] [Google Scholar]
- 14.Murry CE, Richard VJ, Jennings RB, Reimer KA. Preconditioning with ischemia: Is the protective effect mediated by free radical induced myocardial stunning? Ciculation. 1988;78(Suppl II):77. [Google Scholar]
- 15.Tanaka M, Fujiwara H, Yamasaki K, Sasayama S. Superoxide dismutase and N-2-mercaptopropionyl glycine attenuate infarct size limitation effect of ischemic preconditioning in the rabbit. Cardiovasc Res. 1994;28:980–6. doi: 10.1093/cvr/28.7.980. [DOI] [PubMed] [Google Scholar]
- 16.Osada M, Takeda S, Sato T, Komori S, Tamura K. The protective effect of preconditioning on reperfusion induced arrhythmias is lost by treatment with superoxide dismutase. Jpn Circ. 1994;58:259–63. doi: 10.1253/jcj.58.259. [DOI] [PubMed] [Google Scholar]
- 17.Iwamoto T, Miura T, Adachi T, et al. Myocardial infarct size limiting effect of ischemic preconditioning was not attenuated by oxygen free-radical scavengers in the rabbit. Circulation. 1991;83:1015–22. doi: 10.1161/01.cir.83.3.1015. [DOI] [PubMed] [Google Scholar]
- 18.Roth E, Jaberansari MT. Reactive oxygen species in early and delayed cardiac adaptation. Exp Clin Cardiol. 2001;6:81–6. [PMC free article] [PubMed] [Google Scholar]
- 19.Roth E, Jaberansari MT, Jorgensen JJ. Should reperfusion injury be always regarded as deleterious? The up-side of oxidative stress. Perfusion. 2000;13:370. [Google Scholar]
- 20.Richard V, Tron C, Thuillez C. Ischemic preconditioning is not mediated by oxygen derived free radicals in rats. Cardiovasc Res. 1993;27:2016–21. doi: 10.1093/cvr/27.11.2016. [DOI] [PubMed] [Google Scholar]
- 21.Roth E, Hejjel L. Oxygen free radicals in heart disease. In: Pugsley MK, editor. Cardiac Development Guide. Totowa: Humana Press Inc; 2003. pp. 47–66. [Google Scholar]
- 22.Maulik N, Sato M, Price BD, Das DK. An essential role of NFkB in tyrosine kinase signaling of p38 MAP kinase regulation of myocardial adaptation to ischemia. FEBS Lett. 1998;429:365–9. doi: 10.1016/s0014-5793(98)00632-2. [DOI] [PubMed] [Google Scholar]
- 23.Morgan EN, Boyle EM, Jr, Yun W, et al. An essential role for NF-kappaB in the cardioadaptive response to ischemia. Ann Thorac Surg. 1999;68:377–82. doi: 10.1016/s0003-4975(99)00646-3. [DOI] [PubMed] [Google Scholar]
- 24.Dana A, Jonassen AK, Yamashita N, Yellon DM. Adenosine A1 receptor activation induces delayed preconditioning in rats mediated by manganese superoxide dismutase. Circulation. 2000;101:2841. doi: 10.1161/01.cir.101.24.2841. [DOI] [PubMed] [Google Scholar]
- 25.Shinmura K, Tang XL, Wang Y, et al. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proc Natl Acad Sci USA. 2000;97:10197–202. doi: 10.1073/pnas.97.18.10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo Y, Jones WK, Xuan Y-T, et al. The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene. Proc Natl Acad Sci USA. 1999;96:11507. doi: 10.1073/pnas.96.20.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zahler S, Kupatt C, Becke BF. Endothelial preconditioning by transient oxidative stress reduces inflammatory responses of cultured endothelial cells to TNF-alpha. FASEB. 2000;14:555–64. doi: 10.1096/fasebj.14.3.555. [DOI] [PubMed] [Google Scholar]
- 28.Marber MS, Latchman DS, Walker JM, Yellon DM. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–72. doi: 10.1161/01.cir.88.3.1264. [DOI] [PubMed] [Google Scholar]
- 29.Carter DA. Modulation of cellular AP-1 DNA binding activity by heat shock proteins. FEBS Lett. 1997;416:81–5. doi: 10.1016/s0014-5793(97)01174-5. [DOI] [PubMed] [Google Scholar]
- 30.Maulik N, Sato M, Price BD, Das DK. An essential role of NFkB in tyrosine kinase signaling of p38 MAP kinase regulation of myocardial adaptation to ischemia. FEBS Lett. 1998;429:365–9. doi: 10.1016/s0014-5793(98)00632-2. [DOI] [PubMed] [Google Scholar]
- 31.Downey JM, Liu GS, Thornton JD. Adenosine and the anti infarct effects of preconditioning. Cardiovasc Res. 1993;27:3–8. doi: 10.1093/cvr/27.1.3. [DOI] [PubMed] [Google Scholar]
- 32.Schultz JEJ, Hsu AK, Nagase H, Gross GJ. TAN-67, a δ1-opioid receptor agonist, reduces infarct size via activation of Gi/o proteins and KATP channels. Am J Heart Circ Physiol. 1998;43:H909–14. doi: 10.1152/ajpheart.1998.274.3.H909. [DOI] [PubMed] [Google Scholar]
- 33.Eskildsen-Helmond YEG, Gho BCG, Bezstarosti K, et al. Exploration of possible roles of phospholipase D and protein kinase C in the mechanism of preconditioning in the myocardium. Ann N Y Acad Sci. 1996;793:210–25. doi: 10.1111/j.1749-6632.1996.tb33516.x. [DOI] [PubMed] [Google Scholar]
- 34.Speechly-Dick ME, Mocanu MM, Yellon DM. Protein kinase C. Its role in ischemic preconditioning in the rat. Circ Res. 1994;276:H1229–35. doi: 10.1161/01.res.75.3.586. [DOI] [PubMed] [Google Scholar]
- 35.Bolli R, Manchikalapudi S, Tang XL, et al. The protective effects of late preconditioning against myocardial stunning in conscious rabbits are mediated by nitric oxide synthase: evidence that nitric oxide acts both as a trigger and as a mediator of the late phase of ischemic preconditioning. Circ Res. 1997;81:1094–107. doi: 10.1161/01.res.81.6.1094. [DOI] [PubMed] [Google Scholar]
- 36.Vondriska T, Zhang J, Song C, et al. PKCepsilon-SRC modules direct signal transduction in NO-induced cardioprotection: Complex formation as a means for cardioprotective signaling. Circ Res. 1999;88:1306–13. doi: 10.1161/hh1201.092994. [DOI] [PubMed] [Google Scholar]
- 37.Xuan Y-T, Tang X-L, Qiu Y, et al. Biphasic response of cardiac NO synthase to ischaemic preconditioning in conscious rabbits. Am J Physiol. 2000;279:H2360–71. doi: 10.1152/ajpheart.2000.279.5.H2360. [DOI] [PubMed] [Google Scholar]
- 38.Valen G, Yan Z-Q, Hansson GK. Nuclear factor kappa-B and the heart. J Am Coll Cardiol. 2001;38:307–14. doi: 10.1016/s0735-1097(01)01377-8. [DOI] [PubMed] [Google Scholar]
- 39.Mercurio F, Manning AM. NF-kB as a primary regulator of the stress response. Oncogene. 1999;18:6163–71. doi: 10.1038/sj.onc.1203174. [DOI] [PubMed] [Google Scholar]
- 40.Deutsch E, Berger M, Kussmaul WG, Hirshfeld JW, Herrmann HC, Laskey WK. Adaptation to ischemia during percutaneous transluminal coronary angioplasty. Clinical, hemodynamic, and metabolic features. Circulation. 1990;82:2044–51. doi: 10.1161/01.cir.82.6.2044. [DOI] [PubMed] [Google Scholar]
- 41.The ISIS-4 investigators A randomized factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. Lancet. 1995;345:669–85. [PubMed] [Google Scholar]
- 42.Claeys MJ, Vrints CJ, Bosmans JM, Conraads VM, Snoeck JP. Aminophylline inhibits adaptation to ischemia during angioplasty. Role of adenosine in ischemic preconditioning. Eur Heart J. 1996;17:539–44. doi: 10.1093/oxfordjournals.eurheartj.a014906. [DOI] [PubMed] [Google Scholar]
- 43.Leesar MA, Stoddard M, Ahmed M, Broadbent J, Bolli R. Preconditioning of human myocardium with adenosine during coronary angioplasty. Circulation. 1997;95:2500–7. doi: 10.1161/01.cir.95.11.2500. [DOI] [PubMed] [Google Scholar]
- 44.Yellon DM, Alkhulaifi AM, Pugsley WB. Preconditioning the human myocardium. Lancet. 1993;342:276–7. doi: 10.1016/0140-6736(93)91819-8. [DOI] [PubMed] [Google Scholar]
- 45.Perrault LP, Menaschè P, Bel A, et al. Ischemic preconditioning in cardiac surgery: A word of caution. J Thorac Cardiovasc Surg. 1996;112:1378–86. doi: 10.1016/s0022-5223(96)70155-1. [DOI] [PubMed] [Google Scholar]
- 46.Jenkins DP, Pugsley WB, Alkhulaifi AM, Kemp M, Hooper J, Yellon DM. Ischemic preconditioning reduces troponin T release in patients undergoing coronary artery bypass surgery. Heart. 1997;77:314–8. doi: 10.1136/hrt.77.4.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mentzer RM, Rahko PS, Molina-Viamonte V, et al. Safety, tolerance, and efficacy of adenosine as an additive to blood cardioplegia in humans during coronary artery bypass surgery. Am J Cardiol. 1997;79:38–43. doi: 10.1016/s0002-9149(97)00262-2. [DOI] [PubMed] [Google Scholar]
- 48.Menasché P, Jamieson WRE, Flameng W, Davis MK. Acadesine: A new drug that may improve myocardial protection in coronary artery bypass grafting (CABG) J Thorac Cardiovasc Surg. 1995;110:1096–106. doi: 10.1016/s0022-5223(05)80179-5. [DOI] [PubMed] [Google Scholar]