

Abstract
During ischemia-reperfusion, reactive oxygen species are generated along the mitochondrial respiratory chain and induce lipid peroxidation, protein oxidation and DNA damage. Single-strand DNA breaks are the most potent activators of poly(ADP-ribose) polymerase (PARP); prolonged action of PARP culminates in intracellular oxidized nicotinamide adenine dinucleotide (NAD+) and ATP depletion. The integrity of cellular components and the myocardial energy metabolism can be preserved by using PARP inhibitors under conditions of ischemia and reperfusion. Oxidative stress is capable of activating the phosphoinositol-3-kinase–Akt/protein kinase B signalling pathway, which is further enhanced if treated with PARP inhibitors. Akt, in turn, promotes the survival of cardiomyocytes by inhibiting apoptotis, and causing metabolic adjustment and vasodilation in the jeopardized myocardium.
Keywords: Akt, Energy metabolism, Heart, Ischemia-reperfusion, Poly(ADP-ribose) polymerase
Ischemia-reperfusion is associated with enhanced formation of reactive oxygen species (ROS) and peroxynitrite, which initiate lipid peroxidation (1), protein oxidation (2) and the formation of DNA breaks. Poly(ADP-ribose) polymerase (PARP) is a protein-modifying and nucleotide-polymerizing enzyme that is abundantly present in the nucleus. Single-strand DNA breaks are the obligatory triggers of PARP activation, which can be induced by a variety of environmental stimuli and free radicals, most notably hydroxyl radical and peroxynitrite. In response to DNA damage, PARP becomes activated and, by cleaving nicotinamide adenine dinucleotide (oxidized form [NAD+]) as a substrate, catalyzes the building of homopolymers of adenosine diphosphate ribose units, resulting in a substantial depletion of intracellular NAD+. Because nicotinamide adenine dinucleotide (reduced form [NADH]) functions as an electron carrier in the mitochondrial respiratory chain, NAD+ depletion is coupled with a rapid fall in intracellular ATP levels. Thus, excessive activation of PARP leads to ATP depletion, which may ultimately cause cell death (3,4). As a consequence, inhibition of PARP can improve the recovery of different cells from oxidative injury (5).
In heart tissue, a dominant fraction of energy production occurs in the mitochondria; therefore, protection against oxidative damage of mitochondria can be a very important step in the normalization of cardiac energy production. Previous data showed that PARP inhibitors were capable of reducing the oxidative damage of cellular components without any obvious scavenger activity (6). Although necrosis is responsible for a large proportion of cell loss during cardiac ischemia-reperfusion (7), it has been proven that apoptosis also occurs (8). Therefore, apoptosis may provide a new target for cardioprotection during an evolving acute myocardial infarction in humans.
Previous results indicate that the growth factor-associated kinase Akt is phosphorylated following ischemia-reperfusion in cardiomyocytes in a phosphoinositol-3-kinase (PI3-kinase)-dependent manner (9). The PI3-kinase pathway is one of several signal transduction pathways implicated in cell survival. Akt, in turn, phosphorylates a number of downstream targets leading to the inactivation of glycogen synthase kinase-3, the pro-apoptotic Bcl-2 family member BAD (10), caspase-9 and Forkhead transcription factor, and the activation of nuclear factor kappa B (11) and endothelial nitric oxide synthase (12).
In this study, we investigated the effects of a known PARP inhibitor (4-hydroxyquinazoline) and an experimental compound (HO-3089) on the cardiac pathophysiology under conditions of ischemia-reperfusion in an isolated heart perfusion system, including monitoring of the myocardial energy metabolism and cardiac contractile function, and measuring of the infarct size. Furthermore, we studied the effects of PARP inhibitors on the ischemia-reperfusion induced oxidative myocardial injury, ie, lipid peroxidation and protein oxidation. We found that ischemia-reperfusion activated Akt; therefore, we have assessed the ability of PARP inhibitors to influence the phosphorylation state of Akt.
MATERIALS AND METHODS
Chemicals:
4-Hydroxyquinazoline was purchased from Sigma-Aldrich Chemical Co (Hungary). HO-3089 was synthesized in the Institute of Organic and Medicinal Chemistry (University of Pécs, Hungary). All other reagents were of the highest purity commercially available.
Animals:
Male Wistar rats weighing 300 g to 350 g were used for this study. Rats were handled in accordance with the Guide for the Care and Use of Laboratory Animals (13). Rats were anesthetized with 200 mg/kg ketamine intraperitoneally and heparinized with sodium heparin (100 IU/rat intraperitoneally).
Heart pefusion:
Isolated rat hearts were perfused with a modified phosphate-free Krebs-Henseleit buffer according to the Langendorff method at a constant pressure of 70 mmHg at 37°C as previously described (14). The perfusion medium contained 118 mM NaCl, 5 mM KCl, 1.25 mM CaCl2, 1.2 mM MgSO4, 25 mM NaHCO3, 11 mM glucose and 0.6 mM octanoic acid. The perfusate was adjusted to pH 7.40 and bubbled with 95% O2/5% CO2 through a glass oxygenator. After a washout period (nonrecirculating period of 15 min), hearts were either perfused for 10 min (baseline) and freeze-clamped to obtain ‘nor-moxic’ hearts, or perfused for 10 min, freeze-clamped, and then subjected to 30 min global ischemia by closing the aortic influx and reperfused for either 45 min or 90 min. PARP inhibitors were administered (100 μM 4-hydroxyquinazoline or 25 μM HO-3089) into the medium at the beginning of baseline perfusion. During ischemia, hearts were submerged into perfusion buffer at 37°C. At the end of the perfusion, hearts were freeze-clamped.
Nuclear magnetic resonance (NMR) spectroscopy:
31P NMR spectra were recorded with a Varian UNITYINOVA 400 WB instrument (Varian Inc, USA). Detailed technical data were published previously (6,14).
Heart function:
A latex balloon was inserted into the left ventricle through the mitral valve and filled to achieve an end-diastolic pressure of 8 mmHg to 12 mmHg. All measurements were performed at the same balloon volume. Hearts were selected on the basis of the stability of high-energy phosphates (assessed by NMR) during a control period of 15 min before the experiment. The duration of normoxia, ischemia and reperfusion was 10 min, 30 min and 45 min, respectively. PARP inhibitors were added to the perfusion medium after the 15 min control period. Functional data of the hearts (left ventricular developed pressure [LVDP], rate-pressure product [RPP] and dP/dt) were monitored during the entire perfusion.
Infarct size:
Hearts was removed from the Langendorff apparatus. Both the auricles and the aortic root were excised, and the ventricles were kept overnight at −4°C. Frozen ventricles were sliced into uniform sections of about 2 mm to 3 mm thickness. The slices were incubated in 1% triphenyl tetrazolium chloride at 37°C in 0.2 M Tris buffer (pH=7.40) for 30 min (15). The normal myocardium was stained brick red, while the infarcted portion remained unstained. Infarct size was measured by the volume and weight method as previously described (16).
Lipid peroxidation:
Lipid peroxidation was estimated from the formation of thiobarbituric acid reactive substances (TBARS). Cardiac tissue was homogenized in 6.5% trichloroacetic acid, and a reagent containing 15% trichloroacetic acid, 0.375% thiobarbituric acid and 0.25% HCl was added, mixed thoroughly, heated for 15 min in a boiling water bath, cooled, centrifuged, and the absorbance of the supernatant was measured at 535 nm against a blank that contained all the reagents except the tissue homogenate. Using malondialdehyde standard, TBARS were calculated as nmol/g wet tissue.
Determination of protein carbonyl content:
Fifty milligrams of freeze-clamped perfused heart tissue were homogenized with 1 mL of 4% perchloric acid, and the protein content was collected by centrifugation. The protein carbonyl content was determined using the 2,4-dinitrophenylhydrazine method (17).
Western blot analysis:
The procedures for Western blots have been described in detail elsewhere (9). Samples were probed with antibodies recognizing both the nonphosphorylated and the phosphorylated Akt-1 (1:1000 dilution, Cell Signaling Technology, USA) and visualized by enhanced chemiluminescence.
Statistical analysis:
Statistical analysis was performed by analysis of variance and all data were expressed as mean ± SEM. Significant differences were evaluated using unpaired Student’s t tests; P values below 0.05 were considered to be significant.
RESULTS AND DISCUSSION
Effects of PARP inhibitors on the energy metabolism, contractile function and cell death of hearts during ischemia-reperfusion
The energy metabolism of Langendorff perfused hearts was monitored in the magnet of the NMR spectroscope, making it possible to detect changes in high-energy phosphate intermediates. Ischemia induced a rapid decrease in creatine phosphate and ATP levels and a fast evolution of inorganic phosphate. Under our experimental conditions, high-energy phosphate intermediates only partially recovered in untreated hearts during the 45 min reperfusion phase; on the other hand, HO-3089 and 4-hydroxyquinazoline facilitated the recovery of creatine phosphate (Figure 1) and ATP (data not shown). These data show that each PARP inhibitor could significantly improve the final recovery of high-energy phosphate intermediates (61±3% for 4-hydroxyquinazoline-treated hearts and 49±5% for HO-3089-treated hearts versus 23±5% creatine phosphate recovery for untreated hearts; percentages indicate percentage of normoxic level. PARP inhibitors also promoted the faster and more complete reuse of inorganic phosphate during reperfusion (28±3% for 4-hydroxyquinazoline-treated hearts and 35±3% for HO-3089-treated hearts versus 58±4% for untreated hearts, given as the percentage of end-ischemic value).
Figure 1).
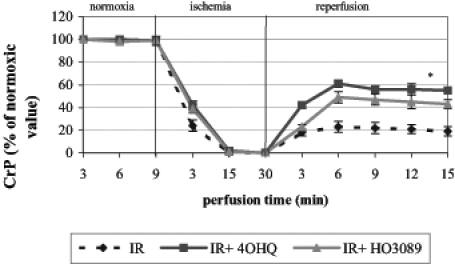
Effect of poly(ADP-ribose) polymerase inhibitors on the recovery of creatine phosphate (CrP) after ischemia-reperfusion (IR) in Langendorff perfused hearts. *P<0.05. IR+4OHQ IR in the presence of 100 μM 4-hydroxyquinazoline; IR+HO3089 IR in the presence of 25 μM HO-3089
To evaluate the effect of PARP inhibitors on the post-ischemic myocardial functional recovery, isolated hearts were perfused in the absence or presence of 100 μM 4-hydroxy-quinazoline or 25 μM HO-3089. At the end of the equilibration period, LVDP was 135±16 mmHg, RPP was 3.4±0.15×104 mmHg/min, dP/dt was 1310±196 mmHg/s and the average heart rate was 217±19 beats/min. Figure 2 shows the maximal percentage recovery of LVDP, RPP and dP/dt during reperfusion compared with the initial values. Both PARP inhibitors significantly improved the recovery of all parameters, indicating that the preservation of the integrity of energy metabolism was accompanied by better functional performance.
Figure 2).
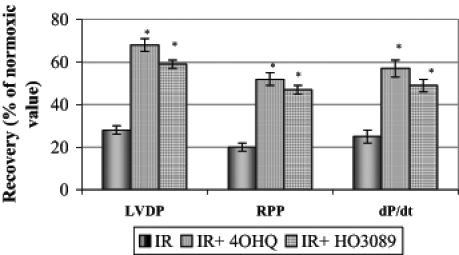
Effect of poly(ADP-ribose) polymerase inhibitors on the maximal percentage recovery of left ventricular developed pressure (LVDP), rate-pressure product (RPP) and dP/dt during the 45 min reperfusion period after ischemia. *P<0.05. IR Ischemia-reperfusion in untreated hearts; IR+4OHQ IR in hearts treated with 100 μM 4-hydroxyquinazo-line; IR+HO3089 IR in hearts treated with 25 μM HO-3089
Triphenyl tetrazolium chloride staining of the myocardium after 90 min of postischemic reperfusion revealed that PARP inhibitors were capable of significantly diminishing the infarct size compared with untreated hearts (Figure 3).
Figure 3).
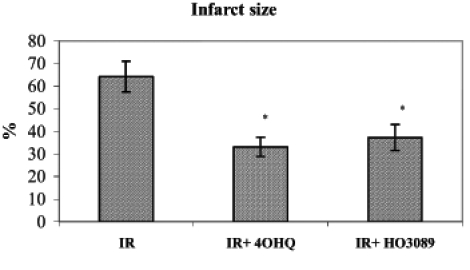
Effect of poly(ADP-ribose) polymerase inhibitors on infarct size after ischemia-reperfusion (IR) in Langendorff perfused hearts. *P<0.05. IR+4OHQ IR in the presence of 100 μM 4-hydroxyquina-zoline; IR+HO3089 IR in the presence of 25 μM HO-3089
Effects of PARP inhibitors on cardiac oxidative damage and ischemia-reperfusion-related Akt phosphorylation
Lipid peroxidation induced by ischemia-reperfusion in Langendorff perfused hearts was characterized by the formation of TBARS. Under our experimental conditions, ischemia-reperfusion increased the amount of TBARS compared with the normoxic conditions (Figure 4). In normoxic hearts, PARP inhibitors did not have significant effects on TBARS. When ischemia-reperfusion occurred in the presence of PARP inhibitors, the formation of TBARS was significantly lower than in the hearts subjected to ischemia-reperfusion alone, indicating that PARP inhibitors prevented the ischemia-reperfusion-induced lipid peroxidation.
Figure 4).
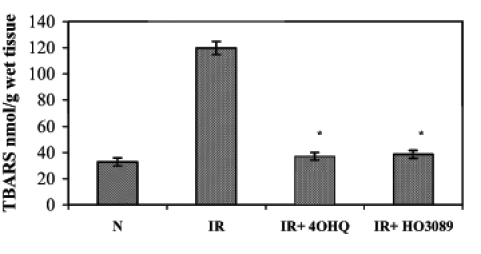
Effect of poly(ADP-ribose) polymerase inhibitors on ischemia-reperfusion (IR)-induced lipid peroxidation in Langendorff perfused hearts. The figure demonstrates the quantity of thiobarbituric acid reactive substances (TBARS) in various samples. *P<0.05. IR+4OHQ IR in the presence of 100 μM 4-hydroxyquinazoline; IR+HO3089 IR in the presence of 25 μM HO-3089; N Normoxia
ROS formation in the ischemia-reperfusion cycle can also trigger the oxidation of proteins, which can be characterized by the quantity of protein-bound aldehyde groups. Figure 5 shows that ischemia-reperfusion significantly elevated the level of protein oxidation; nevertheless, the administration of PARP inhibitors during the ischemia-reperfusion cycle prevented the increase in protein-bound aldehyde groups.
Figure 5).
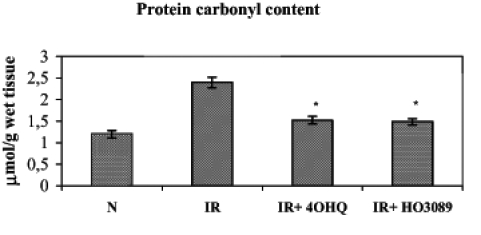
Effect of poly(ADP-ribose) polymerase inhibitors on ischemia-reperfusion (IR)-induced protein oxidation in Langendorff perfused hearts. The figure demonstrates the protein carbonyl content. *P<0.05. IR+4OHQ IR in the presence of 100 μM 4-hydroxyquina-zoline; IR+HO3089 IR in the presence of 25 μM HO-3089; N Normoxia
In addition, moderate Akt kinase phosphorylation occurred in our experimental setting as a result of ischemia-reperfusion. On the other hand, 4-hydroxyquinazoline and HO-3089 treatment further enhanced Akt activation (Figure 6).
Figure 6).
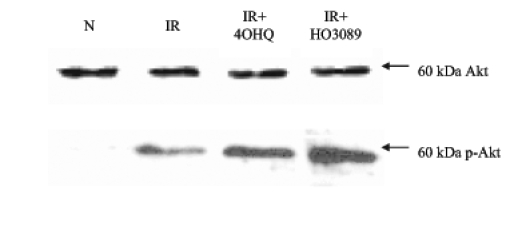
Effect of poly(ADP-ribose) polymerase (PARP) inhibitors on Akt phosphorylation. Ischemia-reperfusion (IR) without any treatment triggered moderate phosphorylation of Akt (p-Akt). Both PARP inhibitors could further enhance the phosphorylation. *P<0.05. IR+4OHQ IR in the presence of 100 μM 4-hydroxyquinazoline; IR+HO3089 IR in the presence of 25 μM HO-3089; N Normoxia
CONCLUSIONS
Taken together, our data show that the investigated PARP inhibitors preserved the myocardial energy metabolism and attenuated the overall oxidative injury of postischemic rat hearts. Stress-induced activation of Akt has been reported to occur in a number of cell types as a result of a variety of cellular stresses (18,19). In the present study, we have demonstrated that ischemia-reperfusion led to the phosphorylation of Akt (activated), while the administration of PARP inhibitors further enhanced the activation of this pathway in some way. Although little is known about the precise triggers of ischemia-reperfusion-related signalling pathways, it has been proposed that oxidative stress, mediated by ROS, may play an important role in this process (20,21). Our findings suggest that Akt activation occurs as a response to PARP inhibitor treatment and might play an important role in promoting cell survival. However, further studies are required to delineate the role of Akt activation and the detailed signalling mechanisms under conditions of various treatment agendas.
Footnotes
Paper was presented at the Fourth International Symposium on Myocardial Cytoprotection, Pécs, Hungary, September 25 to 27, 2003
REFERENCES
- 1.Cuzzocrea S, Riley DP, Caputi AP, Salvemini D. Antioxidant therapy: A new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev. 2001;53:135–59. [PubMed] [Google Scholar]
- 2.Gilad E, Zingarelli B, Salzman AL, Szabo C. Protection by inhibition of poly (ADP-ribose) synthetase against oxidant injury in cardiac myoblasts in vitro. J Mol Cell Cardiol. 1997;29:2585–97. doi: 10.1006/jmcc.1997.0496. [DOI] [PubMed] [Google Scholar]
- 3.Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA. 1999;96:13978–82. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pieper AA, Walles T, Wei G, et al. Myocardial postischemic injury is reduced by polyADPripose polymerase-1 gene disruption. Mol Med. 2000;6:271–82. [PMC free article] [PubMed] [Google Scholar]
- 5.Thiemermann C, Bowes J, Myint FP, Vane JR. Inhibition of the activity of poly(ADP-ribose) synthetase reduces ischemia-reperfusion injury in the heart and skeletal muscle. Proc Natl Acad Sci USA. 1997;94:679–83. doi: 10.1073/pnas.94.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halmosi R, Berente Z, Osz E, Toth K, Literati-Nagy P, Sumegi B. Effect of poly(ADP-ribose) polymerase inhibitors on the ischemia-reperfusion-induced oxidative cell damage and mitochondrial metabolism in Langendorff heart perfusion system. Mol Pharmacol. 2001;59:1497–505. doi: 10.1124/mol.59.6.1497. [DOI] [PubMed] [Google Scholar]
- 7.Buja LM, Eigenbrodt ML, Eigenbrodt EH. Apoptosis and necrosis. Basic types and mechanisms of cell death. Arch Pathol Lab Med. 1993;117:1208–14. [PubMed] [Google Scholar]
- 8.Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM. Apoptosis in human acute myocardial infarction. Circulation. 1997;95:320–3. doi: 10.1161/01.cir.95.2.320. [DOI] [PubMed] [Google Scholar]
- 9.Mockridge JW, Marber MS, Heads RJ. Activation of Akt during simulated ischemia/reperfusion in cardiac myocytes. Biochem Biophys Res Commun. 2000;270:947–52. doi: 10.1006/bbrc.2000.2522. [DOI] [PubMed] [Google Scholar]
- 10.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–9. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 11.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 12.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–5. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 13.National Institutes of Health . NIH Publication No 85-23. Bethesda: National Institutes of Health; 1985. Guide for the Care and Use of Laboratory Animals. (revised 1996). [Google Scholar]
- 14.Szabados E, Literati-Nagy P, Farkas B, Sumegi B. BGP-15, a nicotinic amidoxime derivate protecting heart from ischemia reperfusion injury through modulation of poly(ADP-ribose) polymerase. Biochem Pharmacol. 2000;59:937–45. doi: 10.1016/s0006-2952(99)00418-9. [DOI] [PubMed] [Google Scholar]
- 15.Fishbein MC, Meerbaum S, Rit J, et al. Early phase acute myocardial infarct size quantification: Validation of the triphenyl tetrazolium chloride tissue enzyme staining technique. Am Heart J. 1981;101:593–600. doi: 10.1016/0002-8703(81)90226-x. [DOI] [PubMed] [Google Scholar]
- 16.Chopra K, Singh M, Kaul N, Andrabi KI, Ganguly NK. Decrease of myocardial infarct size with desferrioxamine: Possible role of oxygen free radicals in its ameliorative effect. Mol Cell Biochem. 1992;113:71–6. doi: 10.1007/BF00230887. [DOI] [PubMed] [Google Scholar]
- 17.Oliver CN, Ahn BW, Moerman EJ, Goldstein S, Stadtman ER. Age-related changes in oxidized proteins. J Biol Chem. 1987;262:5488–91. [PubMed] [Google Scholar]
- 18.Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278:29655–60. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- 19.Takano T, Sada K, Yamamura H. Role of protein-tyrosine kinase syk in oxidative stress signaling in B cells. Antioxid Redox Signal. 2002;4:533–41. doi: 10.1089/15230860260196335. [DOI] [PubMed] [Google Scholar]
- 20.Takano H, Zou Y, Hasegawa H, Akazawa H, Nagai T, Komuro I. Oxidative stress-induced signal transduction pathways in cardiac myocytes: Involvement of ROS in heart diseases. Antioxid Redox Signal. 2003;5:789–94. doi: 10.1089/152308603770380098. [DOI] [PubMed] [Google Scholar]
- 21.Steenbergen C. The role of p38 mitogen-activated protein kinase in myocardial ischemia/reperfusion injury; relationship to ischemic preconditioning. Basic Res Cardiol. 2002;97:276–85. doi: 10.1007/s00395-002-0364-9. [DOI] [PubMed] [Google Scholar]