

Abstract
Experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS), is mediated by autoantigen-specific T-helper1 (Th1) cells. IL-12, an inducer of Th1 cell development, exerts immunomodulatory effects in EAE. Programmed death-1 (PD-1) and PD-1 ligand (PD-L), new members of the B7 superfamily of costimulatory molecules, play a critical role in regulating EAE. Whether interaction of IL-12 and the PD-1/PD-L pathway regulates EAE is unclear. We have previously shown IL-12 suppresses EAE induced by MOG35-55 in C57BL/6 mice, but not in IFN-γ-deficient mice, suggesting that IFN- γ is required for inhibitory effects of IL-12 on EAE. In the current study, PD-L1 expression is up-regulated following IL-12 treatment in wild-type mice, but not in IFN-γ-deficient EAE mice. Similarly, IL-12 induces IFN-γ and PD-L1 expression in cultured MOG-specific T cells from wild-type mice but not from IFN-γ-deficient mice. Furthermore, PD-L1 expression increased specifically in CD11b+ antigen presenting cells (APCs) after IL-12 administration. These data suggest that one mechanism of IL-12 suppression of EAE is mediated by PD-1/PD-L signaling downstream of IFN-γ induction in CD11b+ APCs. Regulation of PD-1/PD-L1 may have potential therapeutic effects for EAE and MS.
Keywords: EAE, Autoimmunity, PD-1/PD-L, Cytokines, Antigen presenting cells
1. Introduction
Experimental autoimmune encephalomyelitis (EAE) is a T cell dependent autoimmune disease in the central nervous system that is used as a model for the human inflammatory demyelination mediated neurodegenerative disease, multiple sclerosis (MS) (Wekerle et al., 1994). EAE is mediated by CD4+ T-helper1 (Th1) cells following immunization with myelin protein antigens. An important aspect of the pathogenesis of EAE, with potential for therapeutic intervention, is the role of cytokines in the inflammatory process (Begolka et al., 1998; Steinman, 1999). Interleukin-12 (IL-12), a heterodimeric cytokine produced by activated antigen presenting cells (APCs), is a secreted protein with diverse roles in cellular differentiation and is especially important in regulating immune responses (Trinchieri et al., 2003). IL-12 is composed of a 40 KDa heavy chain (p40) and a 35 KDa light chain (p35) (Kobayashi et al., 1989). The major stimulus for its production is the interaction of CD40 ligand on T cells with CD40 on APCs (Gately et al., 1991). IL-12 regulates the development of naïve CD4+ T cells to either Th1 or Th2 cells, which is a crucial event for effective acquired immunity. IL-12 promotes the growth and cytotoxicity of natural killer (NK) cells and the activation of macrophages by stimulating IFN-γ secretion (Trinchieri and Scott, 1995). Th1 cells produce IFN-γ and promote cell-mediated immunity essential for the response against intracellular pathogens, viruses and bacteria, and IL-12 is the main cytokine that regulates Th1 differentiation (Mullen et al., 2001).
IL-12 has been considered an important factor in the pathogenesis of MS (Gran et al., 2004). However, IL-12 is not strictly required for the induction of EAE and the role of IL-12 and its receptors appears complex (Cua et al., 2003). Our previous work indicated that IL-12p40-deficient mice were resistant to EAE induction while IL-12p35-deficient mice are susceptible to EAE (Gran et al., 2002). We also found that IL-12 receptor β1-deficient mice are completely resistant to induced EAE while IL-12 receptor β2-deficient mice develop very severe EAE (Zhang et al., 2003; Zhang et al., 2003). IL-12 has diverse effects on cellular differentiation and is especially important in regulating immune responses. The immunomodulatory role of IL-12 appears very complex in the pathogenesis of MS and the induction of EAE (Gran et al., 2004). We have previously shown that IL-12 suppresses EAE through induction of IFN-γ (Gran et al., 2004). The precise mechanisms involved in the IFN-γ mediated immunomodulatory role of IL-12 in EAE are still unclear.
Effector T cell function is regulated by a number of nonmutually exclusive mechanisms. Negative signals delivered to activated T cells by the interaction of the classical costimulatory molecules CD28/CTLA-4 and their ligands B7-1/B7-2 are essential contributors to these mechanisms (Greenwald et al., 2001; Sharpe and Freeman, 2002). Recently, programmed death-1 (PD-1), a novel negative regulatory molecule and a new member in the B7-CD28 superfamily, has been implicated in the regulation of effector T cell function (Agata et al., 1996). PD-1 is a 50-55 kDa type I transmembrane receptor protein originally identified in a T cell line undergoing activation-induced cell death, but further studies have shown that its expression is associated with lymphocyte activation rather than cell death, and it has recently been recognized as a costimulatory molecule thought to produce an inhibitory signal (Okazaki et al., 2002). Two ligands, PD-L1 and PD-L2, which belong to the B7 superfamily, have been identified for binding PD-1. The engagement of PD-1 with PD-L can negatively regulate autoreactive T and B cells and have critical and nonredundant functions in the maintenance of tolerance, and is involved in the onset of a variety of autoimmune diseases (Latchman et al., 2001). Recent studies indicated that blockade of PD-1/PD-L significantly augments EAE. Furthermore, PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. PD-L1 expression is suggested to be an important marker of “primed” macrophages and is induced by low doses of IFN-γ in monocytes/macrophages (Liang et al., 2003; Salama et al., 2003; Loke and Allison, 2003; Brown et al., 2003). PD-L1 therefore plays a general role in down-regulating activated T cells. PD-1 pathway mediated suppression of EAE occurs early, with loss of inhibitory effects in later stages of disease (Salama et al., 2003; Zhu et al., 2006).
In this study, we demonstrated that IL-12 treatment induced up-regulation of the PD-1/PD-L pathway mediated by IFN-γ signaling during the induction phase of EAE resulted in suppression of clinical disease severity of EAE in wild type mice. Lack of disease suppression in IFN-γ-deficient mice indicated a critical role for IFN-γ signaling in up-regulation of the PD-1/PD-L pathway and the subsequent immunosuppression induced by IL-12. We further found that IL-12 treatment increased the number of APCs expressing PD-L1, and that PD-L1 expression was upregulated specifically in CD11b+ APCs but not in CD11b- APCs. These data may aid in designing novel therapeutic strategies for the treatment of multiple sclerosis.
2. Materials and Methods
2.1. Mice and EAE induction
Eight- to ten-week-old homozygous IFN-γ-deficient C57BL/6 mice and their wild-type controls were purchased from The Jackson Laboratory (Bar Harbor, ME). To induce EAE, mice were each injected s.c. with 300 μg of myelin oligodendrocyte glycoprotein (MOG) peptide 35-55 (MEVGWYRSPFSRVVHLYRNGK) (Protein Chemistry Laboratory, University of Pennsylvania) in CFA containing 4mg/ml Mycobacterium tuberculosis H37Ra (Difco, Detroit, MI) at two sites in the back. Pertussis toxin (200 ng) (List Biological Laboratory Inc., Denver, CO) was given i.p. on days 0 and 2 post immunization (p.i.). EAE was scored according to a 0-5 scale (Benson et al., 2000) as follows: limp tail or waddling gait with tail tonicity, 1; waddling gait with limp tail (ataxia), 2; ataxia with partial limb paralysis, 2.5; full paralysis of one limb, 3; full paralysis of one limb with partial paralysis of second limb, 3.5; full paralysis of two limbs, 4; moribund, 4.5; and death, 5. The animal protocol was approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University, Philadelphia, PA.
2.2. MOG-specific T cell culture in vitro
Single cell suspensions of mononuclear cells (MNCs) from inguinal and popliteal lymph nodes were prepared on day 10 post-immunization (p.i.) as previously described (Gran et al., 2002). Cells were cultured at a cell density of 5×106 cells/well in completed culture medium DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% FCS (Invitrogen), 2mM L-glutamine (Invitrogen), 100 μg/ml penicillin/streptomycin (Invitrogen), and 1 mM 2-ME (Sigma, St. Louis, MO) in 5 cm2 tissue culture dishes containing MOG35-55 at a final concentration of 25 μg/ml, with or without 20 ng/ml recombinant murine IL-12 (rmIL-12) (R&D Systems Inc.). Supernatants were collected after 48 h for IFN-γ, TNF-α, IL-2, IL-4, and IL-5 production assays.
2.3. T cell proliferation assay
Single cell suspensions of MNCs from the spleen were prepared on day 7 post-immunization (p.i.) as previously described (Gran et al., 2002). Cells were cultured at a cell density of 1×105 cells/well in completed culture medium DMEM in triplicate wells of microtiter 96-well plates. The cells were stimulated by adding 25 μg/ml MOG35–55 in the culture medium, or were cultured in the absence of MOG35-55 as control. After 60 hrs of incubation, the cells were pulsed for 12 hrs with 1 μCi of [3H]methylthymidine. Cells were harvested and counts read with a beta counter (Microbeta; Applied Biosystems, Foster City, CA). The results are expressed as counts per minute.
2.4. Measurement of cytokine production
Single cell suspensions of MNCs from the spleen were prepared on day 7 post-immunization (p.i.) as previously described [11]. Cells were cultured at a cell density of 2×106 cells/ml in completed culture medium in triplicate wells of microtiter 24-well plates containing MOG35–55 at a final concentration of 25μg/ml with/without 20 ng/ml rmIL-12. Supernatants were collected after 48 h. Quantitative ELISA for IFN-γ, TNF-α, IL-2, IL-4, and IL-5 was performed using paired mAbs according to the manufacturer's recommendation (BD PharMingen, San Jose, CA). Where indicated, brefeldin A (Invitrogen) was added to cells cultured with or without MOG35–55 for 48 h during the last 4 hrs of culture. Production of cytokines was determined by Flow Cytometry using the Mouse Th1/Th2 Cytokine Cytometric Bead Array Kit (BD PharMingen) according to the instruction manual.
2.5. Quantification of PD-1, PD-L1, PD-L2 and IFN-γ mRNA expression by real-time RT-PCR
Quantification was performed as described (Livak and Schmittgen, 2001) with modifications. Total RNA was extracted from spleen tissue and cultured splenocytes using TRIzol Reagent (Invitrogen) according to the manufacturer's instructions. From sorted cell populations, total cellular RNA was extracted using the Absolutely RNA Nanoprep Kit (Stratagene, La Jolla, CA) according to the manufacturer's instruction manual. The purity and integrity of RNA were determined by absorbance at A260/280 and gel electrophoresis. Contamination by genomic DNA was excluded by real-time PCR amplification of RNA by β-actin primers. RNA was dissolved in 20 μl of RNase-free water. RNA (5 μg) was used to generate cDNA using Superscript II RNase H- Reverse Transcriptase Kit (Invitrogen). Random hexamers were used to prime RNA samples for reverse transcription. Quantitative real-time PCR (RT-PCR) was performed in an ABI 7000 sequence detection system using gene-on-demand assay products (Applied Biosystems). β-actin housekeeping gene was amplified in each sample as endogenous control. Reaction mixtures for RT-PCR had a final volume of 50 μl consisting of 0.4 μl of cDNA, 2.5 μl primer pair mix (5 pmol/μl each primer), 25 μl of the master mix and 22.1μl DEPC H2O. Amplification conditions were: 1 cycle of 2 min at 50 °C, 10 min at 95 °C, 40 cycles of 15 sec at 95 °C, 30 sec at 60 °C and 30 sec at 70 °C, and 1 cycle of 10 min at 70 °C. The endpoint used in RT-PCR quantification, CT, is defined as the PCR cycle number that crossed the signal threshold. CT values range from 0 to 40. Quantification of gene expression was performed using the comparative CT methods (Sequence Detector User Bulletin 2: Applied Biosystems) and reported as the fold difference relative to the housekeeping gene. To calculate the fold change (increase or decrease), the CT of the housekeeping gene (β-actin) was subtracted from the CT of the target gene to yield the ΔCT. Change in expression of the normalized target gene as a result of an experimental manipulation was expressed as 2-ΔΔCT where ΔΔCT=ΔCT samples – ΔCT controls. Data were analyzed using GeneScan software (Applied Biosystems) (Song et al., 2002; Ingham et al., 2001).
2.6. Flow Cytometry assay
Single cell suspensions of mononuclear cells (MNCs) from the spleen of MOG-immunized mice were washed twice in FACS buffer (PBS containing 1% FCS and 0.1% sodium azide). After washing and blocking with CD16/CD32 antibodies, cells were incubated with antibodies to murine CD4, CD8, CD11b, PD-1, PD-L1 (all from BD Pharmingen), or PD-L2 (eBioscience, San Diego, CA). Data were acquired on a FACSCalibur (Becton-Dickinson, Mountain View, CA) and analyzed using CellQuest software (Becton-Dickinson) and FlowJo software (FlowJo, Ashland, OR).
2.7. Sorting of CD11b+ and CD 11b- APC populations by Flow Cytometry
Single cell suspensions of mononuclear cells (MNCs) from the spleen of MOG-immunized mice were washed twice in FACS buffer (PBS containing 1% FCS and 0.1% sodium azide). After blocking with CD16/CD32 antibodies, cells were incubated with antibodies to murine CD11b and PD-L1 (BD Pharmingen). CD11b+ and CD11b- cells were sorted by flow cytometry to >95% purity using FACSAria (BD Biosciences). A total of 10,000 CD11b+ and CD11b- cells were sorted into an Eppendorf tube containing Solution L, prepared by mixing 0.7 μl β-mercaptoethanol (14.2 M) with 100 μl Lysis Buffer from Absolutely RNA Nanoprep Kit (Stratagene) (Storek et al., 2004). RNA was extracted using the nanoprep RNA purification method according to the instruction manual in the Absolutely RNA Nanoprep Kit, and cDNA was synthesized using Superscript II RNase H- Reverse Transcriptase Kit (Invitrogen) and random hexamers were used to prime RNA samples for reverse transcription. Quantitative RT-PCR was performed as described above.
2.8. Statistical analysis
Statistical analysis was performed using unpaired two sided Student's t-test or Mann-Whitney rank sum test for comparisons between two groups. The data are presented as mean ± S.D. We have calculated significance values from at least three separate experiments. A value of p<0.05 was considered significant.
3. Results
3.1. IL-12 suppresses EAE and decreases the proliferative response of MOG35-55-specific T cells in C57BL/6 wild type mice, but not in IFN-γ-deficient mice
To investigate the regulatory effect of IL-12 on severity of active EAE, female C57BL/6 wild type mice were immunized with MOG35-55 peptide in CFA, and were treated with rmIL-12 injected intraperitoneally at 100 ng/200 μl/day from day 0 to day 10 post-immunization (p.i.), or with PBS as control. Clinical disease severity of EAE was scored daily by double blinded method according to a 0-5 severity scale (Benson et al., 2000). Compared with the PBS control group, the severity of EAE in the IL-12 treated group was significantly reduced (Fig. 1 A), suggesting a suppressive effect of IL-12 on EAE in C57BL/6 wild type mice.
Figure 1.
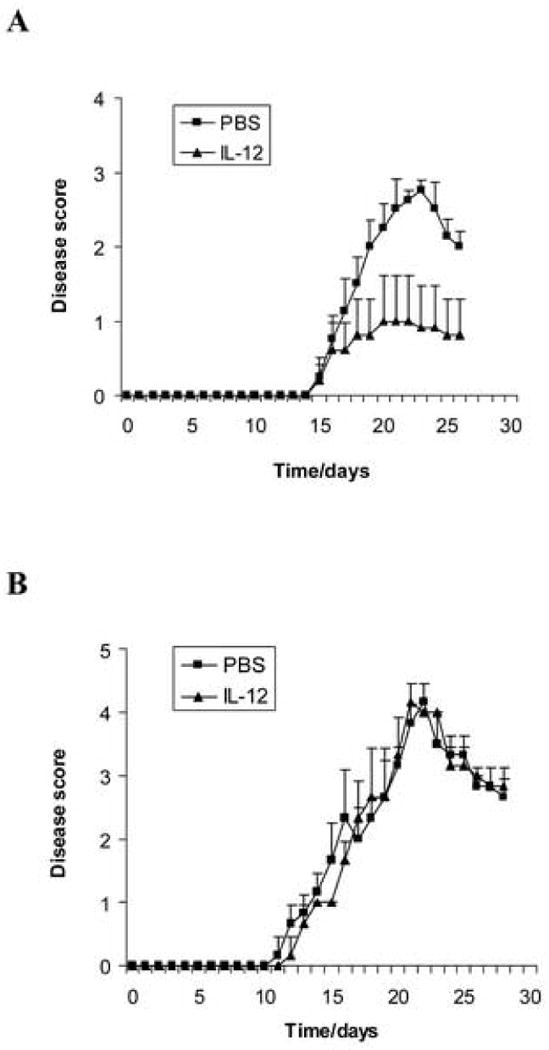
IL-12 suppresses EAE in wild type mice but not in IFN-γ-deficient mice. Female C57BL/6 wild type mice (A) and IFN-γ-deficient mice (B) were immunized with 300 μg MOG35-55 peptide in CFA. The number of mice in each group was 5. rmIL-12 (100 ng/mouse, dissolved in 200 μl PBS) was injected intraperitoneally (i.p.) every day from day 0 to day 10 post-immunization. PBS (200 μl/mouse) only was injected i.p. as control. Clinical disease severity of EAE was scored daily by double blinded method according to a 0-5 severity scale. Data represent the mean clinical scores ± SEM. There is a significant difference in clinical disease score of EAE between the rmIL-12-treated group and the PBS control group in C57BL/6 wild type mice (p<0.01) but there is no significant difference between the two groups in IFN-γ-deficient mice (p>0.05). Experiments were repeated three times and similar results were obtained. One representative experiment is shown.
To determine whether the suppression of EAE induced by IL-12 is IFN-γ-dependent, EAE was induced in IFN-γ-deficient mice (GKO C57BL/6 mice) with MOG35-55 immunization, and treated with IL-12 i.p. and with PBS as control, as described above. The results showed that IFN-γ-deficient mice developed severe EAE after immunization with MOG35-55. There is no significant difference of the clinical disease scores between the IL-12 treated group and the PBS controls (Fig. 1 B), suggesting that IL-12 treatment did not suppress severity of EAE in IFN-γ-deficient mice. IFN-γ is therefore required for the suppression of EAE induced by IL-12.
We also observed the T cell response to antigen MOG35-55 in wild type EAE mice treated with IL-12 or with PBS as control. MOG-specific T cells were isolated from spleen in the immunized mice on day 7 p.i., and the proliferative response to antigen MOG35-55 was measured. Compared with the control normal mice without immunization, T cells from the immunized mice both treated with IL-12 or PBS have a significant proliferative response to antigen MOG35-55, suggesting that these T cells were antigen MOG35-55 specific. Compared with the PBS group, the proliferation of MOG-specific T cells to antigen MOG35-55 was significantly decreased in the IL-12 treated group (** p<0.01) (Fig. 2 A), indicating IL-12 suppresses the T cell response to antigen MOG35-55 in wild type EAE mice. We also isolated MOG-specific T cells from immunized IFN-γ-deficient mice to compare T cell responses to antigen MOG35-55 in mice treated with IL-12 or with PBS as control. The proliferation of MOG-specific T cells to antigen MOG35-55 was not significantly changed in the IL-12 treated group compared with the PBS group, indicating that IL-12 treatment did not suppress the T cell response to antigen MOG35-55 in the IFN-γ-deficient EAE mice (Fig. 2 B).
Figure 2.
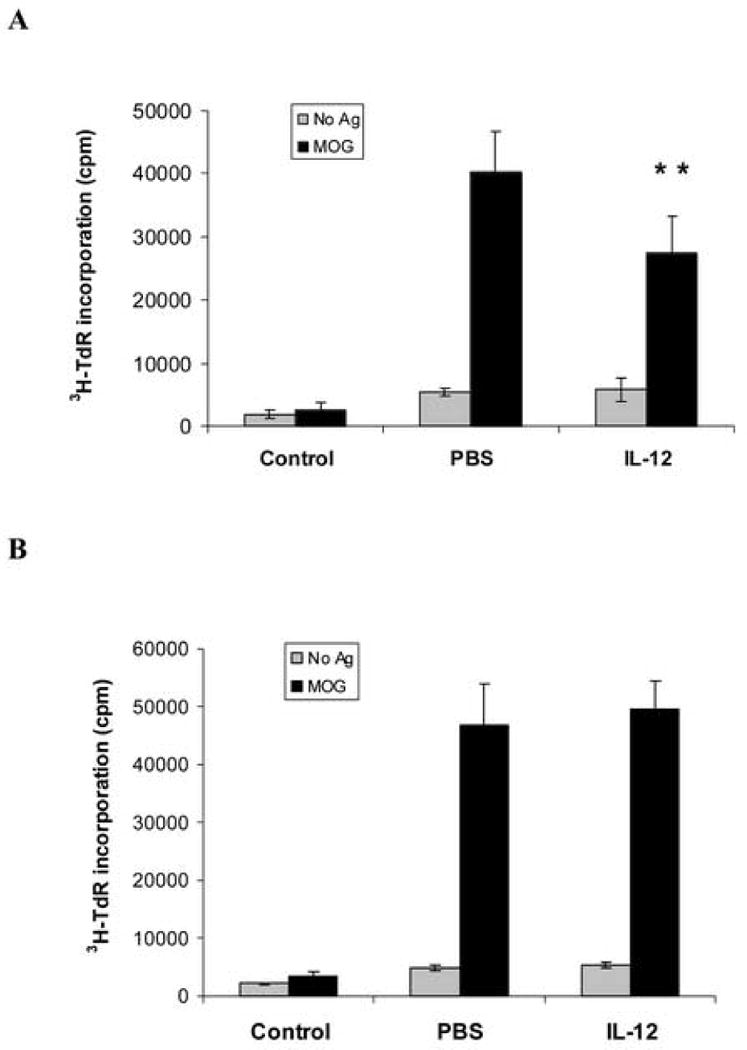
IL-12 treatment decreases the proliferative response of MOG35-55-specific T cells from wild-type EAE mice, but not from IFN-γ-deficient EAE mice. C57BL/6 female mice (A), or IFN-γ-deficient EAE mice (B), were immunized with 300 μg MOG35-55 peptide in CFA, and were sacrificed on day 7 p.i. Spleen cells (4 ×105/ well) were isolated from immunized mice treated with rmIL-12 or PBS, and also from mice without MOG immunization as control, and were cultured with antigen MOG35-55 at 25 μg/ml or without antigen for 72 hours. Results of proliferative responses were expressed by [3H] methylthymidine incorporation as counts per minute (cpm). Data represent mean and SD of triplicate values. There are significant proliferative responses of T cells to antigen MOG both from PBS and rmIL-12 treated groups. (A) Compared with the PBS group, proliferation of T cells to MOG from the rmIL-12 treated group is significantly decreased (** p<0.01). (B) There is no significant difference between the PBS and rmIL-12 treated groups from the IFN-γ-deficient EAE mice (p>0.05). One representative experiment of three is shown.
3.2. IL-12 treatment increases IFN-γ and PD-L1 gene expression in spleen of EAE mice in vivo
IL-12 signaling is important in Th1 cell differentiation and production of IFN-γ from T cells and NK cells activated by antigens or mitogens. To investigate the effect of IL-12 on IFN-γ gene expression in the spleen, we isolated total RNA from spleen in EAE mice treated with IL-12 and with PBS control on day 7 p.i., synthesized cDNA and performed quantitative RT-PCR to measure the level of IFN-γ gene expression. Compared with the PBS control group, IFN-γ gene expression was significantly increased in the EAE mice treated with IL-12 (** p<0.01) (Fig. 3 A, B), showing that IL-12 treatment induced IFN-γ gene expression in spleen of EAE mice at the early phase after immunization.
Figure 3.
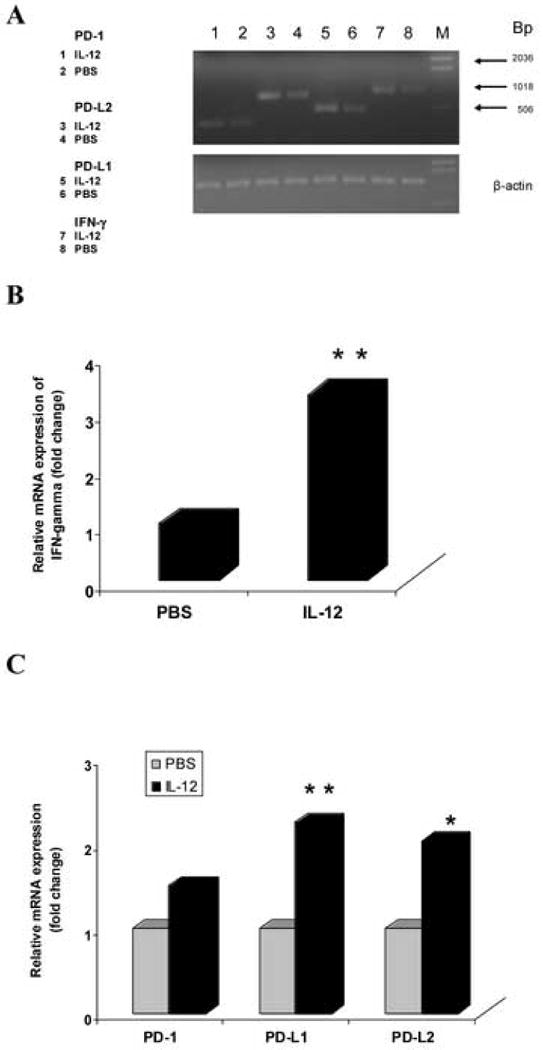
IL-12 treatment increases IFN-γ, PD-L1, and PD-L2 gene expression in spleen of EAE mice. Total RNA was extracted from spleen in C57BL/6 EAE mice treated with rmIL-12 or PBS (n = 5 mice/group) on day 7 p.i., and 5 μg RNA was used to generated cDNA. Quantitative RT-PCR was performed. β-actin was amplified in each sample as endogenous control. Each sample was run in triplicate. Relative quantization was used to calculate the results of the quantization assays with the comparative CT methods. Quantization of gene expression was reported as the fold difference relative to the housekeeping gene. Data were analyzed using GeneScan software. Electrophoresis of final products of PCR of IFN-γ, PD-1, PD-L1, PD-L2 gene and β-actin gene as endogenous control in 2.5% agarose-TBE gel is shown (A). There is a significant difference of IFN-γ gene expression between the PBS and rmIL-12-treated groups (** p<0.01) (B). Compared with the PBS group, PD-L1 and PD-L2 gene expression were increased significantly in the rmIL-12 treated group (** p<0.01, * p<0.05), and there is an increasing tendency of PD-1 gene expression but no significant difference (p>0.05) (C). One representative experiment of three is shown.
The engagement of the PD-1 receptor with its two ligands PD-L1 and PD-L2, which delivers an inhibitory signal, negatively regulates autoreactive T cell activation and response. To examine whether the PD-1/PD-L pathway is involved in the suppression of EAE induced by IL-12, we observed the effect of IL-12 on the gene expression of PD-1, PD-L1, and PD-L2 in the spleen of EAE mice. We extracted total RNA from spleen in EAE mice treated with IL-12 or with PBS as controls on day 7 p.i., synthesized cDNA and performed quantitative RT-PCR to measure the level of PD-1, PD-L1, and PD-L2 gene expression. Compared with the PBS control group, PD-L1 gene expression was significantly increased in EAE mice treated with rmIL-12 (** p<0.01), and the level of PD-L2 gene expression in the rmIL-12 treated group is also higher than controls (* p<0.05). However, the level of PD-1 gene expression has only an increased tendency in the rmIL-12 treated group, with no significant difference between the rmIL-12 treated and PBS control groups (p>0.05) (Fig. 3 A, C). This suggests that IL-12 treatment promoted PD-L1 and PD-L2 gene expression, and the PD-1/PD-L pathway is activated and up-regulated by IL-12 in EAE mice in vivo.
3.3. IL-12 treatment induces IFN-γ and PD-L1 gene expression in MOG-specific T cells in vitro
We found that IL-12 treatment increased the expression of IFN-γ and the PD-1/PD-L pathway in EAE mice in vivo. We further examined how IL-12 exerts function in autoreactive T cells in vitro. To answer this question, we investigated the effects of IL-12 treatment on the expression of IFN-γ and the PD-1/PD-L pathway in MOG-specific T cells in vitro. MOG-specific T cells isolated from inguinal and popliteal lymph nodes were prepared on day 10 p.i. from C57BL/6 mice immunized with MOG35-55, and cultured with MOG35-55 stimulation at a final concentration of 25 μg/ml, and co-cultured with or without 20 ng/ml rmIL-12. Supernatant was collected for IFN-γ, TNF-α, IL-2, IL-4, and IL-5 production assays. Total RNA was extracted from the cultured MOG-specific T cells for quantitative RT-PCR to measure the levels of IFN-γ and PD-1/PD-L gene expression. The results showed that MOG-specific T cells activated by antigen MOG35-55 stimulation produced a significantly higher level of cytokines IL-2, TNF-α, and IFN-γ than resting T cells without antigen MOG35-55 stimulation (## p<0.01, # p<0.05), but the level of IL-4 and IL-5 production did not change significantly (p>0.05) (Fig. 4). Compared with the stimulated MOG-specific T cells without IL-12 co-culture, the MOG-specific T cells co-cultured with IL-12 produced a higher level of IFN-γ (** p<0.01). Production of other cytokines, IL-2, IL-4, IL-5 and TNF-α, did not change significantly (p>0.05) (Fig. 4). Quantitative RT-PCR showed that the levels of IFN-γ and PD-L1 gene expression were increased in MOG-specific T cells cultured with IL-12, compared with T cells cultured without IL-12 (** p<0.01, * p<0.05) (Fig. 5 A, B). PD-1 and PD-L2 gene expression did not change significantly in the two groups (data not shown). This suggested that IL-12 treatment could induce expression of IFN-γ and PD-L1 genes in MOG-specific T cells in vitro, consistent with results from EAE mice in vivo.
Figure 4.
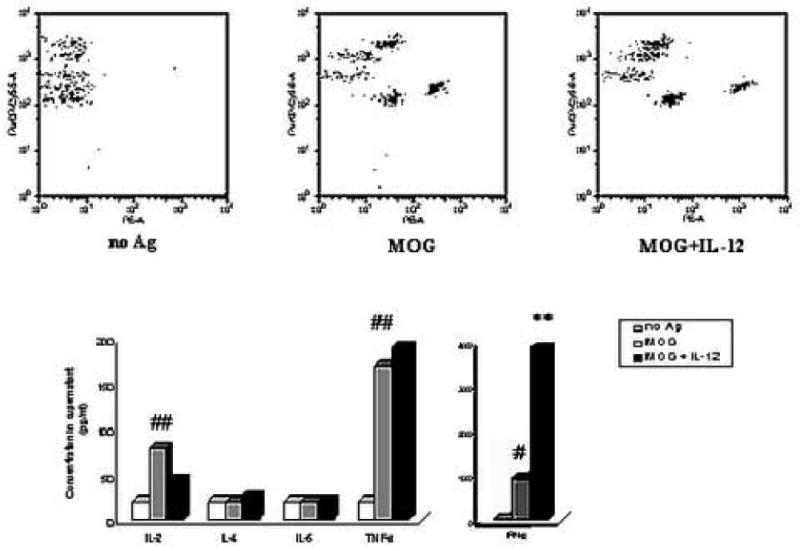
IL-12 treatment induces MOG-specific T cells to produce IFN-γ in vitro. MOG-specific T cells isolated from inguinal and popliteal lymph nodes were prepared on day 10 p.i. from C57BL/6 mice immunized with 300 μg MOG35-55 peptide in CFA. MOG-specific T cells were cultured without any antigen as control (no Ag), or with MOG35-55 stimulation alone at a final concentration of 25 μg/ml (MOG), or with MOG stimulation and co-cultured with 20 ng/ml rmIL-12 (MOG + IL-12). Supernatant was harvested after 48 hours for determining the production of cytokines by Flow Cytometry using the Mouse Th1/Th2 Cytokine Cytometric Bead Array Kit. Results of cytokine production were expressed as concentration in supernatant. Data represent mean of triplicate values. Compared with the “no Ag” group, production of IL-2, TNF-α and IFN-γ in the “MOG” group were significantly increased (## p<0.01, # p<0.05), but production of IL-4 and IL-5 did not change (p>0.05). Compared with the “MOG” group, production of IFN-γ in the “MOG + IL-12” group was significantly increased (** p<0.01), but production of IL-2, IL-4, IL-5 and TNF-α did not change (p>0.05). The figure is a representative of three experiments.
Figure 5.
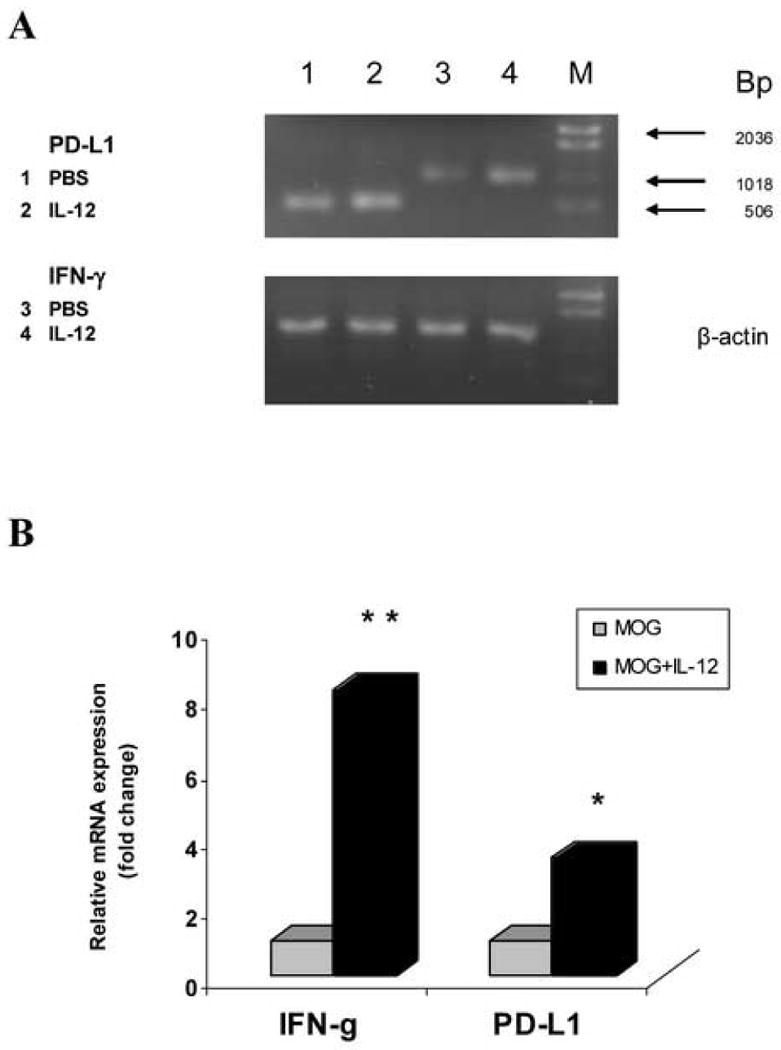
IL-12 treatment induces gene expression of IFN-γ and PD-L1 in MOG-specific T cells in vitro. MOG-specific T cells isolated from inguinal and popliteal lymph nodes were prepared on day 10 p.i. from C57BL/6 mice immunized with 300 μg MOG35-55 peptide. Total RNA was extracted from MOG-specific T cells cultured with MOG35-55 stimulation alone at a final concentration of 25 μg/ml (MOG), or cultured with MOG stimulation and co-cultured with 20 ng/ml rmIL-12 (MOG + IL-12). 5 μg RNA was used to generate cDNA. Quantitative RT-PCR was performed. β-actin was amplified in each sample as endogenous control. Each sample is triplicate. Quantization of gene expression was reported as the fold difference relative to the housekeeping gene. Electrophoresis of final products of PCR of IFN-γ, PD-L1 gene and β-actin as endogenous control in 2.5% agarose-TBE gel is shown (A). There is a significant difference of IFN-γ gene expression between the “MOG” group and the “MOG + IL-12” group (** p<0.01). Compared with the “MOG” group, PD-L1 gene expression was increased significantly in the “MOG + IL-12” group (* p<0.05) (B). One representative experiment of three is shown.
3.4. IL-12 treatment does not increase PD-L gene expression in spleen of IFN-γ-deficient EAE mice
We demonstrated that IL-12 suppressed severity of EAE in C57BL6 wild type mice, but not in IFN-γ-deficient mice, which suggested that the suppression of EAE induced by IL-12 is IFN-γ dependent. We further found that IFN-γ production and PD-L gene expression were increased by IL-12 treatment both in vivo and in vitro from C57BL/6 wild type mice. To determine whether IL-12-induced increase of PD-L gene expression is mediated by IFN-γ, we extracted total RNA from spleen in IFN-γ-deficient EAE mice treated with IL-12 or with PBS as controls, synthesized cDNA and performed quantitative RT-PCR to measure the levels of PD-1, PD-L1, and PD-L2 gene expression. Compared with PBS controls, the levels of PD-1, PD-L1 or PD-L2 gene expression were not significantly increased in the IFN-γ-deficient EAE mice treated with IL-12 (p>0.05) (Fig. 6 A, B), which suggested that IFN-γ was required for the increase of PD-L gene expression induced by IL-12 treatment in EAE mice in vivo.
Figure 6.
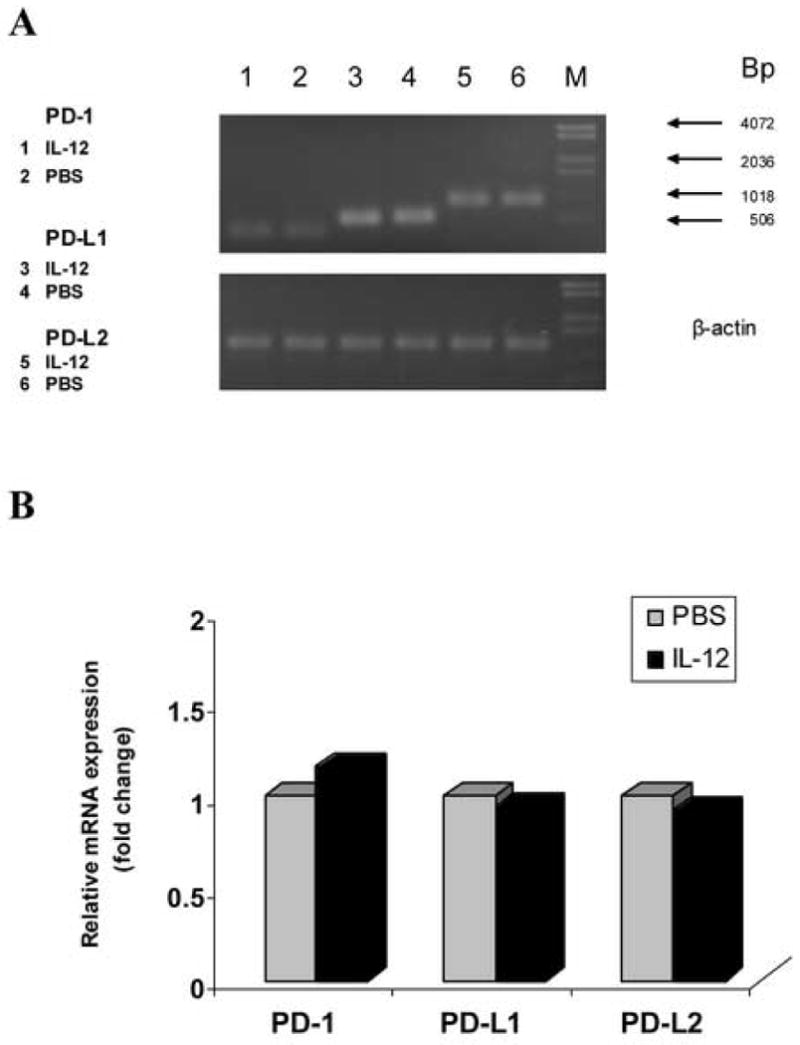
IL-12 treatment does not increase PD-L gene expression in spleen of IFN-γ-deficient EAE mice. Total RNA was extracted from spleen of IFN-γ-deficient EAE mice treated with rmIL-12 or PBS on day 7 p.i., and 5 μg RNA was used to generated cDNA. Quantitative RT-PCR was performed. β-actin was amplified in each sample as endogenous control. Each sample is triplicate. Quantization of gene expression was reported as the fold difference relative to the housekeeping gene. Data were analyzed using GeneScan software. Electrophoresis of final products of PCR of PD-1, PD-L1, PD-L2 gene and β-actin as endogenous control in 2.5% agarose-TBE gel is shown (A). There is no significant difference in PD-1, PD-L1 and PD-L2 gene expression between the PBS and rmIL-12-treated groups (p>0.05) (B). One representative experiment of three is shown.
3.5. IL-12 treatment does not induce PD-L1 gene expression in MOG-specific T cells from IFN-γ-deficient mice in vitro
Our findings showed that increase of PD-L gene expression induced by IL-12 treatment in EAE mice is IFN-γ dependent in vivo. We further determined whether PD-L1 gene expression induced by IL-12 treatment in MOG-specific T cells in vitro is also mediated by IFN-γ. To answer this question, we observed the effects of IL-12 on the gene expression of the PD-1/PD-L pathway in MOG-specific T cells in vitro. IFN-γ-deficient mice were immunized with MOG35-55 and MOG-specific T cells isolated from inguinal and popliteal lymph nodes were prepared from these mice on day 10 p.i. These T cells were cultured with antigen MOG35-55 stimulation at a final concentration of 25 μg/ml, and co-cultured with/without rmIL-12 for 72 hours. Total RNA was extracted, cDNA synthesized, and quantitative RT-PCR was performed to measure levels of PD-1 and PD-L gene expression. Compared with the cultured MOG-specific T cells without IL-12, PD-1, PD-L1 or PD-L2 gene expression were not increased in the MOG-specific T cells cultured with IL-12 (p>0.05) (Fig. 7 A, B). The data in vitro were consistent with the results in vivo, confirming that IL-12 treatment promoted PD-L1 gene expression in EAE mice through induction of IFN-γ. Up-regulation of the PD-1/PD-L pathway by IL-12 in suppression of EAE is mediated by IFN-γ.
Figure 7.
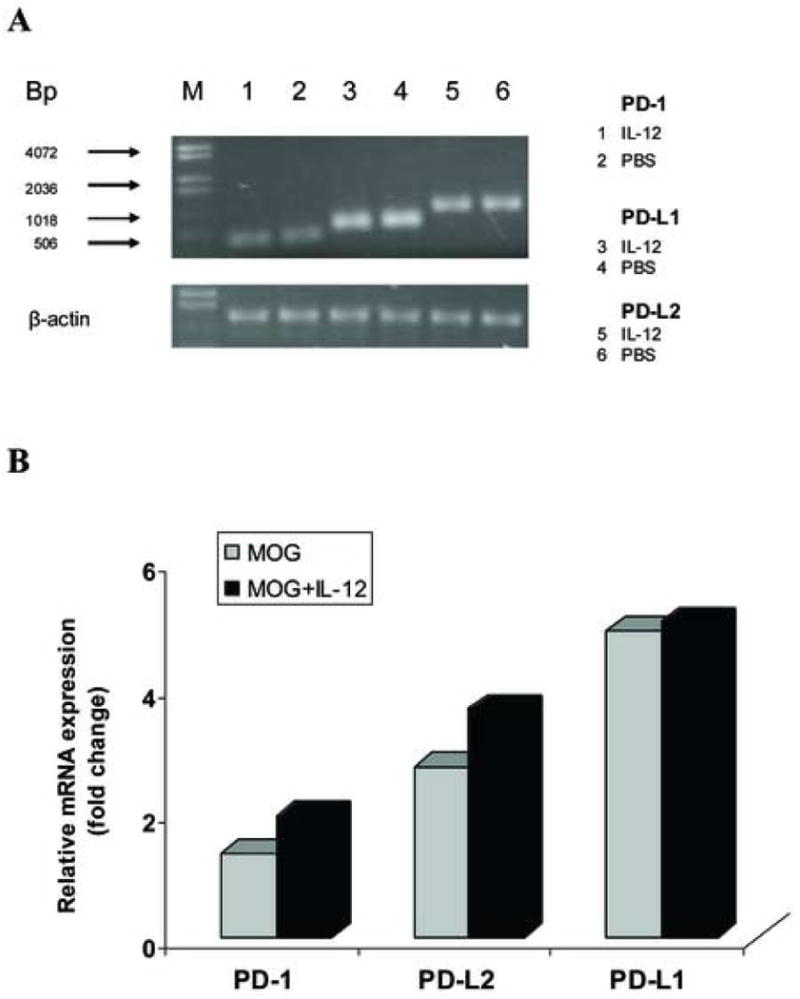
IL-12 treatment does not induce PD-L1 gene expression in MOG-specific T cells from IFN-γ-deficient mice in vitro. MOG-specific T cells isolated from inguinal and popliteal lymph nodes were prepared on day 10 p.i. from IFN-γ-deficient mice immunized with 300 μg MOG35-55 peptide. Total RNA was extracted from MOG-specific T cells cultured with MOG35-55 stimulation alone at a final concentration of 25 μg/ml (MOG), or cultured with MOG stimulation and co-cultured with 20 ng/ml rmIL-12 (MOG + IL-12). 5 μg RNA was used to generate cDNA. Quantitative RT-PCR was performed. β-actin was amplified in each sample as endogenous control. Each sample is triplicate. Quantization of gene expression was reported as the fold difference relative to the housekeeping gene. Electrophoresis of final products of PCR of PD-1, PD-L1 and PD-L2 gene and β-actin as endogenous control in 2.5% agarose-TBE gel is shown (A). There is no significant difference in PD-1, PD-L1 and PD-L2 gene expression between the “MOG” group and the “MOG + IL-12” group (p>0.05) (B). One representative experiment of three is shown.
3.6. IL-12 treatment increases the number of APCs positive for PD-L1 expression in EAE mice
IL-12 treatment induces production of Th1 cytokine IFN-γ, which mediates up-regulation of PD-L expression, and finally results in suppression of EAE. To determine whether IL-12 treatment increases PD-L1 expression in EAE mice by increasing the number of APCs expressing PD-L1, we isolated splenocytes from PBS- and IL-12-treated EAE mice. Stained with fluorescent PD-L1 and CD11b monoclonal antibodies, cells were assayed by flow cytometry to measure the level of PD-L1 expression and percentage of APCs that were positive for PD-L1 expression. The cells were also stained with isotype as control. Compared with the isotype, the percentage of PD-L1+ cells in EAE mice treated both with PBS and with IL-12 were increased significantly, which demonstrated that the cell populations negative for PD-L1 expression were getting activated and PD-L1 was expressed during the inflammation. Compared with the PBS control group, an increased number of APCs were positive for PD-L1 expression in the IL-12-treated group (p<0.05) (Fig. 8). This suggested that IL-12 treatment enhanced not only PD-L1 mRNA expression but protein synthesis as well, indicating that IL-12 treatment upregulates both transcription and translation of PD-L1 expression in the suppression of EAE.
Figure 8.
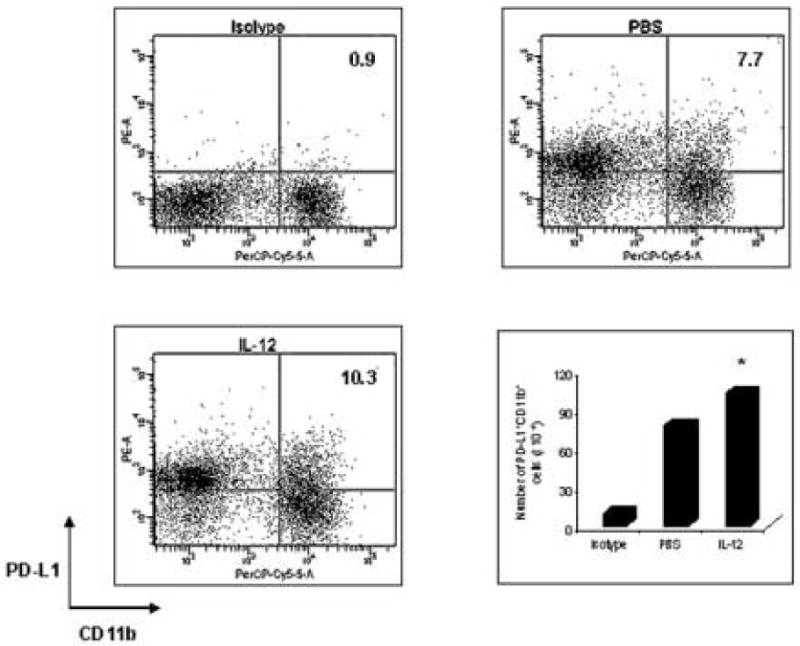
IL-12 treatment increases the number of APCs with PD-L1 expression in EAE mice. C57BL/6 female mice were immunized with 300 μg MOG35-55 peptide, and were sacrificed on day 7 p.i. Spleen cells (1 ×106/tube) were isolated from PBS treated EAE mice or rmIL-12-treated EAE mice. After being washed with staining buffer and blocked with CD16/32 (FcγIII/II receptor) monoclonal antibody, these cells were stained with R-PE-Conjugated PD-L1 monoclonal antibody and PerCP-Cy5.5-Conjugated CD11b monoclonal antibody for 30 min. The cells were also stained with isotype as control. Cells were measured by flow cytometry. Data were acquired on a FACSCalibur and analyzed using CellQuest software and FlowJo software. Fluorescence intensity was measured to determine the percentage of CD11b+ cells that were positive for PD-L1 expression. Compared with the PBS group, the number of PD-L1+ CD11b+ cells in the rmIL-12 treated group is significantly increased (* p<0.05). The figure is a representative of three experiments.
3.7. Increase of PD-L1 gene expression induced by IL-12 treatment is specifically in CD11b+ APCs but not in CD11b- APCs
We further investigated in which subset of APCs PD-L1 expression was increased by IL-12 treatment. We hypothesized that the level of PD-L1 expression was increased within specific APC populations. To test this hypothesis, we sorted CD11b+ and CD11b- APCs to >95% purity using flow cytometric sorting techniques. The number of CD11b+ and CD11b- APCs collected was 1 × 104. Using the nanoprep RNA purification method, total RNA was extracted, cDNA was synthesized, and RT-PCR was performed to quantify levels of PD-L1 gene expression. In CD11b+ APC populations, the level of PD-L1 gene expression in IL-12-treated EAE mice was significantly increased 3 fold compared with PBS controls (** p<0.01) (Fig.9 A, B). In CD11b-APC populations, the level of PD-L1 gene expression in IL-12-treated EAE mice was not changed compared with PBS controls (p>0.05) (Fig.9 A, B). This showed that IL-12 treatment increased the level of PD-L1 gene expression specifically in CD11b+ APCs.
Figure 9.
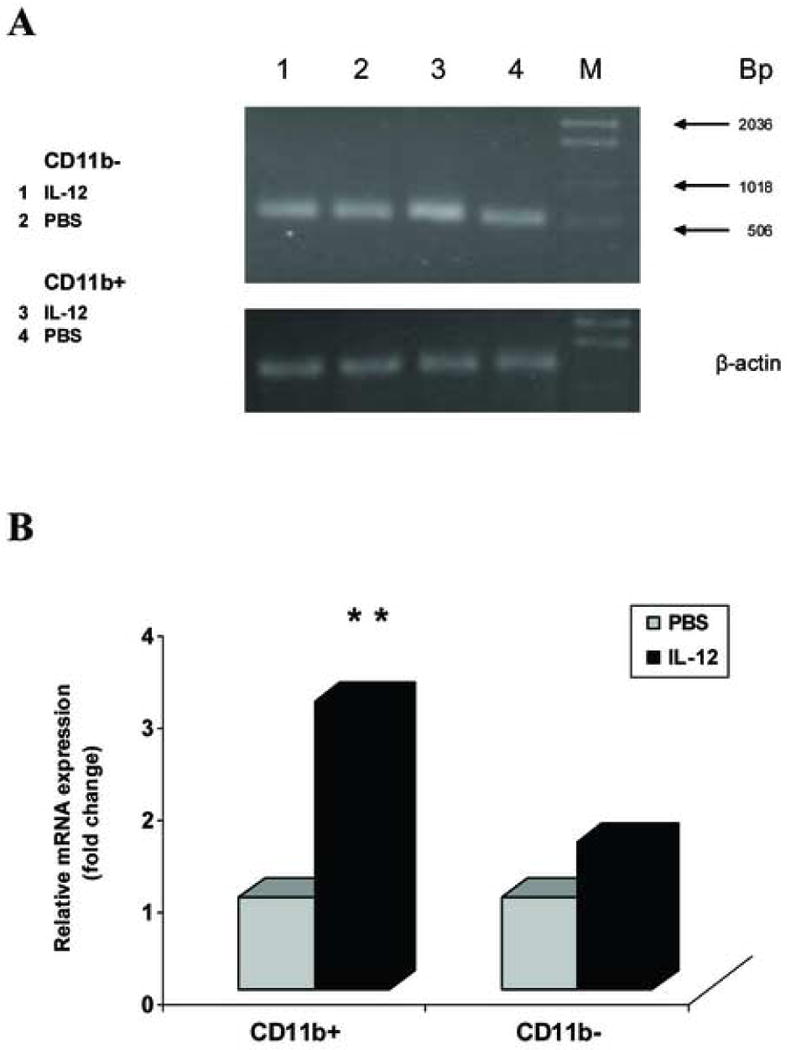
Expression of PD-L1 is increased in CD11b+ APCs but not CD11b- APCs treated with rmIL-12. Spleen cells (1 ×106/tube) were isolated from the PBS- and rmIL-12-treated EAE mice. CD11b+ and CD11b- cells were sorted by Flow cytometry to >95% purity and a total of 10,000 CD11b+ and CD11b- cells were used for RNA extraction. cDNA was synthesized and quantitative RT-PCR was performed. β-actin was amplified in each sample as endogenous control. Each sample is triplicate. Quantization of gene expression was reported as the fold difference relative to the housekeeping gene. Electrophoresis of final products of PCR of PD-L1 and β-actin as endogenous control in 2.5% agarose-TBE gel is shown (A). In the CD11b+ cell populations, expression of PD-L1 gene is significantly increased in the rmIL-12-treated group, compared with the PBS control group (** p<0.01). However, in the CD11b- cell populations, there is no significant difference between the PBS and rmIL-12 treated groups (p>0.05) (B). One representative experiment of three is shown.
4. Discussion
IL-12, a member of a group of secreted proteins, was originally discovered in 1989 (Kobayashi et al., 1989). It has diverse roles in cellular differentiation and is especially important in regulating Th1 responses, acting as a link between innate and adaptive immunity (Ma and Trinchieri, 2001; Langrish et al., 2004). The role of IL-12 in the pathogenesis of MS and EAE appears very complex and is not yet well understood. IL-12 is known to regulate Th1 development and production of IFN-γ (Trinchieri et al., 2003; Segal et al., 1998) and our previous work has shown that IL-12 has immunomodulatory effects in EAE through IFN-γ signaling (Gran et al., 2004). In the current studies, we determined the immunomodulatory potential of IL-12 in active EAE and further elucidate the possible molecular mechanisms. Our data confirmed the suppressive role of IL-12 in active EAE, as IL-12 administration suppressed clinical severity of EAE in wild type mice, but had no effect on EAE in IFN-γ deficient mice, similar to our prior studies (Gran et al., 2004). The fact that PD-L1 expression was induced by IL-12 treatment in EAE spleen in vivo as well as in MOG-specific T cells in vitro in the current studies suggested that IL-12 suppresses EAE through PD-1/PD-L signaling. This conclusion was further substantiated by the finding that IL-12 treatment failed to up-regulate PD-L1 expression in IFN-γ-deficient spleen cells. Moreover, our data showed that PD-L1 expression was increased in CD11b+ APCs but not CD11b- cells. Together, these results indicate that PD-1/PD-L signaling downstream of IFN-γ in CD11b+ APCs is one mechanism mediating immunomodulatory effects of IL-12 in EAE.
Numerous studies have provided data suggesting that IFN-γ downregulates EAE (Billiau et al., 1988; Lublin et al., 1993). Treatment of EAE animals with IFN-γ antibodies resulted in enhanced disease in both rats and mice while giving exogenous IFN-γ ameliorated disease (Heremans et al., 1996; Duong et al., 1992; Voorthuis et al., 1990). Evidence from studies using IFN-γ cytokine knock out mice has shown that IFN-γ is not essential for the generation or function of anti-MOG35-55 effector cells but that it does play an obligatory role in down-regulating the disease (Krakowski and Owens, 1996; Ferber et al., 1996). Increased IFN-γ production has been found to have significant inhibitory effects on autoreactive T cell proliferative responses (Krakowski and Owens, 1996; Ferber et al., 1996). In addition, high levels of IFN-γ are associated with terminal effector differentiation and death, reducing the survival of Th1 cells. Compared with Th1 cells secreting low levels of IFN-γ, Th1 cells secreting high levels of IFN-γ are short-lived and do not develop into long-term memory Th1 cells efficiently. Therefore, increased IFN-γ production induced by IL-12 may limit their ability to survive, initiate autoimmune demyelination, and develop into long-term memory cells (Willenborg et al., 1999; Wu et al., 2002; Loza et al., 2002). Importantly, we found in this report that the PD-1/PD-L pathway of costimulatory molecules was up-regulated by IL-12-induced IFN-γ production in EAE mice, suggesting this signaling mechanism can be used to drive Th1 regulation.
PD-1 and PD-L are newly described members of the B7-CD28 superfamily of costimulatory molecules, which act as negative regulators of activated T cells (Khoury and Sayegh, 2004). PD-1 is expressed on thymocytes during and after T cell receptor (TCR) β-selection, at transition from the CD4-CD8- to CD4+CD8+ stages, and on mature T and B cells following activation (Yamazaki et al., 2002; Lechner et al., 2001). The ligands for PD-1 are PD-L1 (B7-H1) and PD-L2 (B7-DC) (Dong et al., 1999). PD-L physically binds to PD-1 and inhibits the proliferative response of PD-1+/+ but not PD-1-/- splenic T cells activated by anti-CD3 stimulation, indicating that PD-1—PD-L engagement results in negative signaling (Kohm et al., 2002; Cai et al., 2004). The current findings show that IL-12 administration increases PD-L1 gene expression in the spleens of EAE mice in vivo and also in MOG-specific T cells in vitro, demonstrating that there is more PD-L1 available to bind PD-1, which had stable expression levels. This suggests that PD-1/PD-L1 engagement may be providing similar negative signaling to result in inhibition of T cell responses to antigen MOG35-55, which leads to IL-12-induced suppression of EAE. Recent studies using PD-L1-deficient mice have further suggested a role for PD-L1 in negatively regulating autoimmune diseases. Increased MOG-specific CD4+ T cell responses in PD-L1-deficient mice reflect multiple negative regulatory functions for PD-L1: (1) limiting expansion and/or Th1 differentiation of naïve CD4+ T cells, (2) negatively regulating reactivation of MOG-specific effector CD4+ T cells in the target organ, and (3) limiting expansion of antigen-specific T cells through engagement of PD-1 on Tregs (Latchman et al., 2004).
Consistent with our findings, PD-L1 signaling has been shown to mediate IL-12 effects downstream of IFN-γ in other cell types (Eppihimer et al., 2002; Schoop et al., 2004), as evidenced by observations in microvascular endothelial cells (ECs) in vitro and in vivo. Studies using a dual radiolabeled monoclonal antibody (mAb) technique indicated that PD-L1 expression was significantly increased in ECs and in tissues at 24 hours after IL-12-challenge, with peak levels of PD-L1 occurring 72 hours after IL-12-challenge, but IL-12 was not effective at inducing PD-L1 expression in tissues of IFN-γ-deficient mice. These data showed that elevation of PD-L1 expression was induced by IL-12 through IFN-γ (Eppihimer et al., 2002). Comparable and similar data were observed on murine renal tubular epithelial cells (TEC). Expression of PD-L1 and PD-L2 was studied by flow cytometric analysis and reverse transcription-polymerase chain reaction on TEC and it showed that PD-L1 but not PD-L2 was weakly expressed on unstimulated TEC but up-regulated PD-L1 expression was observed upon stimulation with IFN-γ in a dose-dependent manner. Blockade of PD1/PD-L1 pathway with monoclonal antibodies in antigen presentation assays uncovered an inhibitory role of this ligand system in T cell activation and response. This evidence suggested PD-L1 up-regulated by IFN-γ-stimulation effectively provided inhibitory signaling in classical antigen presenting systems and negatively regulated T cell activation and responses (Schoop et al., 2004). Combined with our current studies, it therefore can be inferred that induction of IFN-γ and up-regulation of the PD-1/PD-L1 pathway by IL-12 might be one commonly used signaling pathway that leads to cell-type specific immune responses.
Although IFN-γ has often been recognized as a proinflammatory mediator (Ferber et al., 1996), several studies, similar to ours, have suggested that IFN-γ may act to induce immunosuppression. IFN-γ-deficient mice or mice administered anti-IFN-γ mAbs had exacerbated disease symptoms in EAE, whereas administration of exogenous IFN-γ reduced the incidence and severity of EAE (Krakowski et al., 1996; Tanuma et al., 1999). Based on these results, it is reasonable to suggest that in the IFN-γ-deficient mice, the level of inducible PD-L1 is reduced and may lead to less inhibition of T cell activation by the PD-L1/PD-1 pathway and therefore, contribute to the severity of the disease. Additional studies of PD-1-deficient mice showed an autoimmune-like disease characterized by infiltration of inflammatory cells in various tissues (Nishimura et al., 2001; Khoury and Sayegh, 2004). Thus, these studies combined with our current results demonstrate that elevation of PD-L1 expression can be an important negative signal pathway in IL-12-induced suppression of EAE mediated by IFN-γ.
In previous work, we observed decreased IL-17 mRNA expression in the spleen following IL-12-induced suppression of EAE (Gran et al., 2004). IL-17, a T cell-derived pro-inflammatory molecule, has been found in many autoimmune diseases such as multiple sclerosis and rheumatoid arthritis (Matusevicius et al., 1999; Ziolkowska et al., 2000). Additional studies showed that IL-17 is expressed in response to signals distinct from those associated with the Th1 or Th2 response, and IL-12 has marginal effects on IL-17 production (Langrish et al., 2005; Aggarwal et al., 2003). Stumhofer found that the development of IL-12-producing T helper cells during chronic inflammation of the central nervous system was negatively regulated by IL-27, and IL-27 inhibited production of IL-17 through activation of the Jak-STAT pathway (Stumhofer et al., 2006). It has been reported that Th1/Th2 cells differentially up-regulate PD-L1 and PD-L2 expression in inflammatory macrophages, indicating that PD-L1 and PD-L2 might have different functions under different Th1/Th2 inflammatory situations, although they both bind PD-1 receptor (Loke et al., 2003). Moreover, PD-L1 up-regulation depends on TLR4 and STAT1/STAT4, whereas PD-L2 expression depends on NF-κB, IL-4Rα and STAT6 (Liang et al., 2003; Loke et al., 2003). Our findings demonstrated that PD-L1 was up-regulated, whereas PD-L2 was not, following IL-12 treatment. Understanding the preferential role of PD-L1 in IL-12-induced suppression of EAE will therefore suggest different downstream signaling pathways and future studies will be done to evaluate the role of TLR4 and Jak/STAT signaling transduction pathways.
Furthermore, we found that expression of PD-L1 induced by IL-12 treatment was increased specifically in CD11b+ antigen presenting cells but not in CD11b- cells. Recent studies showed that CD11b is expressed predominantly on monocytes/macrophages, PMN, and NK cells, and CD11b+ APCs inhibit anti-CD3-induced human T cell proliferation as well as IL-2 release (Wu et al., 2004). In splenic CD11b+ APCs, reproducible low levels of p28 and EB13 mRNA are expressed and IL-27 expression is differentially regulated, which has been shown to be involved in the disease development and progression of EAE (Li et al., 2005). Studies using administration of anti-CD11b in vivo indicated that CD11b+ monocyte/macrophages are capable of inducing a contact sensitivity response and a state of tolerance to antigens (Hammerberg et al., 1996). CD11b+ infiltrating cells are not only important in tolerance induction, but play a prominent regulatory role in the induction of autoantigen-specific Th2 cells as well (Schlecht et al., 2004), while Th1/Th2 balance is involved in pathogenesis of EAE. In our report, one intriguing possibility might be that higher PD-L1 levels in CD11b+ monocyte/macrophages inhibits Th1 cells and stimulates Th2 cells. PD-L1 may signal APCs that influence the function of dendritic cells, and transduce negative signals to influence the activation of macrophages upon subsequent interaction with T cells, leading to inhibition of autoreactive T cell response and subsequent suppression of the disease severity of EAE.
In summary, we demonstrate for the first time that suppressive effects of IL-12 on induced EAE can be mediated by promoting expression of the PD-1/PD-L pathway of costimulatory molecules through induction of IFN-γ. These findings indicate that costimulatory molecules providing negative signals have a crucial role in regulating autoimmune disease by cytokine signaling, which is important for understanding the precise mechanisms of cell-mediated autoimmunity. Promotion of the PD-1/PD-L1 pathway in APCs therefore may provide a rationale for developing new strategies for therapeutic intervention in autoimmune diseases such as multiple sclerosis.
Acknowledgments
We thank Drs. Jorge Roman-Blas, Roberto Buccafusca, Tarik Touil, and Kathryn Scott for technical help in RT-PCR experiments and Dr. Dan Rosson for his assistance with flow cytometry work. This work was supported by grants from the National Institute of Health and the National Multiple Sclerosis Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8:765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Begolka WS, Vanderlugt CL, Rahbe SM, Miller SD. Differential expression of inflammatory cytokines parallels progression of central nervous system pathology in two clinically distinct models of multiple sclerosis. J Immunol. 1998;161:4437–4446. [PubMed] [Google Scholar]
- Benson JM, Campbell KA, Guan Z, Gienapp IE, Stuckman SS, Forsthuber T, Whitacre CC. T-cell activation and receptor downmodulation precede deletion induced by mucosally administered antigen. J Clin Invest. 2000;106:1031–1038. doi: 10.1172/JCI10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, Carton H. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-gamma. J Immunol. 1988;140:1506–1510. [PubMed] [Google Scholar]
- Brown J, Dorfman DM, Ma F, Sullivan EL, Munoz O, Wood CR, Greenfield EA, Freeman GJ. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–1266. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- Cai G, Karni A, Oliveira EML, Weiner HL, Hafler DA, Freeman GJ. PD-1 ligands, negative regulators for activation of naïve, memory, and recently activated human CD4+ T cells. Cellul Immunol. 2004;230:89–98. doi: 10.1016/j.cellimm.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- Duong TT, St Louis J, Gilbert JJ, Finkelman FD, Strejan GH. Effect of anti-interferon-gamma and anti-interleukin-2 monoclonal antibody treatment on the development of actively and passively induced experimental allergic encephalomyelitis in the SJL/J mouse. J Neuroimmunol. 1992;36:105–115. doi: 10.1016/0165-5728(92)90042-j. [DOI] [PubMed] [Google Scholar]
- Eppihimer MJ, Gunn J, Freemen GJ, Greenfield EA, Chernova T, Erickson J, Leonard JP. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation. 2002;9:133–145. doi: 10.1038/sj/mn/7800123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-γ gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- Gately MK, Desai BB, Wolitzky AG, Quinn PM, Dwyer CM, Podlaski FJ, Familletti PC, Sinigaglia F, Chizonnite R, Gubler U. Regulation of human lymphocyte proliferation by a heterodimeric cytokine, IL-12 (cytotoxic lymphocyte maturation factor) J Immunol. 1991;147:874–882. [PubMed] [Google Scholar]
- Gran B, Zhang GX, Rostami AM. Role of the IL-12/IL-23 system in the regulation of T-cell responses in central nervous system inflammatory demyelination. Crit Rev Immunol. 2004;24:111–128. doi: 10.1615/critrevimmunol.v24.i2.20. [DOI] [PubMed] [Google Scholar]
- Gran B, Zhang GX, Yu S, Li J, Chen XH, Ventura ES, Kamoun M, Rostami AM. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002;169:7104–7110. doi: 10.4049/jimmunol.169.12.7104. [DOI] [PubMed] [Google Scholar]
- Gran B, Chu N, Zhang GX, Yu S, Li Y, Chen XH, Kamoun M, Rostami AM. Early administration of IL-12 suppresses EAE through induction of interferon-γ. J Neuroimmunol. 2004;156:123–131. doi: 10.1016/j.jneuroim.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14:145–155. doi: 10.1016/s1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- Hammerberg C, Duraiswamy N, Cooper KD. Reversal of immunosuppression inducible through ultraviolet-exposed skin by in vivo anti-CD11b treatment. J Immunol. 1996;157:5254–5261. [PubMed] [Google Scholar]
- Heremans H, Dillen C, Groenen M, Martens E, Billiau A. Chronic relapsing experimental autoimmune encephalomyelitis (CREAE) in mice: enhancement by monoclonal antibodies against interferon-gamma. Eur J Immunol. 1996;26:2393–2398. doi: 10.1002/eji.1830261019. [DOI] [PubMed] [Google Scholar]
- Ingham DJ, Beer S, Money S, Hamsen G. Quantitative real-time PCR assay for determining transgene copy number in transformed plants. Biotechniques. 2001;31:132–134. 136–140. doi: 10.2144/01311rr04. [DOI] [PubMed] [Google Scholar]
- Khoury SJ, Sayegh MH. The roles of the new negative T cell costimulatory pathways in regulating autoimmunity. Immunity. 2004;20:529–538. doi: 10.1016/s1074-7613(04)00116-5. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Fitz L, Ryan M, Hewrick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med. 1989;170:827–845. doi: 10.1084/jem.170.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–4716. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996;26:1641–1646. doi: 10.1002/eji.1830260735. [DOI] [PubMed] [Google Scholar]
- Langrish CL, McKenzie BS, Wilson NJ, Malefyt RW, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rew. 2004;202:96–105. doi: 10.1111/j.0105-2896.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner O, Lauber J, Franzke A, Sarukhan A, von Boehmer H, Buer J. Fingerprints of anergic T cells. Curr Biol. 2001;8:587–595. doi: 10.1016/s0960-9822(01)00160-9. [DOI] [PubMed] [Google Scholar]
- Li J, Gran B, Zhang GX, Rostami AM, Kamoun M. IL-27 subunits and its receptor (WSX-1) mRNAs are markedly up-regulated in inflammatory cells in the CNS during experimental autoimmune encephalomyelitis. J Neurol Sci. 2005;232:3–9. doi: 10.1016/j.jns.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, Sharpe AH. Regulation of PD-1, PD-L1, PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33:2706–2716. doi: 10.1002/eji.200324228. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quatitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci. 2003;100:5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loza MJ, Perussia B. Peripheral immature CD2-/low T cell development from type 2 to type 1 cytokine production. J Immunol. 2002;169:3061–3068. doi: 10.4049/jimmunol.169.6.3061. [DOI] [PubMed] [Google Scholar]
- Lublin FD, Knobler RL, Kalman B, Goldhaber M, Marini J, Perrault M, D'Imperio C, Joseph J, Alkan SS, Korngold R. Monoclonal anti-gamma interferon antibodies enhance experimental allergic encephalomyelitis. Autoimmunity. 1993;16:267–274. doi: 10.3109/08916939309014645. [DOI] [PubMed] [Google Scholar]
- Ma X, Trinchieri G. Regulation of interleukin-12 production in antigen-presenting cells. Adv Immunol. 2001;79:55–92. doi: 10.1016/s0065-2776(01)79002-5. [DOI] [PubMed] [Google Scholar]
- Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–104. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, Kung AL, Cereb N, Yao TP, Yang SY, Reiner SL. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 2001;292:1907–1910. doi: 10.1126/science.1059835. [DOI] [PubMed] [Google Scholar]
- Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai A, Minato N, Honjo T. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Iwai Y, Homjo T. New regulatory co-receptors: inducible co-stimulator and PD-1. Curr Opin Immunol. 2002;14:779–782. doi: 10.1016/s0952-7915(02)00398-9. [DOI] [PubMed] [Google Scholar]
- Salama AD, Chitnis T, Imitola J, Akiba H, Tushima F, Azuma M, Yagita H, Sayagh MH, Khoury SJ. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med. 2003;198:71–78. doi: 10.1084/jem.20022119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlecht G, Loucka J, Najar H, Sebo P, Leclerc C. Antigen targeting to CD11b allows efficient presentation of CD4+ and CD8+ T cell epitopes and in vivo Th1-polarized T cell priming. J Immunol. 2004;173:6089–6097. doi: 10.4049/jimmunol.173.10.6089. [DOI] [PubMed] [Google Scholar]
- Schoop R, Wahl P, Hir ML, Heemann U, Wang M, Wuethrich RP. Suppressed T-cell activation by IFN-γ-induced expression of PD-L1 on renal tubular epithelial cells. Nephrol Dial Transplant. 2004;19:2713–2720. doi: 10.1093/ndt/gfh423. [DOI] [PubMed] [Google Scholar]
- Segal BM, Dwyer BK, Shevach EM. An interleukin (IL)-10/IL-12 immunoregulatory circuit controls susceptibility to autoimmune disease. J Exp Med. 1998;187:537–546. doi: 10.1084/jem.187.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- Song P, Cai C, Skokut M, Kosegi B, Petolino J. Quantitative real-time PCR as a screening tool for estimating transgene copy number in WHISKERS™-derived transgenic maize. Plant Cell Rep. 2002;20:948–954. [Google Scholar]
- Steinman L. Assessment of animal models for MS and demyelinating disease in the design of rational therapy. Neuron. 1999;24:511–514. doi: 10.1016/s0896-6273(00)81107-1. [DOI] [PubMed] [Google Scholar]
- Storek J, Zhao Z, Lin E, Berger T, McSweeney PA, Nash RA, Akatsuka Y, Metcalf MD, Lu H, Kalina T. Recovery from and consequences of severe iatrogeniclymphopenia (induced to treat autoimmune dieases) Clin Immunol. 2004;113:285–298. doi: 10.1016/j.clim.2004.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, John LM, Villarino AV, Huang Q, Yoshimura A, Sehy D, Saris CJ, O'shea JJ, Hennighausen L, Ernst M, Hunter CA. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nature Immunology. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- Tanuma N, Shin T, Kogure K, Matsumoto Y. Differential role of TNF-alpha and IFN-gamma in the brain of rats with chronic relapsing autoimmune encephalomyelitis. J Neuroimmunol. 1999;96:73–79. doi: 10.1016/s0165-5728(99)00018-1. [DOI] [PubMed] [Google Scholar]
- Trinchieri G, Scott P. Interleukin-12; a proinflammatory cytokine with immunoregulatory functions. Res Immunol. 1995;146:423–431. doi: 10.1016/0923-2494(96)83011-2. [DOI] [PubMed] [Google Scholar]
- Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity. 2003;19:641–644. doi: 10.1016/s1074-7613(03)00296-6. [DOI] [PubMed] [Google Scholar]
- Voorthuis JA, Uitdehaag BM, De Groot CJ, Goede PH, von der Meide PH, Dijkstra CD. Suppression of experimental allergic encephalomyelitis by intraventricular administration of interferon-gamma in Lewis rats. Clin Exp Immunol. 1990;81:183–188. doi: 10.1111/j.1365-2249.1990.tb03315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wekerle H, Kojima K, Lannes-Vieira J, Lassmann H, Linington C. Animal models. Ann Neurol. 1994;361:S47–53. doi: 10.1002/ana.410360714. [DOI] [PubMed] [Google Scholar]
- Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-γ plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1999;157:3223–3227. [PubMed] [Google Scholar]
- Wu CY, Kirman JR, Rotte MJ, Davey DF, Perfetto SP, Rhee EG, Freidag BL, Hill BJ, Douek DC, Seder RA. Distinct lineages of T(H)1 cells have differential capacities for memory cell generation in vivo. Nat Immunol. 2002;3:852–858. doi: 10.1038/ni832. [DOI] [PubMed] [Google Scholar]
- Wu H, Rodgers JR, Perrard XY, Perrard JL, Prince JE, Abe Y, Davis BK, Dietsch G, Smith CW, Ballantyne CM. Deficient of CD11b or CD11d results in reduced staphylococcal enterotoxin-induced T cell response and T cell phenotypic Changes. J Immunol. 2004;173:297–236. doi: 10.4049/jimmunol.173.1.297. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, Shin T, Tsuchiya H, Pardoll DM, Okumura K, Azuma M, Yagita H. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002;169:5538–5545. doi: 10.4049/jimmunol.169.10.5538. [DOI] [PubMed] [Google Scholar]
- Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, Kamoun M, Rostami AM. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-β2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. 2003;170:2153–2160. doi: 10.4049/jimmunol.170.4.2153. [DOI] [PubMed] [Google Scholar]
- Zhang GX, Yu S, Gran B, Li J, Siglienti I, Chen X, Calida D, Ventura E, Kamoun M, Rostami AM. Role of IL-12 receptor β1 in regulation of T cell response by APC in experimental autoimmune encephalomyelitis. J Immunol. 2003;171:4485–4492. doi: 10.4049/jimmunol.171.9.4485. [DOI] [PubMed] [Google Scholar]
- Zhu B, Guleria I, Khosroshahi A, Chitnis T, Imitola J, Azuma M, Yagita H, Sayegh MH, Khoury SJ. Differential role of programmed death-ligand 1 and programmed death-ligand 2 in regulating the susceptibility and chronic progression of experimental autoimmune encephalomyelitis. J Immunol. 2006;176:3480–3489. doi: 10.4049/jimmunol.176.6.3480. [DOI] [PubMed] [Google Scholar]
- Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W. High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000;164:2832–2838. doi: 10.4049/jimmunol.164.5.2832. [DOI] [PubMed] [Google Scholar]