

Abstract
Traditionally, the elucidation of genes involved in maturation and aging has been studied in a temporal fashion by examining gene expression at different time points in an organism's life as well as by knocking out, knocking in, and mutating genes thought to be involved. Here, we propose an in silico method to combine clinical electronic medical record (EMR) data and gene expression measurements in the context of disease to identify genes that may be involved in the process of human maturation and aging. First we show that absolute lymphocyte count may serve as a biomarker for maturation by using statistical methods to compare trends among different clinical laboratory tests in response to an increase in age. We then propose using the rate of decay for absolute lymphocyte count across 12 diseases as a proxy for differences in aging. We correlate the differing rates with gene expression across the same diseases to find maturation/aging related genes. Among the 53 genes with strongest correlations between expression profile and change in rate of decay, we found genes previously implicated in the process of aging, including MGMT (DNA repair), TERF2 (telomere stability), POLD1 (DNA replication and repair), and POLG (mtDNA replication).
1. Introduction
The integration of bioinformatics, basic science, and statistical methods has been recognized as being essential to the progression of translational research. Advances made in the understanding of biological systems using such an integrated approach can have a direct impact at both the bench and bedside to further our understanding of human disease [1]. Techniques like gene expression microarray measurement and analysis, which has been used extensively with research involving model organisms, can be extremely informative about diseases, aging and other biological processes. There have been many innovative ways of integrating these microarrays with various data sets to identify genes and their potential function, but most of these methods have led to a reductionist approach to the study of disease, where novel subtypes and features observed in microarray analyses are used to describe singular disorders [2].
Against this reductionist trend, recent work has focused on the use of measurements across a variety of disorders to find the common elements across disease. Daniel Rhodes and colleagues searched for commonalities in cancer in 2004 [3]. After collecting 40 available sets of microarray data with over 3,700 samples of cancer, Rhodes calculated a genome-wide signature representative of neoplastic transformation. Andrea Bild and colleagues linked cellular models with disease samples to find commonly deregulated biological pathways across cancers that correspond with worsening survival [4]. Eran Segal and colleagues used microarray-based expression measurements annotated with both biological and clinical conditions to create modules which were examined across types of cancer [5]. In our previous work, we linked gene measurements, as measured by microarrays, to phenotypes and responses to environment, as represented by biomedical concepts in the Unified Medical Language System (UMLS), to create a phenome-genome network [6]. Each of these is an important example of how genome-era measurements can be used to quantitate mechanistic similarities and differences between diseases previously categorized using syndromic or anatomic descriptors.
An often-overlooked area that can contribute to translational research is clinical laboratory data. In the past, data collected during clinical care were prone to transcription errors while transferring information from paper forms to an electronic format. However, as an increasing number of institutions move towards using electronic medical records (EMR) data quality has increased due to elimination of transcription and omission errors [7]. While EMRs have created a structured environment for reporting of laboratory measurements for physicians, they rarely provide data in a manner easily accessible for translational researchers. Even when such data is available for clinical research, it is typically accessed on a disease-by-disease basis. Here, we hypothesize that these laboratory test measurements can provide an important link between gene expression measurements and the physical manifestations of patients.
One type of physical manifestation is aging. The mechanisms of aging, though still far from being determined, are thought to involve three main biological phenomena leading to cellular senescence: DNA damage, telomere shortening and mitochondrial dysfunction [8]. Research into these areas has focused almost exclusively on in vitro and in vivo models, wherein gene expression measurements for different time points in an organism's lifespan as well as the knock-out, overexpression, or mutation of genes suspected of being aging-related remain the gold standard to find such genes. However, the study of aging is difficult as there are many genetic as well as environmental influences that contribute to its progression, not to mention the fact that the mechanisms of aging in model organisms may differ from that of humans.
In this paper, we introduce a novel translational method that uses clinical laboratory measurements in conjunction with gene expression levels to elucidate genes that may be involved in the process of human maturation and aging. After using clinical laboratory measurements to find a biomarker that correlates with an increase in age, we order several diseases based on the accelerated or decelerated change in this biomarker. We then use publicly available gene expression data sets representative of these diseases to find genes changing in expression in the same profile as the rate of change in the biomarker. While we find that our set of genes correlating with the change in our aging biomarker are over-represented with known genes associated with aging, we are releasing this list in the hopes that these results will be validated through biological assays. Finally, this method of incorporating clinical laboratory data with gene expression microarray data is extensible and we believe it will be useful in deciphering and understanding many complex human diseases.
2. Methods
2.1. Data Collection and Processing
Quantitative clinical laboratory data, consisting of 1,104,316 measurements across 656 distinct lab tests, originally obtained at the Lucile Packard Children's Hospital, were collected in a de-identified manner from the Stanford Translational Research Integrated Database Environment (STRIDE). In total, this data represented 4,844 patients across all ages that were diagnosed with one or more of a set of 12 chronic diseases (Table 1). The use of de-identified clinical laboratory data in this manner was approved by the Institutional Review Board of the Stanford University School of Medicine.
Table 1.
List of the twelve diseases, the abbreviations used in this paper, and the GEO data sets used to represent the genome-wide changes in gene expression seen in each disease.
| Disease | Abbreviation | GEO Data Sets |
|---|---|---|
| Autoimmune polyendocrinopathy-candidiasis- ectodermal dystrophy |
AP Dystrophy | 167 |
| Asthma | Asthma | 13, 14, 42, 56, 58, 60 |
| Cystic fibrosis (of pancreas) | Cystic Fibrosis | 567 |
| Juvenile spondyloarthropathy | JS | 711 |
| Crohn's disease | Crohn's | 559, 560 |
| Familial hypercholesterolemia | Fam. Hyperchol. | 279 |
| Down syndrome | Down Syndrome | 681, 682 |
| Insulin dependent diabetes mellitus | Ins. Dep. Diabetes | 10 |
| Ulcerative colitis | Ulcerative Colitis | 559, 560 |
| Duchenne muscular dystrophy | DM Dystrophy | 639 |
| HIV infection | HIV | 171 |
| Neurofibromatosis type 1 | Neurofibromatosis I | 604 |
We applied a filter to restrict laboratory measurements to only those measured between the ages of 0 and 17 years, in order to restrict our analysis to the pediatric samples making up the majority of our data. Although patients with certain diseases, like cystic fibrosis, may be seen at a children's hospital through their adult years, we felt that laboratory measurements collected after the pediatric years were not representative enough to include in our analysis. This filter resulted in 4,086 patients with a distribution of ages between 0 and 17 years, diagnosed with one or more of our set of 12 diseases.
We identified 20 microarray experiments within a 2006 snapshot of the NCBI Gene Expression Omnibus (GEO), an international repository for gene expression data, developed and maintained by the National Library of Medicine [9]. Each experiment studied one of 12 diseases using an experimental design in which normal samples were compared to disease samples. Experiments were manually examined and those lacking normal to disease comparisons as well as those not representative of the clinical diagnosis were excluded. A rank based approach was used for normalization of gene expression due to inconsistencies between microarray platforms as well as inconsistencies in submitted data. The gene expression measurements on each microarray was rank-normalized to numbers between 0 and 1, depending on the relative ranking of the expression level of a gene compared to all the other measured genes on that microarray. The mean rank expression for each gene was calculated for control and disease samples, and the difference in these mean rank-normalized expression levels was calculated and assigned to each gene. The mean rank difference for a gene between control and disease states describes relative change of expression for that gene. GEO data sets (GDS) were merged across similar series of microarray types. For example, GDS559 has the title “Inflammatory bowel disease (HG- U133A)” and GDS560 has the title “Inflammatory bowel disease (HG-U133B)”. Since the A and B chips are from the same series of microarray, these two data sets were combined, and multiple measurements for a gene were averaged. In this example, the group of microarrays labeled by the submitter as “ulcerative colitis” was compared to the group of microarrays labeled as “control” and the difference in mean rank-normalized expression measurements was assigned to the disease ulcerative colitis. Finally, genes missing measurements in 2 or more of the 12 diseases were dropped. This yielded a matrix of 4,956 genes across 12 diseases.
2.2. Finding Biomarkers for Maturation Using Analysis of Variance
Each de-identified laboratory measurement was associated with a measurement and the age of a patient when the test was obtained, as an integer. To find a biomarker representative of maturation, we examined trends within laboratory measurements that corresponded to the age of the patient when the measurement was made. We averaged laboratory measurements across each distinct lab for individual ages within a patient's clinical history to generate a laboratory profile for that patient at that specific age. This resulted in 8,500 distinct laboratory profiles distributed between age 0 and 17 years.
We examined the variance of individual laboratory measurements within each age group binned by year (Mean Square Error, or MSE) and the variance of the laboratory measurement means between distinct age groups (Mean Square Between, or MSB) to determine whether or not maturation had an effect on the laboratory measurement. This was done for each distinct lab test separately by using one-way analysis of variance (ANOVA). In order to show that the null hypothesis was false and that a biomarker was indeed indicative of maturation, we needed to show that the MSB was significantly larger than the MSE. For each distinct laboratory we calculated F, the ratio of MSB to MSE. We also calculate a corresponding p-value along with each F as a measure of significance. The laboratory tests with the smallest p-values were taken to be our initial set of prospective biomarkers for human maturation.
2.3. Using Diseases to Model Maturation
Rather than looking at gene expression at different time points in an organism's life to study the effects of maturation, we examine maturation in the context of disease. We propose that different diseases exhibit different rates of maturation. Given a set of biomarkers indicative of maturation, we consider them to be proxies for aging, at least for the pediatric age group. We use the measurements of the proxy among patients within our set of 12 diseases as a surrogate for distinct disease-specific rates of maturation. For patients with multiple diagnoses, we assume that their laboratory profile at each age is associated with all previously diagnosed diseases. Multiple diagnoses were not common in these pediatric patients, as expected. We then attempt to fit the biomarker's measurements across all ages for a given disease to an exponential decay model. We arbitrarily chose two models we thought from visual inspection fit the data well, namely a linear model and exponential decay model. We found the error between an exponential decay model and the actual data is less than that of a linear model. The values of the parameters for the curve we fit represent the rate at which the biomarker changes, which we use to represent the rate of accelerating or decelerating of maturation that a disease emulates. Each disease has its own parameters, based on the curve fit to the measurements of a biomarker for patients with that disease. We take the value for these parameters and measure the correlation of these values across the 12 diseases with the changes in rank-normalized gene expression measurements (described above) across the same 12 diseases, using Spearman's rank correlation. We recorded Spearman's ρ as well as the p-value of the correlation, with the null hypothesis of no significant correlation, to inform us of the directionality of both the correlation as well as the significance. Literature search was applied to the most significant genes to determine if they were previously shown be correlated with the aging or maturation.
3. Results
3.1. Clinical Biomarkers for Maturation
After reducing and compiling laboratory measurement data to 8,500 patient profiles representing over 4,000 patients at different time points in their clinical history, one-way analysis of variance (ANOVA) was used to elucidate the differences between laboratory measurements at various ages. This was repeated for all lab tests individually. The result of the ANOVA returned prospective biomarkers that could be indicative of increasing age. Four of the top results were as follows: Total Bilirubin (F = 104.54, p-value = 8.58 × 10−279); Total Serum/Plasma Protein (F = 68.66, p-value = 3.15 × 10−193); Mean Corpuscular Volume (F = 65.46, p-value = 1.58 × 10−201); Absolute Lymphocyte (F = 59.47, p-value = 8.57 × 10−181). The F and p-values show a statistically significant connection between increasing age and the prospective biomarkers.
As shown in Figure 1, for three out of the four labs, the wide distribution of measurements specifically between age 0 and 1 years could unduly influence the F and p-value. To examine this influence, an ANOVA was run again on the same data set with all measurements before age 1 excluded. The results show that Total Bilirubin (F = 1.85, p-value = 1.99 × 10−2), Total Serum/Plasma Protein (F = 7.92, p-value = 1.78 × 10−18), and Mean Corpuscular Volume (F=25.90, p-value = 4.57 × 10−85) were influenced more than Absolute Lymphocyte (F = 44.60, p-value = 1.11 × 10−128). This was also verified by applying Bonferroni correction to a pairwise t-test between all age groups. Absolute lymphocyte returned the highest number of significant pairwise comparisons. Based on these results, we selected absolute lymphocyte count as a proxy for maturation and aging.
Figure 1.
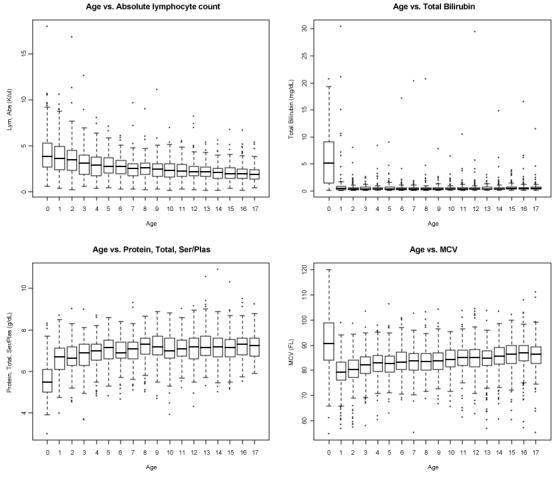
Boxplots showing the distribution of laboratory measurements at different ages. Top left, absolute lymphocyte count; top right, total bilirubin; bottom left, total protein; bottom right, mean corpuscular volume.
3.2. Finding Maturation and Aging Related Genes
There were 4,045 distinct relations between absolute lymphocyte measurements, patient age, and disease identifier. These profiles were distributed across 12 diseases. We used nonlinear least squares to fit the absolute lymphocyte measurements for each disease across all ages to a model of exponential decay:
where α represents the magnitude and λ the rate of decay. We excluded the disease autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, due to a paucity of measurements.
For our purpose, we ignore α and focus on λ as it represents the rate at which the absolute lymphocyte count decreases. Each disease has a distinct λ. A smaller λ, as λ is negative, represents a faster drop in the biomarker. We propose, that if our conjecture holds true and that absolute lymphocyte count is representative of maturation, a change in the biomarker rate of decline could be suggestive of a change in the rate of maturation, so that we can use the same λ to model these differences (Figure 2). We measure the correlation of the set of λ's with the change in rank-normalized expression measurements for each gene, across the same set of diseases. The correlation was done using Spearman's rank correlation. Out of 4956 genes, 53 had p-values less than 0.02 (Table 2)
Figure 2.
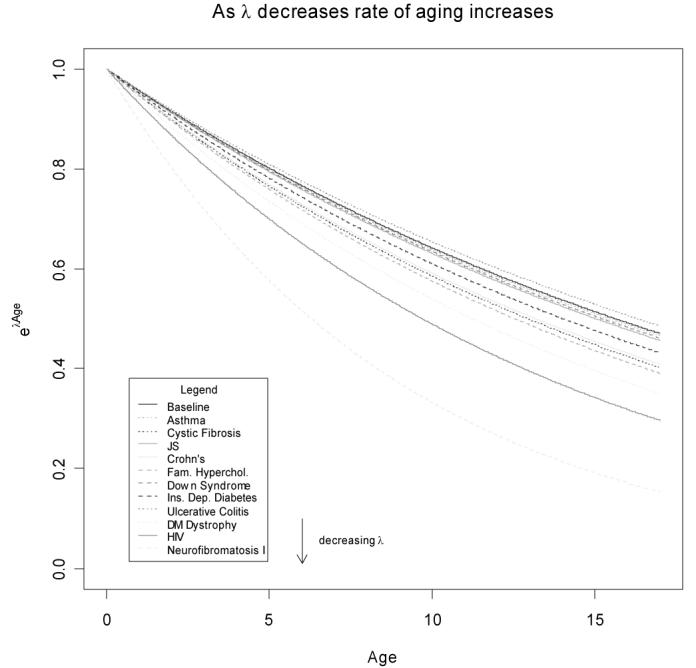
Comparison of different rates of decay across 11 diseases and the baseline.
Table 2.
Genes with the best Spearman's Rank Correlation between λ and expression measurements (p-values < 0.02). Genes in bold are in the GenAge database, known to be involved in aging. Stars indicate genes where evidence exists for involvement in aging, yet not appearing in the GenAge database
| Symbol | Gene name | p-value | Spearman's ρ |
|---|---|---|---|
| PPIC | peptidyl-prolyl isomerase c | 0 | −0.9182 |
| * CYP1B1 | Cytochrome P450, family 1, subfamily B, polypeptide 1 |
0.0025 | −0.8363 |
| TIPARP | TCDD-inducible poly(ADP-ribose) polymerase | 0.0027 | −0.8667 |
| POLD1 |
Polymerase (DNA directed), delta 1, catalytic subunit |
0.0041 | 0.8091 |
| CES2 | carboxylesterase 2 | 0.0044 | −0.8091 |
| * MGMT | O-6-methylguanine-DNA methyltransferase | 0.0048 | 0.8 |
| CENTG2 | centaurin, gamma 2 | 0.0050 | 0.8303 |
| PNN | pinin, desmosome associated protein | 0.0056 | 0.7909 |
| HGF | hepatocyte growth factor | 0.0061 | −0.7909 |
| KLKB1 | kallikrein B, plasma 1 | 0.0061 | −0.7909 |
| POLG | Polymerase (DNA directed), gamma | 0.0061 | −0.7909 |
| GPD1 | glycerol-3-phosphate dehydrogenase 1 | 0.0062 | 0.8182 |
| ICT1 | immature colon carcinoma transcript 1 | 0.0065 | 0.7818 |
| DLG1 | discs, large homolog 1 (Drosophila) | 0.0065 | 0.7818 |
| PUM2 | pumilio homolog 2 (Drosophila) [ | 0.0065 | 0.7818 |
| TNA | C-type lectin domain family 3, member b | 0.0070 | −0.7818 |
| HGD | Homogentisate 1,2-dioxygenase (homogentisate oxidase) |
0.0086 | 0.7636 |
| RBMS2 | RNA binding motif, single stranded interacting protein 2 |
0.0092 | −0.7636 |
| MAPK10 | mitogen-activated protein kinase 10 | 0.0098 | 0.7545 |
| PRC1 | protein regulator of cytokinesis 1 | 0.0108 | −0.8167 |
| MYH10 | myosin, heavy chain 10, non-muscle | 0.0108 | −0.8167 |
| MMP15 | matrix metallopeptidase 15 | 0.0112 | 0.7454 |
| THRB | thyroid hormone receptor, beta | 0.0119 | −0.7454 |
| IL10RA | interleukin 10 receptor, alpha | 0.0126 | 0.7364 |
| GZMM | granzyme M | 0.0126 | 0.7364 |
| RGS9 | regulator of G-protein signalling 9 | 0.0126 | 0.7697 |
| GUCA1A | guanylate cyclase activator 1A | 0.0134 | −0.7364 |
| GFRA2 | GDNF family receptor alpha 2 | 0.0134 | −0.7364 |
| NEUROD1 | neurogenic differentiation 1 | 0.0134 | −0.7364 |
| UEV3 | UEV and lactate/malate dehyrogenase domains | 0.0135 | −0.7782 |
| LRP6 | low density lipoprotein receptor-related protein 6 | 0.0137 | −0.7697 |
| CRYZ | crystallin, zeta | 0.0138 | 0.8 |
| CLCN1 | chloride channel 1 | 0.0142 | 0.7273 |
| EIF3S6 | eukaryotic translation initiation factor 3, subunit 6 48kDa |
0.0142 | 0.7273 |
| MBNL1 | Muscleblind-like (Drosophila) | 0.0148 | 0.7576 |
| EHD1 | EH-domain containing 1 | 0.0148 | 0.7576 |
| PTX3 | pentraxin-related gene | 0.0150 | −0.7273 |
| P2RY2 | purinergic receptor P2Y | 0.0159 | 0.7182 |
| PDK1 | pyruvate dehydrogenase kinase, isozyme 1 | 0.0159 | 0.7182 |
| POU5F1 | POU domain, class 5, transcription factor 1 | 0.0159 | 0.7182 |
| SPOCK2 | sparc/osteonectin, cwcv and kazal-like domains proteoglycan |
0.0159 | 0.7182 |
| ATP1B3 | ATPase, Na+/K+ transporting, beta 3 polypeptide | 0.0168 | −0.7182 |
| CYB5 | Cytochrome b-5 | 0.0168 | −0.7182 |
| CSF1R | colony stimulating factor 1 receptor, | 0.0168 | −0.7182 |
| KIAA0101 | KIAA0101 | 0.0168 | −0.7182 |
| CYP2E1 | Cytochrome P450, family 2, subfamily E, polypeptide 1 |
0.0168 | −0.7182 |
| EXOC7 | exocyst complex component 7 | 0.0171 | 0.7454 |
| SRP68 | signal recognition particle 68kDa | 0.0172 | 0.7833 |
| CETN1 | centrin, EF-hand protein, 1 | 0.0177 | 0.7091 |
| NCL | Nucleolin | 0.0177 | 0.7091 |
| TERF2 | telomeric repeat binding factor 2 | 0.0177 | 0.7091 |
| CSNK1G2 | casein kinase 1, gamma 2 | 0.0197 | 0.7 |
| APOC4 | Apolipoprotein C-IV | 0.0197 | 0.7 |
Spearman's ρ represents how good the gene expression correlates with the changes in λ while also informing us of the directionality of the correlation. A positive Spearman's ρ implies that lower gene expression indicates a faster rate of maturation/aging whereas an increase in gene expression indicates a lower rate of maturation/aging. In contrast, a negative Spearman's ρ implies that a lower gene expression indicates a slower rate of maturation/aging and an increase in gene expression indicates a faster rate of maturation/aging. We investigated the significant genes returned by using literature search. Among the most biologically relevant genes were MGMT, POLD1, POLG and TERF2
4. Discussion
We have shown the ability to use statistical methods to infer biomarkers and predict genes implicated in maturation by integrating clinical laboratory measurements with gene expression measurements. The most significant biomarker from our analysis, absolute lymphocyte count, has not previously been shown to be a biomarker for aging. However, there is evidence that suggests a decrease in lymphocyte function, as well as a decrease in certain lymphocyte cell types, as age increases [10]. We believe that this method of using clinical laboratory measurements can be extended to find trends within complex diseases and other biological phenomena.
We acknowledge the following caveats in the way we proceeded with this research. The clinical laboratory information we used came from patients ranging in age from 0 to 17 years, which only is able to model a certain aspect of aging, namely the process of maturation. We understand that aging revolves around the complete lifespan of an organism and thus our future work will attempt to reproduce these results using a larger data set of clinical laboratory data spanning across more decades of life. We also have speculated that the rate of absolute lymphocyte change is representative of disease-specific rates of maturation, which is currently only conjecture.
There were a handful of diseases that had significantly fewer measurements than other diseases. We excluded the disease autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy but kept the others as there were enough data points to fit an exponential decay curve. Ideally we would gather more measurements from patients having these diseases. However, as these diseases tend to be rarer in comparison to conditions like asthma, they will ultimately be less represented. We also acknowledge that we binned patients with different diseases to identify biomarkers related to aging. However, as clinical data rarely consists of “normal” data we are limited to such analyses. We note, however, that the majority of absolute lymphocyte counts across patients with varying diseases all lay in the normal range. Lastly, we would hope to increase the number of diseases to more than 12 to increase the power of our correlations. We also acknowledge that the sample sizes were not large enough to enable sufficient permutation testing and q-value calculation for our Spearman's rank correlations.
The biological relevance of measuring the correlation between rate of acceleration or deceleration of maturation that a disease emulates to changes in rank-normalized gene expression measurements can be expressed via Spearman's ρ. For example, MGMT, a DNA repair gene, has been implicated in the aging of mice and trials have been underway to determine whether or not transgenic MGMT mice live longer [11]. Given the Spearman's ρ we calculated, we would predict that an increase in MGMT would slow aging and thus increase longevity. Spearman's rank correlation was used to account for the possibilities of non-linear correlations. There remain a plethora of statistical methods which can be applied to examine both linear and non-linear relationships between change in gene expression rank and rates of aging that Spearman's rank correlation may not be capturing.
Out of 53 genes that returned a p-value less than the arbitrary cut-off of 0.02, we found three that were represented in 253 aging-related genes from the curated GenAge database [12]. Using a hypergeometric distribution with 253 known genes involved in aging and the number of genes in the human genome conservatively estimated at 20,000 [13], we find that retrieving 3 aging-related genes out of 53 is statistically significant at p = 0.023. Although this is encouraging, a better validation strategy must be developed. The absence of the 50 remaining genes from our gold standard could be due to GenAge's lack of comprehensiveness as well as may include numerous false positives. Future work revolves around developing better validation strategies as well as increasing sample size to perform more robust analysis including false discovery rates and q-values.
We set out to use a translational approach linking basic science, clinical electronic medical records, and statistical methods to examine phenomena, maturation and aging, which have been and continue to be difficult to study. Our method returned results that were previously known to be aging-related; although that in and of itself is an accomplishment, what is more notable is the fact that we were able to integrate these disparate fields of the study of disease into a cohesive method of research that has been proselytized as being necessary for the advancement of knowledge about human health. These methods can prove to be invaluable in the future of translational research. As more clinical and hospital environments have moved towards EMRs, the amount of patient data available for translational research will only increase. This must be leveraged and used in conjunction with basic science methods in order to explain biological phenomena in humans that cannot be explained by model organisms.
Acknowledgements
The work was supported by grants from the Lucile Packard Foundation for Children's Health, National Library of Medicine (K22 LM008261 and T15 LM007033), National Institute of General Medical Sciences (R01 GM079719), Howard Hughes Medical Institute, and the Pharmaceutical Research and Manufacturers of America Foundation. Stanford Medical School provides the funding for the development of the STRIDE system. Lucile Packard Children's Hospital provides resources and ongoing operational support. We thank Alex Skrenchuk for parallel computer cluster support.
References
- 1.Zerhouni EA. Translational and clinical science--time for a new vision. New Engl J Med. 2005 Oct 13;353(15):1621–3. doi: 10.1056/NEJMsb053723. [DOI] [PubMed] [Google Scholar]
- 2.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–11. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 3.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. P Natl Acad Sci USA. 2004 Jun 22;101(25):9309–14. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006 Jan 19;439(7074):353–7. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 5.Segal E, Friedman N, Koller D, Regev A. A module map showing conditional activity of expression modules in cancer. Nat Genet. 2004 Oct;36(10):1090–8. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- 6.Butte AJ, Kohane IS. Creation and implications of a phenome-genome network. Nat Biotechnol. 2006 Jan;24(1):55–62. doi: 10.1038/nbt1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Payne PR, Johnson SB, Starren JB, Tilson HH, Dowdy D. Breaking the translational barriers: the value of integrating biomedical informatics and translational research. J Invest Med. 2005 May;53(4):192–200. doi: 10.2310/6650.2005.00402. [DOI] [PubMed] [Google Scholar]
- 8.von Zglinicki T, Burkle A, Kirkwood TB. Stress, DNA damage and ageing -- an integrative approach. Exp Gerontol. 2001 Jul;36(7):1049–62. doi: 10.1016/s0531-5565(01)00111-5. [DOI] [PubMed] [Google Scholar]
- 9.Wheeler DL, Church DM, Edgar R, Federhen S, Helmberg W, Madden TL, et al. Database resources of the National Center for Biotechnology Information: update. Nucleic Acids Res. 2004 Jan 1;32(Database issue):D35–40. doi: 10.1093/nar/gkh073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nature Immunol. 2004 Feb;5(2):133–9. doi: 10.1038/ni1033. [DOI] [PubMed] [Google Scholar]
- 11.Anisimov VN. Mutant and genetically modified mice as models for studying the relationship between aging and carcinogenesis. Mech Ageing Dev. 2001 Sep;122(12):1221–55. doi: 10.1016/s0047-6374(01)00262-7. [DOI] [PubMed] [Google Scholar]
- 12.de Magalhaes JP, Toussaint O. GenAge: a genomic and proteomic network map of human ageing. FEBS Lett. 2004 Jul 30;571(1-3):243–7. doi: 10.1016/j.febslet.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Finishing the euchromatic sequence of the human genome. Nature. 2004 Oct 21;431(7011):931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]