

Abstract
To the best of our knowledge, this is the first report on the structure of product-inhibited mammalian peroxidase. Lactoperoxidase is a heme containing an enzyme that catalyzes the inactivation of a wide range of microorganisms. In the presence of hydrogen peroxide, it preferentially converts thiocyanate ion into a toxic hypothiocyanate ion. Samples of bovine lactoperoxidase containing thiocyanate (SCN−) and hypothiocyanate (OSCN−) ions were purified and crystallized. The structure was determined at 2.3-Å resolution and refined to Rcryst and Rfree factors of 0.184 and 0.221, respectively. The determination of structure revealed the presence of an OSCN− ion at the distal heme cavity. The presence of OSCN− ions in crystal samples was also confirmed by chemical and spectroscopic analysis. The OSCN− ion interacts with the heme iron, Gln-105 Nɛ1, His-109 Nɛ2, and a water molecule W96. The sulfur atom of the OSCN− ion forms a hypervalent bond with a nitrogen atom of the pyrrole ring D of the heme moiety at an S–N distance of 2.8 Å. The heme group is covalently bound to the protein through two ester linkages involving carboxylic groups of Glu-258 and Asp-108 and the modified methyl groups of pyrrole rings A and C, respectively. The heme moiety is significantly distorted from planarity, whereas pyrrole rings A, B, C, and D are essentially planar. The iron atom is displaced by ≈0.2 Å from the plane of the heme group toward the proximal site. The substrate channel resembles a long tunnel whose inner walls contain predominantly aromatic residues such as Phe-113, Phe-239, Phe-254, Phe-380, Phe-381, Phe-422, and Pro-424. A phosphorylated Ser-198 was evident at the surface, in the proximity of the calcium-binding channel.
Introduction
Lactoperoxidase (LPO, E.C.1.11.1.7) is a heme containing a ≈68-kDa single-chain glycoprotein. It belongs to mammalian peroxidase superfamily whose other members are myeloperoxidase (MPO), eosinophil peroxidase (EPO), and thyroid peroxidase (TPO). Lactoperoxidase, MPO, and EPO play important roles in strengthening the innate immune system by catalyzing the conversion of halide or thiocyanate ions into potent ions that are toxic to pathogens (1–4). These peroxidases are widely present in mammalian systems. Myeloperoxidase is found in granules of circulating polymorphonuclear neutrophils and monocytes (2,3). Eosinophil peroxidase is an important granule protein of eosinophils (4), whereas LPO is present in exocrine secretions. It provides initial protection against harmful microorganisms (1). Thyroid peroxidase is a different member of the family that plays a critical role in thyroid gland function (5). Lactoperoxidase oxidizes the conversion of halide and pseudohalide ions into potent antimicrobial agents, and thus contributes to the host defense against infections. So far, structural data are available only on MPO (6–8). Crystallization efforts on LPO are underway in several laboratories throughout the world, but these have been largely unsuccessful. Only very recently was the crystal structure of caprine lactoperoxidase (GLPO) reported (9). However, a decade ago, the successful crystallization of buffalo lactoperoxidase was reported (10). The complex catalytic mechanisms involving peroxidases are not fully understood. The structural and chemical basis of preferences for various substrates has also not been explained clearly. Therefore, it is of the utmost importance to determine the crystal structure of LPO from different mammalian species in native and bound forms with various ligands. It is known that many enzymes are inhibited by their products (11). However, no such structure is known for mammalian peroxidases. We report here on the crystal structure of the complex of bovine lactoperoxidase (CLPO) with the CLPO-catalyzed reaction product, the hypothiocyanate (OSCN−) ion. The determination of structure revealed that the OSCN− ion binds to CLPO at the distal heme cavity. It is oriented in such a way that the oxygen atom of the OSCN− ion forms a strong interaction with heme iron. The same oxygen atom is also involved in two other hydrogen bonds with Gln-105 and His-109. The binding of the OSCN− ion to CLPO induces a notable conformational change in the heme group, compared with the native conformation of GLPO (9). The hydrogen-bonded chain of five buried water molecules, W2–W6, is conserved. The shape of the ligand-binding channel is also slightly altered. The presence of phosphorylated Ser-198 and the SCN− ion at the entrance of channel leading to the calcium-binding site produces favorable conditions for calcium absorption.
Materials and Methods
Purification of the protein
Crude CLPO was obtained from Morinaga (Tokyo, Japan). It was dissolved in 50 mM Tris-HCl, pH 8.0, to make a final concentration of 10 mg/mL. The sample was loaded on a CM-Sephadex C-50 (Pharmacia, Uppsala, Sweden) column (10 × 2.5 cm) preequilibrated with 50 mM Tris-HCl, pH 8.0. The elution of LPO was performed with a linear gradient of 0.0–0.5 M NaCl in the same buffer. The protein fractions of Rz value 0.79 or higher were pooled and concentrated, using an Amicon ultrafiltration cell (Millipore, Billerica, MA). The concentrated protein sample was passed through a Sephadex G-100 column (100 × 2 cm), using 50 mM Tris-HCl buffer, pH 8.0. The elution was performed at a flow rate of 6.0 mL/h. The fractions of an Rz value of 0.93 or higher were pooled and dialyzed against deionized water, lyophilized, and stored at 253 K. Protein samples were examined after sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and compared with standard molecular-weight markers. This examination revealed a single band on the SDS-PAGE that corresponded to a molecular mass of ∼68 kDa. It was blotted on a polyvinyl difluoride membrane (Sigma-Aldrich, St. Louis, MO), and was used for N-terminal sequence determination on an automated protein sequencer (PPSQ 21, Shimadzu, Kyoto, Japan). The N-terminal sequence of first 20 residues was obtained as Ser-Trp-Glu-Val-Gly-Cys-Gly-Ala-Pro-Val-Pro-Leu-Val-Thr-Cys-Asp-Glu-Asn-Ser-Pro. It matched with the reported sequence of bovine LPO (12).
Detection of OSCN− ion
To confirm the presence of the OSCN− ion in the present structure, crystals of the CLPO complex with the OSCN− ion were used. First of all, crystals were washed with distilled water and dissolved in normal protein buffer. The solution was incubated with 1 M NaCl for 1 h, and then ultrafiltered using a membrane with a 1-kDa cutoff. The concentration of OSCN− ions in the filtrate was estimated by the oxidation of 2 mol of 5-thio 2-nitrobenzoic acid (NBS) to the disulfide compound 5,5′-dithiobis (2-nitrobenzoic acid) (NBS2) by 1 mol of OSCN− ion (13). The reduction in concentration of NBS after mixing it with the filtrate was determined by the decrease in absorbance at 412 nm, using a spectrophotometer (Perkin-Elmer, Boston, MA), and considering a molar absorption coefficient of 13,600 M−1 cm−1 for NBS (14).
Spectroscopic analysis of CLPO
The spectral changes in heme absorbance at 412 nm were determined for three protein preparations. The first solution was prepared by dissolving protein crystals in distilled water. The second solution was prepared as OSCN− ion free protein by incubating the first solution with 1 M NaCl and ultrafiltering it using a membrane with a 1-kDa cutoff. The third solution was made by adding 1 mM KSCN and 0.2 mM H2O2 in 10 mM potassium phosphate buffer, pH 6.6, to the protein solution (15).
Measurement of activity
The activity assay of the protein sample obtained by dissolving crystals of 0.1 M phosphate buffer, pH 7.0, was performed using the procedure described by Kumar et al. (10). We mixed 3.0 mL of 1 mM 2,2′-azino-bis(3-ethylbenzthiazoline-sulfonic acid, ABTS) in phosphate buffer (0.1 M, pH 6.0) with 0.1 mL of 0.5 mg/mL protein solution containing 0.1% gelatin, to initialize the spectrophotometer (Perkins Elmer). We added 0.1 mL of 3.2 mM hydrogen peroxide to the above solution. Absorbance was measured at 412 nm as a function of the oxidized product of ABTS. There was no change in absorbance. One unit of activity was defined as that amount of enzyme catalyzing the oxidation of 1 μmol of ABTS min−1 at 293 K (molar absorption coefficient, 32,400 M−1 cm−1). The protein samples obtained from the crystals were dialyzed in the presence of 1 M NaCl. These were also used for the activity assay under identical conditions. This sample showed a significant peroxidase activity of 6.0 units/mL.
Phosphoserine detection
The purified sample of LPO was processed on SDS-PAGE, using 10% polyacrylamide gel as described by Laemmli (16). The protein was transferred from SDS-PAGE to a polyvinyl difluoride membrane (Sigma-Aldrich) by Western blotting, and nonspecific binding sites were blocked for 2–4 h at room temperature, using 5% dried milk and Tris-buffered saline (pH 7.4) and 0.1% Tween-20. The membrane was probed overnight at 4°C with primary antiphosphoserine antibody (17) (1:5000) for the serine phosphorylation site in CLPO. A secondary horseradish peroxidase-labeled antibody raised in rabbit (goat anti-rabbit IgG, Jackson Immunochemicals, West Baltimore Pike, West Grove, PA), in combination with an enhanced chemiluminescence detection system (Super Signal West Pico Chemiluminescent Substrate, Thermo Fisher Scientific, Waltham, MA), was used to visualize the primary antibodies.
Crystallization
The purified samples of protein were dissolved in 0.01 M phosphate buffer to a concentration of 25 mg/mL. A reservoir solution containing 0.2 M of ammonium nitrate and 20% (w/v) PEG-3350 was prepared. Six microliters of protein solution were mixed with 6 μL of reservoir solution, to prepare 12 μL of drops for the hanging drop vapor diffusion method. Rectangular dark brown crystals, measuring up to 0.4 × 0.3 × 0.2 mm3, were obtained after 5 days.
X-ray intensity data collection
The X-ray intensity data were collected at 283 K, using a 345 mm diameter MAR Research dtb imaging plate scanner (Marresearch, Norderstedt, Germany) mounted on a Rigaku RU-300 rotating anode x-ray generator (Rigaku) operating at 100 mA and 50 kV. The CuKα radiation was produced using Osmic Blue confocal optics (Rigaku). The data were processed using the programs DENZO and SCALEPACK (18). The space group and unit cell dimensions were P21 and a = 54.5 Å, b = 80.6 Å, c = 77.6 Å, and β = 102.6°. The final data set shows an overall completeness of 99.3%, with an Rsym of 10% for data to 2.3-Å resolution. A summary of final data-collection statistics is given in Table 1.
Table 1.
Data collection and refinement statistics
| Data collection | |
|---|---|
| Space group | P21 |
| Unit cell dimensions | 54.5 |
| a (Å) | 80.6 |
| b (Å) | 77.6 |
| c (Å) | 102.6 |
| β (°) | |
| Number of molecules in the unit cell | 2 |
| Vm (Å3/Da) | 2.5 |
| Solvent content (%) | 52 |
| Resolution range (Å) | 24.5–2.3 |
| Total number of measured reflections | 154,457 |
| Number of unique reflections | 27,641 |
| Completeness of data (%) | 99.3 (99.4) |
| Refinement | |
| Rsym (%) | 10.0 (46.3) |
| I/σ(I) | 9.1 (2.2) |
| Rcryst (%) | 18.4 |
| Rfree (%) (reflections used, 1477) | 22.1 |
| Coordinate error from Luzzati plot | 0.2 |
| Coordinate error from Sigmaa | 0.2 |
| Protein atoms | 4774 |
| Calcium ion | 1 |
| Hypothiocynate (1) atoms | 4 |
| Thiocyanate (1) atoms | 3 |
| NAG residues (8) (N-linked) | 112 |
| MAN residues (2) (N-linked) | 22 |
| Water oxygen atoms | 413 |
| Rmsd in bond lengths (Å) | 0.009 |
| Rmsd in bond angles (°) | 1.7 |
| Rmsd in torsion angles (°) | 18.8 |
| Average B-factor (Å2) | |
| Overall | 28.3 |
| Main chain | 26.3 |
| Side chain | 27.3 |
| Solvent | 38.1 |
| Wilson | 27.1 |
| Residues in most favored regions (%) | 89.4 |
| Residues in additionally allowed regions (%) | 10.2 |
| Residues in generously allowed regions (%) | 0.4 |
| Protein Data Bank code | 3BXI |
Values in parentheses correspond to values in highest resolution shell. Rmsd, root mean-square deviation.
Structure determination and refinement
The structure of the complex of bovine LPO with the hypothiocyanate ion was determined according to the molecular replacement method, using the principle of maximum likelihood in PHASER (19). The native GLPO structure (9) was used as the search model using Protein Data Bank code 2R5L. The rotation and translation search functions were computed using the reflections in a resolution range of 12.0–4.0 Å. It yielded a clear solution with a distinct peak of minimum R value and maximum correlation coefficient. The molecular packing in the unit cell, calculated according to this solution, did not produce unfavorable short contacts. The coordinates were transformed using PHASER, and were subjected to 25 cycles of rigid body refinement with REFMAC (20) from the CCP4i version 6.0.0 program package (21). After the first round of refinement, the Rcryst and Rfree factors reduced to 0.326 and 0.412, respectively (5% of the reflections were used for the calculations of Rfree; these reflections were not included in the refinement). The initial Fourier (|2Fo − Fc|) and difference Fourier (|Fo − Fc|) maps showed good electron densities for the heme group. The coordinates of the heme group were included in further rounds of refinement that were performed with the intermittent manual model building of the protein, using |2Fo − Fc| Fourier and |Fo − Fc| difference Fourier maps, with graphics programs O (22) and coot (23), on a Silicon Graphics O2 Workstation (Sunnyvale, CA). At the end of this refinement, the Rcryst and Rfree factors converged to 0.248 and 0.278, respectively. The difference Fourier map calculated at this stage revealed interpretable electron densities for glycan chains at four sites, with two N-acetylglucosamine (NAG) residues and one mannose (MAN) at Asn-95, two NAG residues at Asn-205, two NAG + one MAN residues at Asn-241, and two NAG residues at Asn-332. Excellent electron densities were also evident at the distal heme cavity for the OSCN− ion (Fig. 1 a) and SCN− ion at the surface in the proximity of Arg-202 (Fig. 1 b), as well as for the calcium ion. A further detailed examination of the electron density maps revealed additional electron densities for five nitrate ions. All of these were included in subsequent rounds of refinement. Further difference |Fo – Fc| Fourier maps indicated additional electron densities for the phosphorylation of Ser-198. The positions of 413 water oxygen atoms were determined using the usual criteria of correctness. The refinement finally converged with Rcryst and Rfree values of 0.184 and 0.221, respectively. The final refinement statistics are included in Table 1. The refined atomic coordinates of this complex were deposited in the Protein Data Bank, with an accession code 3BXI.
Figure 1.
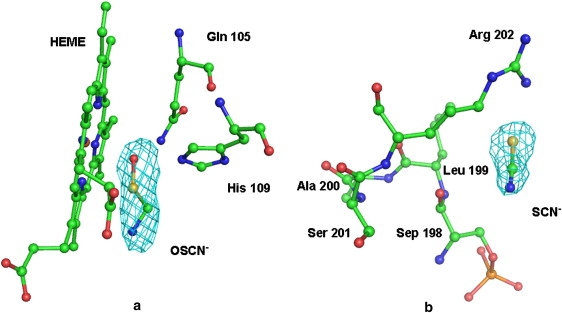
(a) OSCN− fitted into 2.5 σ (|Fo − Fc|) electron density. The heme group and distal Gln-105 and His-109 are also shown. (b) SCN− fitted into 2.5 σ (|Fo − Fc|) electron density. Residues in the vicinity are also indicated.
Results
OSCN− ion in crystals
Oxidation of NBS to NBS2 in the filtrate obtained from crystal solution confirmed the presence of the OSCN− ion in the crystals. The concentration of OSCN− ion was estimated to be 0.1 mM. Because LPO contains a heme group at the active center, the change in absorption of heme with regard to ligand-binding is the indication of interactions involving heme moiety. Spectral studies were performed of CLPO crystal solution, OSCN− ion free solution, and crystal solution containing OSCN− ions. The heme absorption at 412 nm showed identical values with the spectral curves, superimposing well for the samples of crystal solution and protein solution with OSCN− ions. However, the measurements of OSCN− ion free samples showed different values, and the absorption curve did not superimpose on the spectral curves obtained for the OSCN− containing protein solutions. It confirmed the presence of OSCN− ions in the crystals.
Quality of the model
The final model (summarized in Table 1) comprises amino-acid residues from 1–595, corresponding to the complete polypeptide chain of LPO. The entire protein molecule is well-defined in the observed electron density. The structure consists of a covalently linked heme group, one calcium, five nitrate, one thiocyanate, and one hypothiocyanate ions, four glycan chains, and 413 water oxygen atoms. The overall mean B-factor of CLPO is 28.3 Å2, comparable to those observed for other well-refined protein structures (7–9). A Ramachandran plot (24) of the polypeptide chain torsion angles with various allowed regions, as defined in the program PROCHECK (25), shows that 89.4% of residues are present in the most favored regions, whereas 10.2% of residues were found in the additionally allowed regions. Only two residues, Thr-169 and Tyr-172, occur in the generously allowed regions. Both residues belong to a short loop that is involved in an extraordinary intramolecular disulfide bridge between Cys-6 and Cys-167. This intramolecular S-S bond is not present in MPO, because residue Cys-6 is absent.
Overall structure
The determination of crystal structure shows that a hypothiocyanate ion is present at the distal heme cavity. The presence of OSCN− ions in the crystals was also indicated by spectroscopic measurements. The determination of structure also revealed an SCN− ion at the surface, in the proximity of Arg-202. The folding of the polypeptide chain of CLPO is essentially similar to that of GLPO (9). The root mean-square (rms) shift for the Cα traces of two proteins is 0.6 Å. The molecular organization, together with four glycan chains, the heme group, and a bound OSCN− ion, is shown as a ribbon diagram (Fig. 2). The structure shows that the heme group is sandwiched between two antiparallel helices, H2 and H8. The perpendicular distance between the backbone atoms of helices H2 and H8 is increased by 0.4 Å in the complex with the OSCN− ion, compared with the native structure of CLPO. The central core of the molecule contains five long α-helices (H2, H5, H6, H8, and H12), with a covalently attached heme group. The first 75 residues of the N-terminus adopt a flexible nonrepetitive structure. Because of the flexible nature of the N-terminal segment, the rms shift for the Cα atoms of the first 75 residues of CLPO and GLPO is 1.3 Å, i.e., considerably higher than the overall rms shift calculated using all the residues. As seen in Fig. 2, the heme group lies below the triangle formed by helices H2, H5, and H6. A narrow opening is evident from the heme group to the surface of the protein. One side of this channel is composed of helices H8, H11, H12, and H2a, whereas the other side contains helices H2, H3, H4, and H9. The helix H2a is not present in MPO. This channel is used for the diffusion of substrates into the protein core where the heme is located. This is the only opening evident in the structure, insofar as all other sides are tightly packed. Helix H2a (residues 123–132) is a unique feature of LPO and is not possible in the structure of MPO, because the corresponding protein stretch of six residues is absent in the sequence of MPO (6–8). Because this helix is located at the entrance of the substrate-binding channel, it contributes to the overall shaping of the substrate channel in LPO. As a result, the shape of the substrate channel in LPO is considerably different from that of MPO (6–8). The structure of LPO also shows that the lower half of the molecule is packed with short horizontal helices, whereas the upper half contains vertical helices.
Figure 2.
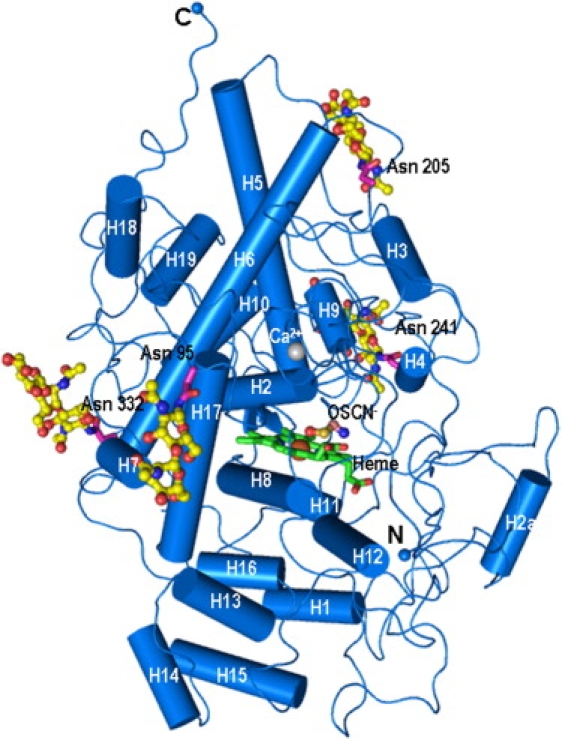
Schematic diagram of bovine lactoperoxidase complex with OSCN− ion, indicating positions of α-helices (cylinders 1–19), heme moiety (green online and medium gray in print), hypothiocyanate ion, and glycan chains (yellow online and light gray in print). Side chains of Asn-95, Asn-205, Asn-241, and Asn-332, covalently linked to glycan chains, are also shown. Figure was drawn using Pymol (39).
Binding of OSCN−
Lactoperoxidase catalyzes the reaction of hydrogen peroxide with thiocyanate, and produces the OSCN− ion, which is toxic to various pathogens. The lactoperoxidase system (LPO-H2O2) is a natural antimicrobial system present in mammalian secretions, including milk. Thiocyanate (SCN−) ions are also present in the milk as a result of the hydrolysis of glucosinolates, as well as from the detoxification of cynogenic glucosides (26). The H2O2 is either supplied exogenously or generated by lactic-acid bacteria, which are also present in the milk (27). In the process of the reaction of the LPO-system, H2O2 is consumed. The reduction in the concentration of H2O2 in milk results in stopping the conversion of SCN− to OSCN−. Therefore, samples of milk generally contain both SCN− and OSCN− ions. Alternately, OSCN− ions could accumulate if the reactive components or pathogens are not present in the milk. In this case, we observed that an OSCN− ion binds to LPO at the distal site, thereby inhibiting its catalytic activity of the enzyme. One bound SCN− ion was also observed at the surface of the protein in the vicinity of helix H3. The determination of structure revealed the position of the OSCN− ion at the distal heme cavity, between the heme moiety and distal His-109 (Fig. 3). The OSCN− ion displaced at least three water molecules from the substrate-binding site, including W1, which interacts with the Fe3+ ion and His-109 Nɛ2 in the native structure (9). The OSCN− ion interacts extensively at this site, indicating it as the preferred binding site for the OSCN− ion. The oxygen atom of the OSCN− ion forms three hydrogen bonds: one with His-109 Nɛ2 (2.7 Å), a second with heme iron (2.7 Å), and the third with Gln-105 Nɛ2 (3.1 Å). Both nitrogen and sulfur atoms of the OSCN− ion form hydrogen bonds with W96 (S-W96 = 2.8 Å, and N-W96 = 3.3 Å). Yet another important interaction was observed between the S atom of the OSCN− ion and the nitrogen atom of the pyrrole ring D of the heme moiety (S-ND = 2.8 Å). This kind of hypervalent bond between sulfur and nitrogen atoms was reported elsewhere (28,29). The binding of the OSCN− ion to the protein and the heme moiety disturbed an important interaction between Gln105 Nɛ2 and the heme nitrogen atom NA, as observed in the native structure, and stated to be one of the characteristic features of LPO structure (9). In our complex under discussion, this distance increased by 0.6 Å, to 3.7 Å. This shows that the binding of the OSCN− ion at the distal heme cavity, and the resulting interactions between the OSCN− ion and the protein heme moiety, displaced three water molecules and several protein/water interactions with the heme moiety. This appears to be the site of inhibition in LPO, and the binding characteristics of the OSCN− ion to LPO display all the features of a competitive inhibitor. Thus the complex of LPO with the OSCN− ion is a complex between the enzyme and the inhibitor. Such types of product inhibitions were observed to occur commonly in those enzymes that form highly reactive intermediates (11). An SCN− ion is also evident in the structure on the surface of the protein in the vicinity of helix H3. It forms an ion pair with the side chain of Arg-202. This site was reported to be occupied by an iodide ion in the native structure of GLPO (9). In contrast, the corresponding site is not formed in MPO, because it contains Asn-186 in place of Arg-202. The binding of the SCN− ion at this position in LPO may be structurally relevant, because this site of SCN− ion-binding is in close proximity to the phosphorylated Ser-198. The presence of the SCN− ion presumably neutralizes the positive charge on the Arg-202 side chain, which might be helpful for the intake of calcium ions. These features are in striking contrast to those present in the structure of MPO (6–8).
Figure 3.

Electron density (|Fo − Fc|) for hypothiocyanate ion bound in distal heme cavity, contoured at 2.5 σ level. Residues Gln-105 and His-109 on distal site and His-351 on proximal site are also shown. Positions of α-helices H2 and H8 with respect to heme group are also indicated. Two opposite orientations (a and b) are shown for clarity of interactions with OSCN− ion.
Heme environment
The heme group is covalently bonded to protein through modified methyl groups on pyrrole rings A and C and the carboxyl groups of Glu-258 and Asp-108, respectively (Fig. 4). The heme moiety is deeply buried in the protein molecule, and also forms a number of noncovalent interactions with the protein atoms. The four pyrrole rings A, B, C, and D of protoporphyrin IX are planar, and the iron atom is shifted by ∼0.2 Å toward the proximal side from the plane made by the nitrogen atoms of four pyrrole rings. This shift is twice the corresponding shift (0.1 Å) observed in the native GLPO (9). The plane calculated using four pyrrole nitrogen atoms and the heme iron atom shows that it forms a bowl-like structure, with the iron atom at the bottom of the bowl when viewed from the distal side. The lines connecting the nitrogen atoms of pyrrole rings A and C, and B and D, are inclined toward the iron atom, with angles ∠NA–Fe–NC and ∠NB–Fe–ND of 167.5° and 165.0°, respectively. The Nɛ2 atom of His-351 on the proximal site is coordinated to the iron atom at a distance of 2.1 Å, whereas the Nδ1 of this residue is hydrogen-bonded to the amide carbonyl oxygen atom of Asn-437.
Figure 4.
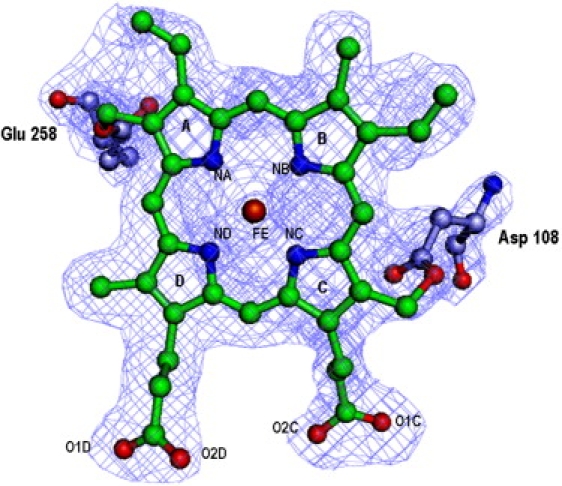
Electron density (|2Fo − Fc|) for heme moiety contoured at 1.2 σ level. Electron densities for residues Asp-108 and Glu-258, covalently linked to the heme group, are also shown. Iron, oxygen, and nitrogen atoms of the heme group are labeled. The four pyrrole rings are also labeled.
Distal heme cavity and substrate channel
The distal heme cavity plays a pivotal role in determining the catalytic activity of LPO. This cavity is connected to the surface through a narrow channel which is supported by the β, γ, and δ carbon atoms of Arg-255 on one side, whereas side chains of Phe-113, Pro-234, Phe-239, Phe-254, Phe-380, Phe-381, Phe-422, and Pro-424 form the remaining sides (Fig. 5). A notable feature of this structure pertains to the residue Phe-254 in CLPO, which is a part of the channel wall. It increases the hydrophobicity of the channel considerably. The corresponding residue is threonine in MPO, whereas both EPO and TPO have a glycine residue at the corresponding position. The distal heme cavity is filled by the side chains of residues Gln-105, His-109, and Arg-255. A chain of five hydrogen-bonded buried water molecules and a side chain of His-266 extend from the distal His-109 to the surface of the protein molecule, presumably to conduct protons away from the distal histidine, and ensuring that Nɛ2 is free to accept a proton from hydrogen peroxide.
Figure 5.
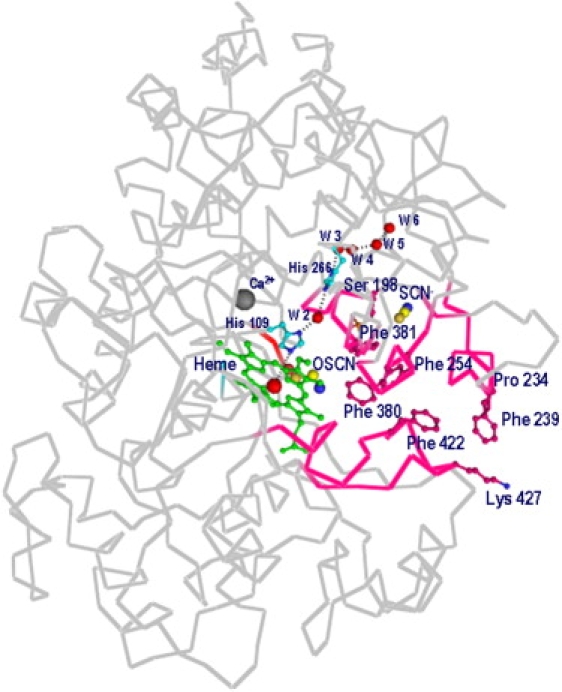
View of substrate channel connecting distal heme cavity to surface of protein. Positions of calcium and OSCN− ions, and of His-109, W2–W6, and His-266, are shown. Position of SCN− ion is also indicated. Channel walls contain predominantly hydrophobic residues, which are also indicated.
Carbohydrate structure
The amino-acid sequence of bovine LPO reveals four potential N-glycosylation sites, with Asn-X-Ser/Thr sequence motifs. These are at Asn-95, Asn-205, Asn-241, and Asn-332 (12). The crystallographic analysis confirmed the glycan attachments at all four sites, and the remarkably good quality of electron densities at these sites enabled us to model at least 10 sugar residues. The electron densities are present for two N-acetylglucosamine (NAG1 and NAG2) residues and a β-1,4-mannose (MAN3) residue at Asn-95, for a disaccharide comprising two NAG (NAG4 and NAG5) residues at Asn-205, for a trisaccharide comprising two NAG (NAG6 and NAG7) residues, and a β-1,4-mannose (MAN8) residue at Asn-241, whereas a disaccharide with two NAG (NAG9 and NAG10) residues is attached to Asn-332. In MPO, out of five potential glycosylation sites, only three were found with attached glycan chains (6,7). The three glycosylated sites in MPO are identical to the second, third, and fourth sites in LPO. The glycosylation at Asn-95 is unique to LPO. Asn-95 is located in a highly positive environment. In addition to the covalent linkage with Asn-95, this carbohydrate moiety forms two hydrogen bonds, with Arg-504 NH1 and Gln-568 Oɛ1 involving acetamido oxygen atoms O7 of carbohydrate residues 1 and 2, respectively. Asn-95 is part of an important helix H2 in the distal side, and supports the heme environment. A well-anchored glycan chain at Asn-95 contributes to the stability of helix H2. This is an important difference between the structures of LPO and MPO that might have some bearing on the properties of the heme group and on the binding characteristics at the distal site.
Discussion
The amino-acid sequences of four mammalian peroxidases indicate that the mode of covalent binding to the heme moiety with two covalent linkages in LPO is similar to those of EPO and TPO, whereas MPO forms three covalent bonds with the heme group, including two identical linkages with those of LPO, EPO, and TPO. This is because residues Glu-258 and Asp-108 form covalent bonds with the heme moiety, and are conserved in all four mammalian peroxidases. A third covalent bond was observed only in MPO through sulfonium ion linkage between the sulfur atom of Met-243 and the terminal β-carbon of the vinyl group on pyrrole ring A. The other three mammalian peroxidases lack the methionine residue at the corresponding positions. The residues at these positions in LPO, EPO, and TPO are Gln-259, Thr-242, and Val-361, respectively. The structure of LPO (9) shows that Gln-259 is involved in an extensive hydrogen-bonded network with Gln-102, Gln-105, and Leu-261 in the back of the distal heme cavity. Gln-105 Oɛ1 forms two hydrogen bonds with Gln-259 Nɛ2 and Gln-102 Nɛ2, whereas Gln-105 Nɛ2 is involved in three hydrogen bonds: two as a donor with Glu-258ɛ1 (3.1 Å) and the heme pyrrole ring A nitrogen atom (3.3 Å), and the third as an acceptor with water molecule W1 (2.8 Å). The interaction between Gln-105 Nɛ2 and pyrrole nitrogen atom NA is recognized as a feature unique to the LPO structure, because the corresponding distance in MPO is 3.9 Å (6,7). In this regard, EPO with Thr-242 and TPO with Val-361 at the position of Gln-259 are expected to induce different possibilities for interactions because of the conserved glutamine residue corresponding to Gln-105 in LPO. Therefore, protein-heme interactions are not identical in the four mammalian peroxidases, because the residues corresponding to Met-243 in MPO, LPO, EPO, and TPO have different residues, i.e., Gln-259, Thr-242, and Val-361, respectively.
In structure under discussion, the position of water molecule W1 in the distal heme cavity is occupied by the oxygen atom of the OSCN− ion. It interacts with Gln-105, His-109, and the heme iron (Fig. 3). The OSCN− ion also forms a hypervalent bond, with an S-N bond distance of 2.8 Å between the sulfur atom of the OSCN− ion and the nitrogen atom ND of the heme group (28,29). This indicates tight binding between the enzyme and the OSCN− ion, suggesting that it is a potent inhibitor. The OSCN− ion is a highly reactive intermediate, and accumulates in the absence of other interactable reactive components. As a result, it forms a stable complex with LPO. Although its precise physiological significance is not clearly understood, it was observed in a number of enzymes, particularly in the case of oxidases, where highly reactive products are formed (11). Because the OSCN− ion is tightly held between the heme group from one side and His-109, water molecule W96, Gln-105, and Arg-255 from the other sides, it produces notable perturbations in the positions of surrounding protein residues and heme moiety. A noteworthy observation pertains to the loss of an important interaction between Gln-105 Nɛ2 and the heme nitrogen atom NA, as observed in the native LPO structure (9). Other structures with small ions at the distal site, such as Cl− (2QRB), I− (2IPS), and PO42− (2EFB), also show interactions between Gln-105 and the heme moiety, whereas larger ligands such as salicylhydroxamic acid (2QPK) and acetyl salicylic acid (2QQT) show distances between Gln-105ɛ2 and NA higher than 3.6 Å, indicating the absence of interactions between the side chain of Gln-105 and the heme group. Thus the binding of larger ligands in LPO converts it to a form similar to that observed in native MPO (6,7). The calculations also show that the heme moiety in the structure of LPO with the OSCN− ion is considerably more planar, compared with those observed in the structures of LPO with Cl− (2QRB), I− (2IPS), and PO42− (2EFB). The heme planarity observed in the present structure is comparable to those observed in complexes with salicylhydroxamic acid (2QPK) and acetyl salicylic acid (2QQT). These observations indicate that a tightly packed ligand with significant interactions at the distal substrate binding site increases the planarity of the heme moiety.
The structure of the complex of MPO with the SCN− ion (8) shows that the nitrogen atom is hydrogen-bonded to the heme iron, whereas the sulfur atom is placed farther away from the heme moiety. The shortest distance between any nitrogen atom of the heme group and the sulfur atom of the SCN− ion is more than 4.5 Å. Although a comparable structure of LPO with the SCN− ion at the distal site is not yet available, the complex of the OSCN− ion with LPO indicates that the disposition of such a ligand in LPO and MPO may differ significantly (Fig. 6). This may be attributable to a significantly different local structural environment in the proximity of the substrate-binding site in LPO, with an extensive hydrogen-bonded network involving residues Gln-259, Gln-105, Gln-102, and Leu-261 (Fig. 7). In contrast, the orientation of the side chain of Gln-91 in MPO is very different from the side chain of the corresponding Gln-105 in LPO, because the residues corresponding to Gln-259 and Leu-261 in LPO are Met-243 and Glu-245, respectively, in MPO, and are unsuitable for generating a hydrogen-bonded network similar to that observed in LPO. Apart from this, in place of the covalent bond formed between Met-243 and the heme moiety in MPO, a hydrogen bond involving Gln-105 Nɛ2 and a pyrrole ring A nitrogen atom was observed in LPO. Some other interactions (Fig. 8) involving pyrrole ring D propionate were also found to be considerably different in LPO and MPO. Yet another striking difference pertains to the conformation of the loop 421–430, in the proximity of the substrate-binding site and the heme moiety. The corresponding loop is short and adopts a very different conformation in MPO (9). These significant differences between the structures of MPO and LPO in the proximity of the heme moiety and the distal site may be responsible for the selection of different substrates. The perturbations in planarities of heme moieties seem to result in different values of the reduction potentials of various intermediates of LPO and MPO (30). The values of standard reduction potentials for compound I (ferric state) were found to be 1.16 V for MPO, 1.10 V for EPO, and 1.09 V for LPO (30,31). The corresponding value for TPO is not known.
Figure 6.
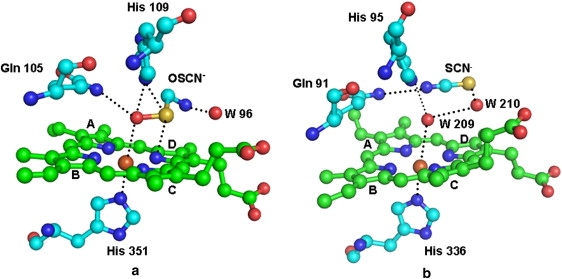
Relative positions of (a) OSCN− ion in LPO and (b) SCN− ion in MPO. The bound water molecule (W209) is present in MPO structure. Corresponding water molecule is displaced by OSCN− ion in LPO. The OSCN− ion seems to bind more preferentially than does the SCN− ion.
Figure 7.
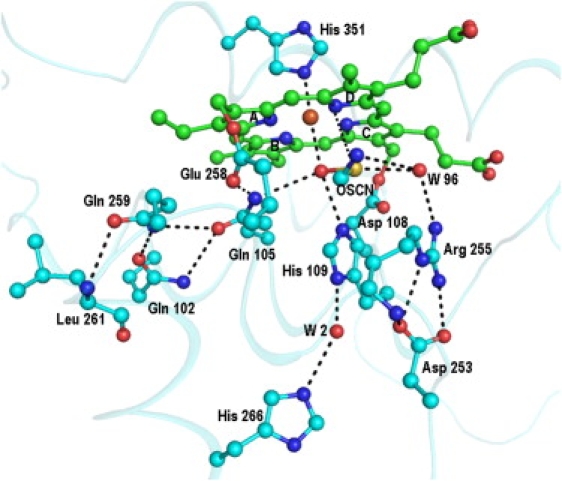
Interactions involving OSCN− ion and heme group in distal heme cavity of LPO, as indicated by dashed line.
Figure 8.
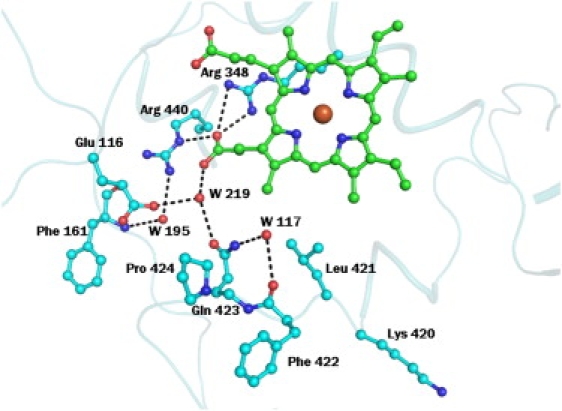
Some interactions are shown between protein residues in proximity of heme group and with heme group, which are unique in LPO.
In contrast to mammalian peroxidases, two binding sites in plant peroxidases were detected: one is close to the γ-heme edge (32–34), whereas the more common second binding site is located near the δ-heme edge in the cavity that connects the distal side of the heme to the surface of the protein (35–38). The latter is similar to the binding site observed in mammalian peroxidases. The environments of distal histidine in the plant peroxidases differ from those of mammalian peroxidases, but the mechanisms of action are similar.
Acknowledgments
The authors acknowledge financial assistance from the Department of Science and Technology (New Delhi, India). A.K.S. thanks the Council of Scientific Industrial Research (New Delhi, India) for a Senior Research Fellowship. T.P.S. thanks the Department of Biotechnology, Ministry of Science and Technology (New Delhi, India) for a Distinguished Biotechnologist Award.
References
- 1.Langbakk B., Flatmark T. Lactoperoxidase from human colostrum. Biochem. J. 1989;259:627–631. doi: 10.1042/bj2590627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrison J.E., Schultz J. Studies on the chlorinating activity of myeloperoxidase. J. Biol. Chem. 1976;251:1371–1374. [PubMed] [Google Scholar]
- 3.Wever R., Kast W.M., Kasinoedin J.H., Boelens R. The peroxidation of thiocyanate catalyzed by myeloperoxidase and lactoperoxidase. Biochim. Biophys. Acta. 1982;709:212–219. doi: 10.1016/0167-4838(82)90463-0. [DOI] [PubMed] [Google Scholar]
- 4.Bolscher B.G., Plat H., Wever R. Some properties of human eosinophil peroxidase, a comparison with other peroxidases. Biochim. Biophys. Acta. 1984;784:177–186. doi: 10.1016/0167-4838(84)90125-0. [DOI] [PubMed] [Google Scholar]
- 5.Kimura S., Kotani T., McBride O.W., Umeki K., Hirai K. Human thyroid peroxidase: complete cDNA and protein sequence, chromosome mapping, and identification of two alternately spliced mRNAs. Proc. Natl. Acad. Sci. USA. 1987;84:5555–5559. doi: 10.1073/pnas.84.16.5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeng J., Fenna R.E. X-ray crystal structure of canine myeloperoxidase at 3 Ǻ resolution. J. Mol. Biol. 1992;226:185–207. doi: 10.1016/0022-2836(92)90133-5. [DOI] [PubMed] [Google Scholar]
- 7.Fiedler T.J., Davey C.A., Fenna R.E. X-ray crystal structure and characterization of halide-binding sites of human myeloperoxidase at 1.8 Å resolution. J. Biol. Chem. 2000;275:11964–11971. doi: 10.1074/jbc.275.16.11964. [DOI] [PubMed] [Google Scholar]
- 8.Blair-Johnson M., Fiedler T., Fenna R. Human myeloperoxidase: structure of a cyanide complex and its interaction with bromide and thiocyanate substrates at 1.9 Å resolution. Biochemistry. 2001;40:13990–13997. doi: 10.1021/bi0111808. [DOI] [PubMed] [Google Scholar]
- 9.Singh A.K., Singh N., Sharma S., Singh S.B., Kaur P. Crystal structure of lactoperoxidase at 2.4 Å resolution. J. Mol. Biol. 2008;376:1060–1075. doi: 10.1016/j.jmb.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Kumar R., Bhatia K.L., Dauter Z., Betzel C., Singh T.P. Purification, crystallization and preliminary x-ray crystallographic analysis of lactoperoxidase from buffalo milk. Acta Crystallogr. 1995;D51:1094–1096. doi: 10.1107/S0907444995004422. [DOI] [PubMed] [Google Scholar]
- 11.Frieden E., Walter C. Prevalence and significance of the product inhibition of enzymes. Nature. 1963;198:834–837. doi: 10.1038/198834a0. [DOI] [PubMed] [Google Scholar]
- 12.Cals M.M., Mailliart P., Brignon G., Anglade P., Dumas B.R. Primary structure of bovine lactoperoxidase, a fourth member of a mammalian heme peroxidase family. Eur. J. Biochem. 1991;198:733–739. doi: 10.1111/j.1432-1033.1991.tb16073.x. [DOI] [PubMed] [Google Scholar]
- 13.Aune T.M., Thomas E.L. Accumulation of hypothiocyanate ion during peroxidase-catalyzed oxidation of thiocyanate ion. Eur. J. Biochem. 1977;80:209–214. doi: 10.1111/j.1432-1033.1977.tb11873.x. [DOI] [PubMed] [Google Scholar]
- 14.Ellman G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 15.Aune T.M., Thomas E.L. Oxidation of protein sulfhydryls by products of peroxidase-catalyzed oxidation of thiocyanate ion. Biochemistry. 1978;17:1005–1010. doi: 10.1021/bi00599a010. [DOI] [PubMed] [Google Scholar]
- 16.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 17.Colyer J., Wang J.H. Dependence of cardiac sarcoplasmic reticulum calcium pump activity on the phosphorylation status of phospholamban. J. Biol. Chem. 1991;266:17486–17493. [PubMed] [Google Scholar]
- 18.Otwinowski Z., Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 19.McCoy A.J., Grosse-Kunstleve R.W., Storoni L.C., Read R.J. Likelihood-enhanced fast translation functions. Acta Crystallogr. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 20.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 21.CCP4. 1994. Collaborative Computational Project. Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D50:760–763. [DOI] [PubMed]
- 22.Jones T.A., Zou J., Cowan S.W., Kjeldgaard M. Improved methods for building models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–118. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 23.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Ramachandran G.N., Sasisekaran V. Conformation of polypeptides and proteins. Adv. Protein Chem. 1968;23:283–438. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 25.Laskowski R.A., Macarthur M., Moss D., Thornton J. PROCHECK: a program to check stereo chemical quality of protein structures. J. Appl. Cryst. 1993;26:283–290. [Google Scholar]
- 26.Reiter B., Harnulv G. Lactoperoxidase antimicrobial system: natural occurrence, biological functions and practical applications. J. Food Prot. 1984;47:724–732. doi: 10.4315/0362-028X-47.9.724. [DOI] [PubMed] [Google Scholar]
- 27.Bjorck L. Antimicrobial effect of the lactoperoxidase system on psychotropic bacteria in milk. J. Dairy Res. 1978;45:109–118. doi: 10.1017/s0022029900016253. [DOI] [PubMed] [Google Scholar]
- 28.Kuti M., Rabai J., Kapovits I., Jalsovszky I., Argay G. Transannular sulfur-nitrogen interaction in 1,5-thiazocine derivatives: an x-ray study. J. Mol. Struct. 1996;382:1–11. [Google Scholar]
- 29.Morokuma K., Hanamura M., Akiba K. Role of sulfur d orbitals in the S-N bond stability of ammonioalkylsulfuranes. Chem. Lett. (Jpn.) 1984;13:1556–1560. [Google Scholar]
- 30.Furtmuller P.G., Arnhold J., Jantschko W., Zederbauer M., Jakopitsch C. Standard reduction potentials of all couples of the peroxidase cycle of lactoperoxidase. J. Inorg. Biochem. 2005;99:1220–1229. doi: 10.1016/j.jinorgbio.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 31.Arnhold J., Furtmuller P.G., Regelsberger G., Obinger C. Redox properties of the couple compound I/native enzyme of myeloperoxidase and eosinophil peroxidase. Eur. J. Biochem. 2001;268:5142–5148. doi: 10.1046/j.0014-2956.2001.02449.x. [DOI] [PubMed] [Google Scholar]
- 32.Sharp K.H., Mewies M., Moody P.C., Raven E.L. Crystal structure of the ascorbate peroxidase-ascorbate complex. Nat. Struct. Biol. 2003;10:303–307. doi: 10.1038/nsb913. [DOI] [PubMed] [Google Scholar]
- 33.Sundaramoorthy M., Kishi K., Gold M.H., Poulos T.L. The crystal structure of manganese peroxidase from Phanerochaete chrysosporium at 2.06 Å resolution. J. Biol. Chem. 1994;269:32759–32767. [PubMed] [Google Scholar]
- 34.Poulos T.L. The Janus nature of heme. Nat. Prod. Rep. 2007;24:504–510. doi: 10.1039/b604195g. [DOI] [PubMed] [Google Scholar]
- 35.Metcalfe C., Macdonald I.K., Murphy E.J., Brown K.A., Raven E.L. The tuberculosis prodrug isoniazid bound to activating peroxidases. J. Biol. Chem. 2008;283:6193–6200. doi: 10.1074/jbc.M707412200. [DOI] [PubMed] [Google Scholar]
- 36.Kunishima N., Fukuyama K., Matsubara H., Hatanaka H., Shibano Y. Crystal structure of the fungal peroxidase from Arthromyces ramosus at 1.9 Å resolution. Structural comparisons with the lignin and cytochrome c peroxidases. J. Mol. Biol. 1994;235:331–344. doi: 10.1016/s0022-2836(05)80037-3. [DOI] [PubMed] [Google Scholar]
- 37.Poulos T.L., Freer S.T., Alden R.A., Edwards S.L., Skogland U. The crystal structure of cytochrome c peroxidase. J. Biol. Chem. 1980;255:575–580. [PubMed] [Google Scholar]
- 38.Henriksen A., Schuller D.J., Meno K., Welinder K.G., Smith A.T. Structural interactions between horseradish peroxidase C and the substrate benzhydroxamic acid determined by x-ray crystallography. Biochemistry. 1998;37:8054–8060. doi: 10.1021/bi980234j. [DOI] [PubMed] [Google Scholar]
- 39.DeLano W.L. DeLano Scientific; San Carlos, CA: 2002. The PyMol Molecular Graphics System.http://www.pymol.org [Google Scholar]