

Abstract
The link between estrogen and the development and proliferation of breast cancer is well documented. Estrogen stimulates growth and inhibits apoptosis through estrogen receptor-mediated mechanisms in many cell types. Interestingly, there is strong evidence that estrogen induces apoptosis in breast cancer and other cell types. Forty years ago, before the development of tamoxifen, high-dose estrogen was used to induce tumor regression of hormone-dependent breast cancer in post-menopausal women. While the mechanisms by which estrogen induces apoptosis were not completely known, recent evidence from our laboratory and others demonstrates the involvement of the extrinsic (Fas/FasL) and the intrinsic (mitochondria) pathways in this process. We discuss the different apoptotic signaling pathways involved in E2 (17β-estradiol)-induced apoptosis, including the intrinsic and extrinsic apoptosis pathways, the NF-κB (nuclear factor-kappa-B)-mediated survival pathway as well as the PI3K (phosphoinositide 3-kinase)/Akt signaling pathway. Breast cancer cells can also be sensitized to estrogen-induced apoptosis through suppression of glutathione by BSO (L-buthionine sulfoximine). This finding has implications for the control of breast cancer with low-dose estrogen and other targeted therapeutic drugs.
Introduction
Breast cancer is one of the most frequently diagnosed cancers among women, with an estimated 184,450 new cases of invasive disease and 40,930 deaths in 2008. There is strong evidence that estrogen plays a role in its development and progression [1]. Breast cancer was first recognized to be estrogen-dependent when the British surgeon George Beatson [2] published his findings of the beneficial effects of oophorectomy in a pre-menopausal patient with advanced breast cancer. Beatson had based his approach on the role of the ovaries in mammalian lactation and presumed that there would be a similar mechanism for breast cancer growth. Since that time, there has been an expanding clinical database that implicates estrogen in the development and progression of breast cancer. Evidence to support this conclusion comes from clinical studies of hormone replacement therapy, which were initially designed to determine the benefits of replacement approaches on post-menopausal women's health [3,4], and the successful clinical strategy of treating breast cancer by blocking estrogen action using the anti-estrogen tamoxifen [5] or preventing estrogen synthesis using aromatase inhibitors (AIs) [6].
Estrogens are a class of sex steroid hormones that are synthesized from cholesterol and are secreted primarily by the ovaries, with secondary contributions from placenta, adipose tissue, testes, and adrenal glands. After menopause, ovarian estrogen biosynthesis is minimal, with circulating estrogens being derived principally from peripheral aromatization of adrenal androgens. Estrogens are essential to the function of the female reproductive system and are required for the proliferation and differentiation of healthy breast epithelium. Estrogens occur naturally in several structurally related forms; however, the predominant intracellular estrogen is 17β-estradiol (E2). In mammary glands, E2 promotes cell proliferation in both normal and transformed epithelial cells by modifying the expression of hormone-responsive genes involved in the cell cycle and/or programmed cell death. In estrogen receptor (ER)-positive MCF-7 human breast cancer cells, one of the principal mechanism by which E2 stimulates growth is through the induction of G1- to S-phase transition. This induction is associated with the rapid and direct upregulation of c-myc, which controls cyclin D1 expression along with activation of cyclin-dependent kinase (CDK) and phosphorylation of retinoblastoma protein [7]. E2 also rapidly activates cyclin E-CDK2 complexes, accelerating the G1-to-S transition [8]. Additionally, E2 has 'non-genomic or membrane-initiated' effects (that is, independent of ER-mediated transcription) that occur within minutes after E2 administration [9-11]. Specifically, ER-α interacts with a number of proteins, including c-Src, the p85 subunit of phosphoinositide 3-kinase (PI3K), caveolin 1, and modulator of non-genomic activity of ER (MNAR) [10,12], epidermal growth factor receptor (EGFR), insulin-like growth factor receptor 1 (IGFR1), and HER2 [13], and it rapidly increases PIP2-phospholipase C activity and activates the mitogen-activated protein kinase (MAPK) and PI3K/Akt pathways [9,12,13]. More importantly, E2 is a potent inhibitor of apoptosis and it regulates the expression of several apoptotic proteins, including Bcl-2 in MCF-7, T47-D, and ZR-75-1 breast cancer cells [14].
Remarkably, there is another dimension to estrogen action which contrasts with its ability to stimulate growth and inhibit apoptosis. Physiologic E2 is also capable of inducing apoptosis in breast cancer cells that have been long-term estrogen-deprived (LTED) or have been treated exhaustively with anti-estrogens [15-23], prostate cancer cells [24], neuronal cells [25], bone-derived cells [26], thymocytes [27], and ER-transfected cells [28,29]. These data are particularly interesting because high-dose estrogen therapy was used as a treatment for post-menopausal patients with metastatic breast cancer from the 1940s until the introduction of the safer anti-estrogen tamoxifen in the 1970s [30]. At that time, however, the mechanism of estrogen-induced tumor regression was not known. In this review, we will discuss the current understanding of estrogen-induced apoptosis in breast cancer and will summarize the possible mechanisms involved in this estrogen-mediated process.
Estrogen-induced apoptosis: laboratory observations
Recent in vitro studies from our laboratory [18,31] and other investigators [19,20,32] have shown that long-term estrogen deprivation of hormone-dependent MCF-7 breast cancer cells causes them to undergo adaptive changes in which estradiol switches from being a proliferative agent to paradoxically inhibiting growth and inducing apoptosis. Interestingly, LTED cells also exhibit enhanced sensitivity to estradiol in that an estradiol concentration that is three logs lower can stimulate proliferation of these cells compared with wild-type MCF-7 cells [19]. The development of hypersensitivity to estradiol as a result of LTED is associated with the upregulation of ER-α and the MAPK, PI3K, and mTOR (mammalian target of rapamycin) growth factor pathways [33]. In contrast, the apoptotic mechanisms of estradiol in LTED cells are thought to involve the death receptors as well as the mitochondrial pathways. Specific molecular events include the activation of the Fas death receptor/Fas ligand (FasL) complex [20], the release of cytochrome c from the mitochondria and alterations in Bcl-2 [18,32], and the downregulation of the anti-apoptotic factor nuclear factor-κ [31,34]. It is important to note that estradiol also induces apoptosis in in vivo models of anti-hormone drug resistance [23,35,36]; however, the mechanisms by which this occurs are not completely known.
Estrogen therapy and breast cancer: clinical observations
Clinical data support the use of high-dose estrogen to treat hormonally sensitive breast tumors [37-41]. In 1944, Sir Alexander Haddow and colleagues [37] published the results of their clinical trial with the synthetic estrogens triphenylchlorethylene, triphenylmethylethylene, and stilbestrol administered at high doses. They found that 10 out of 22 post-menopausal patients with advanced mammary carcinomas, who were treated with triphenylchlorethylene, had significant regression of tumor growth. Five patients out of 14 who were treated with high-dose stilbestrol produced similar responses. Interestingly, the duration of the post-menopausal period was found to be a critical factor affecting the success of this therapy. For example, when the synthetic estrogen diethylstilbestrol (DES) was administered at 15 mg per day, women who had experienced the onset of menopause less than 1 year prior to therapy did not respond to DES; women who had experienced the onset of menopause within 5 years of menopause experienced a 7.9% objective response rate; and women who reached menopause more than 10 years earlier experienced a 22% response rate [41]. Despite the benefits, however, there were significant systemic side effects associated with high-dose estrogen therapy [37].
Cole and colleagues [39] reported the first clinical trial of the anti-estrogen tamoxifen in women with late or recurrent breast cancer and compared their findings with historical data from women receiving DES. They concluded that the levels of response were similar for DES and tamoxifen; however, tamoxifen had a lower incidence of side effects. Ingle and colleagues [30] compared tamoxifen with DES directly and noted that response rates were similar but tamoxifen had fewer side effects. Based on these data, the use of high-dose estrogen for treatment of advanced breast cancer fell out of favor, and tamoxifen became the standard first-line endocrine therapy. The Ingle study [30] that compared DES-treated and tamoxifen-treated patients was followed up but surprisingly showed a survival advantage for DES-treated patients [41]. Another small trial was conducted by Lonning and colleagues [40] in post-menopausal patients with advanced breast cancer exposed to multiple endocrine therapies and revealed a 31% objective response rate with DES therapy. More recently, Ellis and colleagues [42] reported that a daily dose of 6 mg of E2 could stop the growth of tumors or even cause them to shrink in about 25% of women with metastatic breast cancer that had developed resistance to standard anti-hormonal therapy. These clinical observations that estrogen can induce tumor regression after several years of anti-hormonal therapy provide a clue that the adaptation of cancer cells to low levels of estrogen might sensitize cells to the apoptotic effect of estrogen. While the mechanisms by which estrogen exerts its pro-apoptotic/anti-tumor effect are not known, a growing body of evidence suggests the involvement of the extrinsic (death receptor) and intrinsic (mitochondrial) pathways in this process.
Two main pathways involved in apoptosis regulation
Apoptosis is a form of programmed cell death that plays a critical role in the maintenance of tissue homeostasis [43]. It is a highly regulated physiologic mechanism that removes excess or damaged cells [43]. The dysregulation of apoptosis is a hallmark of cancer, with both the loss of pro-apoptotic signals and the gain of anti-apoptotic mechanisms contributing to tumorigenesis [44]. The induction of apoptosis in many cell types is achieved through the activation of the extrinsic and the intrinsic pathways [45]. The extrinsic pathway (Figure 1) is initiated by the interaction between specific ligands and surface receptors, such as CD95/Fas/Apo1, tumor necrosis factor (TNF) receptor 1 (TNFR1), TNF receptor 2 (TNFR2), and death receptors 3–6 (DR3–6) [46], which are able to deliver a death signal from the extracellular microenvironment to the cytoplasm. Binding of the ligand to the receptor induces receptor multimerization, binding of Fas-associated death domain (FADD) adapter protein, formation of the death-induced signaling complex (DISC) which recruits the initiator caspases 8 and 10, and subsequently activation of the effector caspases 3 and 7 [46]. In the intrinsic pathway (Figure 1), the integrity of the mitochondrial membrane is controlled primarily by a balance between the antagonistic actions of the proapoptotic and antiapoptotic members of the Bcl-2 family [47] (please see Table 1 for a detail description of common abbreviations used in apoptosis). Bcl-2 family proteins comprise three principal subfamilies: (a) anti-apoptotic members, including which possess the Bcl-2 homology (BH) Bcl-2/Bcl-xL, domains BH1, BH2, BH3, and BH4; (b) pro-apoptotic members, such as Bax, Bak, and Bok, which have the BH1, BH2, and BH3 domains; and (c) BH3-only proteins, such as Bid, Bim, Bad, Bik, and Puma, which generally possess only the BH3 domain [47]. The Bcl-2 family of proteins regulates apoptosis by altering mitochondrial membrane permeabilization and controlling the release of cytochrome c. Several lines of evidence demonstrate that the Bcl-2 family functions are controlled by growth factor signaling pathways, including the PI3K/Akt, the JAK (Janus kinase)/Stat (signal transducer and activator of transcription), and the Ras/MAPK pathways [48]. Phosphorylation and dephosphorylation of the members of the Bcl-2 family of proteins by the above pathways regulate the stabilization of mitochondrial homeostasis [48].
Figure 1.

The two main pathways involved in apoptosis regulation. (a) The extrinsic pathway begins outside the cell through the activation of receptors on the cell surface by specific molecules known as pro-apoptotic ligands, including CD95L/FasL (receptor CD95/Fas). Once activated, the death domains of these receptors bind to the adapter protein Fas-associated death domain (FADD), resulting in the assembly of death-induced signaling complex (DISC) and recruitment and assembly of initiator caspases 8 and 10. The two caspases are stimulated and processed, releasing active enzyme molecules into the cytosol, where they activate caspases 3, 6, and 7, thereby converging on the intrinsic pathway. (b) The intrinsic (mitochondrial) pathway is initiated in response to cellular signals resulting from DNA damage, a defective cell cycle, detachment from the extracellular matrix, hypoxia, loss of cell survival factors, or other types of severe cell stress. This triggers activation of specific members of the pro-apoptotic Bcl-2 protein family involved in the promotion of apoptosis, Puma and Noxa, which in turn activate the pro-apoptotic proteins Bax or Bak. These two proteins move to the mitochondrial membrane and disrupt the anti-apoptotic function of the Bcl-2 family proteins, allowing for permeabilization of the mitochondrial membrane. Apaf-1, apoptotic protease activating factor 1; Bad, Bcl-2/Bcl-XL-associated death domain protein; Bak, Bcl-2 homologous antagonist-killer protein; Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma-2; Bcl-XL, Bcl-2-related gene, long form; Bid, Bcl-2-interacting domain; Bim, Bcl-2-interacting mediator of cell death; Casp, caspase; Cyt c, cytochrome c; E2, 17β-estradiol; ER, estrogen receptor; ERE, estrogen-responsive element; FasL, Fas ligand; FLIP, FLICE-inhibitory protein; IAP, inhibitor of apoptosis; Noxa, phorbol-12-myristate-13-acetate-induced protein 1; Puma, p53-upregulated modulator of apoptosis.
Table 1.
Description of common abbreviations used in apoptosis and signal transduction
| Abbreviation | Meaning | Synonyms |
| Bad | Bcl-2/Bcl-XL-associated death domain protein | BH3-only member of the Bcl-2 family |
| Bak | Bcl-2 homologous antagonist-killer protein | Multi-BH domain pro-apoptosis protein |
| Bax | Bcl-2-associated X protein | Multi-BH domain pro-apoptosis protein |
| Bcl-2 | B-cell lymphoma-2 | Defining member of the family; originally characterized as an oncogene |
| Bcl-XL | Bcl-2-related gene, long form | Bcl-XS is a shorter splice variant that is pro-apoptotic |
| Bim | Bcl-2-interacting mediator of cell death | BH3-only member of the Bcl-2 family |
| IκB | Inhibitor of NF-κB | Interacts with NF-κB |
| IKK | IκB kinase | Phosphorylates IκB to promote its degradation |
| MDM2 | Murine double minute | Negative regulator of the p53 tumor suppressor |
| NF-κB | Nuclear factor-kappa type B | Originally linked with enhancement of immunoglobulin kappa light-chain gene |
| p53 | 53 kDa protein | Tumor-suppressor protein |
| PDK-1 | 3-phosphoinositide-dependent protein kinase 1 | Master kinase that is crucial for the activation of Akt/PKB |
| PI3K | Phosphoinositide 3-kinase | Phosphatidylinositol 3-kinase; PI 3-kinase; PtdIns3K |
| PKB | Protein kinase B | Akt; RACK (related to A and C kinase); has PH domain |
| PMAIP-1/Noxa | Phorbol-12-myristate-13-acetate-induced protein 1 | BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis |
| PUMA | p53-upregulated modulator of apoptosis | BH3-only member of the Bcl-2 family |
BH, Bcl-2 (B-cell lymphoma-2) homology.
Signaling pathways implicated in estrogen-induced apoptosis
The extrinsic (receptor-mediated) pathway
Mechanistic studies have used either LTED MCF-7 breast cancer cells [18,20,31,34] or selective ER modulator (SERM) (tamoxifen or raloxifene)-stimulated tumor models [23,35,36,49-51] to demonstrate the involvement of the Fas/FasL death signaling pathway in the paradoxical apoptotic/anti-tumor effects of E2. Song and colleagues [20] were the first to demonstrate that E2 caused apoptosis in breast cancer cells that were adapted to grow in an E2-free environment for prolonged periods. They reported that their LTED cells, which were derived by growing wild-type MCF-7 breast cancer cells under long-term (6 to 24 months) estrogen-deprived conditions, expressed high levels of Fas compared with the parental MCF-7 cells and that treatment of these cells with E2 resulted in a marked increase in FasL. This finding was confirmed by Osipo and colleagues [35], who reported that physiologic levels of E2 induced regression of tamoxifen-resistant breast cancer tumors by inducing Fas expression and suppressing the anti-apoptotic/pro-survival factors nuclear factor-kappa-B (NF-κB) and HER2/neu. A similar finding was reported by Liu and colleagues [49] in raloxifene-resistant MCF-7 cells in vitro and in vivo. In addition, Tonetti and colleagues [50,51] previously reported that stable overexpression of protein kinase C-alpha (PKC-α) in hormone-responsive T47D:A18 breast cancer cells (T47D:A18/PKC-α) produced a hormone-independent/tamoxifen-resistant and E2-inhibitory phenotype in vivo [50,51]. Using the T47D:A18/PKC-α-overexpressing tumor model, they further demonstrated that E2-induced regression and apoptosis were due to increased expression of Fas/FasL proteins and downregulation of the pro-survival Akt pathway [36]. In all of these model systems, the ER-α was shown to be critical for E2-induced tumor regression and apoptosis. Blockade of the ER-α signaling pathway using the pure anti-estrogen fulvestrant completely inhibited the apoptotic effect of E2 [20,35,36,49].
It is worth noting that a putative estrogen-responsive element (ERE) has been identified in the promoter region of the FasL gene [52], suggesting direct estrogen effects on FasL expression. In addition, a number of transactivating factors have been identified as regulators of FasL gene expression, including activator protein 1 (AP-1) [53] and specificity protein 1 (Sp-1) [54]. Sp-1 is involved in the transcriptional regulation of many genes and has also been identified to be important in the regulation of FasL gene expression and apoptosis. Indeed, this transcription factor is able to activate FasL promoter via a distinct recognition element, and inducible FasL promoter activation is abrogated by the expression of the dominant-negative mutant form of Sp-1 [54]. Functional studies have demonstrated that Sp-1 is a crucial effector of E2 signal in enhancing FasL gene expression. For instance, it is well known that ERs can transactivate gene promoters without directly binding to DNA but instead through interaction with other DNA-bound factors in promoter regions lacking TATA box. This has been investigated extensively in relation to protein complexes involving Sp-1 and ER-α at GC boxes, which are classic binding sites for members of the Sp-1 family of transcription factors. Sp-1 protein plays an important role in the regulation of mammalian and viral genes, and recent results have shown that E2 responsiveness of c-fos, cathepsin D, retinoic acid, and insulin-like grow factor-binding protein 4 gene expression in breast cancer cells is linked to specific GC-rich promoter sequences that bind ER/Sp-1 complex in which only Sp-1 protein binds DNA [55-59]. Thus, it is possible that, when E2 upregulates FasL production in these different model systems, an apoptotic signal is initiated by FasL binding on Fas receptor.
The intrinsic (mitochondrial) pathway
Over the last several years, there has been accumulating evidence that, apart from the extrinsic/receptor-mediated pathway, the mitochondrial (intrinsic) pathway plays a role in E2-induced apoptosis. Indeed, we [18] have previously reported that, in our LTED breast cancer cell line, MCF-7:5C, E2 treatment markedly increased the expression of several pro-apoptotic proteins, including, Bax, Bak, Bim, Noxa, Puma, and p53, and that blockade of Bax and Bim expression using short interfering RNAs (siRNAs) almost completely reversed the apoptotic effect of E2 in these cells. E2 treatment also led to a loss of mitochondrial potential and a dramatic increase in the release of cytochrome c from the mitochondria, which resulted in activation of caspases 7 and 9 and cleavage of poly(ADP-ribose)polymerase (PARP). Furthermore, over-expression of anti-apoptotic Bcl-xL completely blocked E2-induced apoptosis in MCF-7:5C cells. Interestingly, microarray analysis of wild-type MCF-7 cells and LTED MCF-7:5C cells revealed significant differences in the gene expression profile between the two cell lines following E2 treatment (Figure 2a). In particular, E2 treatment caused a marked increase in several pro-apoptotic genes in MCF-7:5C cells compared with wild-type MCF-7 cells (Figure 2b).
Figure 2.

Gene expression profile of wild-type MCF-7 cells and long-term estrogen-deprived (LTED) MCF-7:5C breast cancer cells following 17β-estradiol (E2) treatment. Cells were treated with 1 nM E2 for 48 hours, and RNA was hybridized to the Affymetrix Human Genome U133 Plus 2.0 Arrays (Affymetrix, Santa Clara, CA, USA). (a) Hierarchical clustering dendogram of E2-regulated genes in MCF-7 and MCF-7:5C cells. Microarray expression data for each cell line were first filtered for minimal intra-replicate standard deviation (<0.25) and a standard deviation between all samples of at least 0.25. This generated a total of 2,743 genes. In addition, genes displaying a minimum of twofold upregulation or downregulation by E2 in either MCF-7 or MCF-7:5C cells were extracted, revealing a set of 539 differentially expressed, E2-regulated genes. (b) E2 regulation of pro-apoptotic and anti-apoptotic genes in MCF-7 cells (top panel) and MCF-7:5C cells (bottom panel). Bak, Bcl-2 homologous antagonist-killer protein; Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma-2; Bim, Bcl-2-interacting mediator of cell death; GADD45β, growth arrest and DNA damage; p53, 53 kDa protein.
In two other estrogen-deprived breast cancer lines, LTED and E8CASS, basal Bcl-2 level was greatly elevated and knockdown of Bcl-2 expression with siRNA markedly sensitized these cells to the apoptotic action of E2 [32]. A similar finding was reported for another LTED breast cancer cell line, MCF-7:2A, which expressed elevated basal levels of Bcl-2 and was initially resistant to E2-induced apoptosis [34]. We found that suppression of Bcl-2 expression in these cells enhanced the apoptotic effect of E2 by almost fivefold [34], thus suggesting an important role for this protein in the apoptotic action of E2. Currently, there is renewed interest in developing small-molecule inhibitors of Bcl-2 [60] as anti-cancer cell and anti-angiogenic agents. The Bcl-2 antisense oligonucleotide, Oblimersen (Genasense; Genta Incorporated, Berkeley Heights, NJ, USA), which works by blocking Bcl-2 protein production, is now in a phase III clinical trial [61].
Inhibition of the survival pathways Akt and nuclear factor-kappa-B
The existence of various checkpoints in apoptosis reveals a complex balance between cell survival and cell death in cells. Two of the main signaling pathways involved in cell survival are the Akt and NF-κB signaling pathways (Figure 3). The PI3K/Akt/protein kinase B (PKB) pathway plays a central role in a variety of cellular processes, including cell growth, proliferation, motility, and survival in both normal and tumor cells. It impinges upon a remarkable array of intracellular events that influence either directly or indirectly whether a cell will undergo apoptosis. Many of the transforming events in breast cancer are a result of enhanced signaling of the PI3K/Akt pathway. Akt, also called PKB, is the human homologue of the viral oncogene v-akt [62], which regulates multiple targets, including several apoptotic genes. In a series of publications [63,64], Akt was found to mediate phosphorylation and hence inactivation of pro-apoptotic factors like Bad, which controls the release of cytochrome c, procaspase 9, and Forkhead transcription factors. Akt also activates anti-apoptotic genes, including cyclic-AMP response element-binding protein (CREB) and IκB (inhibitor of NF-κB) kinase (IKK), the primary regulator of NF-κB activity.
Figure 3.

Summary of some of the key processes regulated in the cytoplasm, at the mitochondria, in the nucleus, or in the cytosol by the PI3K/Akt pathway in controlling apoptosis. The positive events controlled either directly or indirectly by PI3K/Akt are indicated by arrows, whereas blocked lines represent events that have inhibitory effects. Bad, Bcl-2/Bcl-XL-associated death domain protein; Bax, Bcl-2-associated X protein; Bcl-XL, Bcl-2-related gene, long form; Bim, Bcl-2-interacting mediator of cell death; FasL, Fas ligand; IAP, inhibitor of apoptosis; IκB, inhibitor of nuclear factor-kappa-B; IKK, IκB (inhibitor of nuclear factor-kappa-B) kinase; Mcl-1, myeloid cell leukemia 1; Mdm2, murine double minute; NF-κB, nuclear factor-kappa-B; p53, 53 kDa protein; PDK-1, phosphoinositide-dependent protein kinase 1; PI3K, phosphoinositide 3-kinase; PKB/Akt, protein kinase B.
Several groups have demonstrated that E2 can also inhibit the P13K/Akt signaling pathway and consequently induce apoptosis of cancer cells. In tamoxifen-resistant PKC-α-overexpressing cells, E2-induced tumor regression is associated with the downregulation of phosphorylated Akt [36]. In addition, in LTED MCF-7:5C and MCF-7:2A breast cancer cells, the basal level of phosphorylated Akt is markedly upregulated and E2 treatment significantly reduces its expression (Figure 4). There is also evidence that, in MCF-7.beclin-overexpressing cells, E2 treatment significantly reduces Akt phosphorylation, which is associated with a decrease in cell proliferation [65]. Akt, therefore, is considered a rational target for cancer therapies and inhibitors of the PI3K/Akt pathway have been identified [66].
Figure 4.
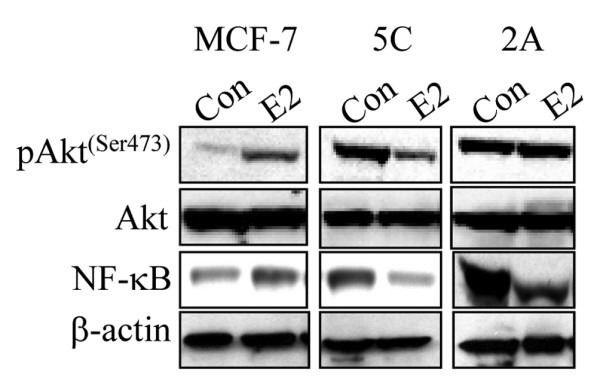
E2 (17β-estradiol) regulation of survival pathways in wild-type MCF-7 cells and long-term estrogen-deprived MCF-7:5C and MCF-7:2A breast cancer cells. Cells were treated with 1 nM E2 for 72 hours, and protein lysates were analyzed by Western blot for p-Akt, Akt, and nuclear factor-kappa-B (NF-κB). β-actin was used as a loading control.
NF-κB is one of the most studied transcription factors in mammalian cells. Its family is composed of five members: RELA (p65), RELB, REL (cRel), NF-κB1 (p50 and its precursor p105), and NF-κB2 (p52 and its precursor p100) [67]. These proteins form homodimeric and heterodimeric complexes, and the activity of these proteins is regulated by two major pathways: the classical or canonical NF-κB activation pathway [67] and the non-canonical NF-κB activation pathway [67]. One of the most documented functions of NF-κB is its ability to promote cell survival through the induction of target genes (Figure 3), the products of which inhibit the apoptotic machinery in normal and malignant cells [68]. Indeed, overall reduction in NF-κB activity is associated with an increased apoptotic index in many cell types [68]. Furthermore, NF-κB activation has been shown to inhibit p53-dependent apoptosis following expression of the oncogene AP12/MALT1 [69]. Thus, blocking this signaling pathway might be a promising option to improve the efficacy of conventional anti-cancer therapies.
Several studies have shown that E2 can inhibit the activity of NF-κB and thereby increase apoptosis. For example, Osipo and colleagues [35] reported that, in tamoxifen-resistant MCF-7 tumors, E2 treatment almost completely down-regulated the level of the NF-κB p65 subunit protein, which correlated with the anti-proliferative and pro-apoptotic effects of E2 in this model system. These investigators also reported that cyclooxygenase 2 (COX-2), an NF-κB-responsive gene, was markedly reduced in E2-treated tamoxifen-stimulated MCF-7 tumors [35]. They concluded from this finding that E2-induced apoptosis and tumor regression in tamoxifen-resistant MCF-7 tumors occurred, in part, through suppression of the pro-survival/anti-apoptotic factor NF-κB. It should be noted that NF-κB expression is also markedly elevated in raloxifene-resistant MCF-7 breast cancer cells [49] and LTED breast cancer cells (Figure 4) and its downregulation by E2 is associated with the suppression of proliferation and the induction of apoptosis [31,32,70].
Glutathione suppression and estrogen-induced apoptosis
Previous studies have reported that, apart from its action on the mitochondria, Bcl-2 functions as an anti-oxidant to block apoptosis and that Bcl-2 protein levels and glutathione (GSH) intracellular concentration are coordinately regulated, with a decrease in either favoring cell death [71]. It is believed that one mechanism by which Bcl-2 may function as an anti-oxidant is through upregulation of GSH, leading to rapid detoxification of reactive oxygen species and inhibition of free radical-mediated mitochondrial damage. Bcl-2 also has the ability to shift the entire cellular redox potential to a more reduced state which is independent of its effect on GSH levels [72].
GSH is a water-soluble tripeptide composed of glutamine, cysteine, and glycine. It is the most abundant intracellular small-molecule thiol present in mammalian cells, and it serves as a potent intracellular anti-oxidant, protecting cells from toxins such as free radicals [73]. Changes in GSH homeostasis have been implicated in the etiology and progression of a variety of human diseases, including breast cancer [74], and studies have shown that elevated levels of GSH prevent apoptotic cell death whereas depletion of GSH facilitates apoptosis [75]. L-buthionine sulfoximine (BSO) is a specific γ-glutamylcysteine synthetase inhibitor that blocks the rate-limiting step of GSH biosynthesis and, in doing so, depletes the intracellular GSH pool in both cultured cells and whole animals [73].
Recently, we reported that GSH participates in retarding apoptosis in anti-hormone-resistant LTED MCF-7:2A human breast cancer cells and that depletion of this molecule by BSO, a potent inhibitor of GSH biosynthesis, sensitized these resistant cells to E2-induced apoptosis [34]. GSH levels were elevated approximately 60% in MCF-7:2A cells compared with wild-type MCF-7 cells and these cells failed to undergo apoptosis following 1 week of E2 treatment. In the presence of BSO (100 μM), however, 1 nM E2 caused a dramatic increase in apoptosis which was observed as early as 48 hours, with maximum induction observed at day 7. The apoptotic effect of E2 plus BSO in MCF-7:2A cells was associated with a marked decreased in Bcl-2 and phosphorylated Bcl-2 protein levels, mitochondrial membrane disruption and cytochrome c release, caspase 7 activation, and PARP cleavage [34]. It is important to note that the concentration of BSO (100 μM) used in this study is clinically achievable without significant side effects [76]. Furthermore, early-phase clinical trials of BSO at doses resulting in both peripheral and tumor GSH depletion show that BSO can be safely administered with melphalan (L-PAM) to patients with refractory disease [77,78]. Thus, it is possible that future clinical studies of BSO infusions combined with low-dose estrogen hold the promise of improving disease control for patients with anti-hormone-resistant ER-positive metastatic breast cancer.
c-Jun N-terminal kinase signaling pathway
There is also evidence that E2 induces apoptosis by regulating the c-Jun N-terminal kinase (JNK) pathway. JNKs are a group of MAPKs that bind the NH2-terminal activation domain of the transcription factor c-jun and phosphorylate c-jun on amino acid residues Ser-63 and Ser-73 [79]. JNKs are stimulated by multiple factors, including cytokines, DNA-damaging agents, and environmental stresses, and are important in controlling programmed cell death or apoptosis. The inhibition of JNKs has been shown to enhance chemotherapy-induced inhibition of tumor cell growth, suggesting that JNKs may provide a molecular target for the treatment of cancer [79]. Recently, Altiok and colleagues [80] reported that, under low growth-stimulated conditions, high concentrations (1 μM) of E2 induced apoptosis and concomitantly increased phosphorylation of c-jun in ER-positive MCF-7 breast cancer cells but not in ER-negative MDA-MB 231 cells, thus suggesting an ER-mediated event. Interestingly, when the JNK signaling pathway was disrupted by the JNK inhibitor SP600125, the ability of E2 to inhibit the growth of MCF-7 cells and to induce apoptosis was completely blocked. More recently, we reported that JNK activation (as measured by the increased levels of phospho-JNK1/2 and the JNK substrate phospho-c-Jun) was increased by low-dose E2 in the presence of BSO in anti-hormone-resistant MCF-7:2A cells [34]. While the exact mechanism by which JNK promotes apoptosis is not currently known, the phosphorylation of transcription factors such as c-jun and p53, as well as pro- and anti-apoptotic Bcl-2 family members, has been suggested to be of importance [81]. The treatment with BSO plus E2 markedly increased phosphorylated c-jun in MCF-7:2A cells and decreased phosphorylated Bcl-2 in these cells. These findings thus suggest that BSO plus E2 might mediate their apoptotic effect, in part, through activation of JNK.
Clinical exploitation of estrogen-induced apoptosis
Laboratory studies uniformly demonstrate that low concentrations of estrogen can cause apoptotic tumor cell death following profound estrogen deprivation with anti-hormones. The question that now needs to be answered is how can this new laboratory knowledge be translated into patient care?
Recently, Ellis and colleagues [42] reported that low-dose E2 (6 mg daily: 2 mg three times a day) produced a 25% response rate for patients with ER-positive AI-resistant advanced breast cancer. This number is slightly lower than the 31% objective response rate reported by Lonning and colleagues [40] with DES (5 mg three times a day) in post-menopausal women heavily pre-treated with endocrine therapy. The Lonning study [40] recruited patients with advanced breast cancer who were previously treated with exhaustive endocrine therapy. Of the 32 patients enrolled, four patients obtained complete response (CR) and six patients obtained partial response. In contrast, the Ellis study [42] recruited patients who were treated with an AI with 24 or more weeks of progression-free survival or who had a relapse after 2 or more years of adjuvant AI. Interestingly, there were no CRs in the Ellis study [42]. Clinical observations suggest that the duration of the post-menopausal period is one of the crucial factors affecting the success of low-dose estrogen therapy. In other words, the longer the estrogen deprivation period, the higher the likelihood of a response to low-dose estrogen. The fact that there were four CRs in the Lonning study [40] but none in the Ellis study [42] suggests the need for extensive estrogen blockade or withdrawal to get the best effects from low-dose estrogen.
Estrogen and bone remodeling
In addition to its role in female sexual development and reproductive physiology, estrogen plays a key role in bone cell metabolism. Estrogen contributes to the strength of a woman's skeleton by maintaining bone density. Bone is a dynamic tissue that is constantly being reshaped by osteoblasts, which build bone, and osteoclasts, which resorb bone [82]. This dynamic process is called remodeling. Osteoblasts are derived from pleiotropic mesenchymal stem cells in the bone marrow, whereas osteoclasts are multinuclear macrophage-like cells derived from hematopoietic stem cells also in the bone marrow. Bone resorption and deposition are tightly coupled, and their balance defines both bone mass as well as quality. The regulation of bone remodeling is complex; however, estrogen is thought to play a key role in this process [82]. Estrogen inhibits bone remodeling and bone resorption and enhances bone formation. Conversely, loss of estrogen, due to menopause or surgical oophorectomy, leads to an increased rate of remodeling and tilts the balance between bone resorption and formation in favor of the former [83]. Estrogen deficiency in post-menopausal women frequently leads to osteoporosis, the most common skeletal disorder. The imbalance in bone turnover that is induced by estrogen deficiency in women and female rodents can be ameliorated with estrogen/progestin hormone therapy or SERMs [84].
The main effect of estrogen on bone remodeling is to decrease activation frequency and subsequently decrease the numbers of osteoclasts and osteoblasts. Its effects on osteoclasts are mainly indirect and mediated by products secreted by the osteoblast. These products include RANK-L (the ligand of the receptor activator of NF-κB), colony-stimulating factor 1 (CSF-1), and osteoprotegerin (OPG). They regulate the differentiation of osteoclast precursors to osteoclasts and then modulate the activity of the mature osteoclasts and regulate its rate of apoptosis. Estrogen also decreases the secretion of the pro-inflammatory cytokines interleukin (IL) 1, IL-6, and TNF-α by marrow monocytes, resulting in decreased production of OPG and RANK-L by the osteoblasts, thereby decreasing the rate of production of osteoclasts, their activity, and their survival [82]. There is also evidence that estrogen has direct effects on osteoclast lineage cells. It induces apoptosis of these cells and it suppresses RANK-L-induced osteoclast differentiation by blocking RANK-L/macrophage colony-stimulating factor (M-CSF)-induced AP-1-dependent transcription through a reduction of c-jun activity [85]. Moreover, estrogen has been shown to inhibit the activity of mature osteoclasts through direct, receptor-mediated mechanisms. Indeed, a recent study by Nakamura and colleagues [86] reported that estrogen, acting via the ER-α, induced apoptosis in osteoclasts through activation of the Fas/FasL system and that this leads to suppression of bone resorption through truncating the short life span of differentiated osteoclasts.
Future perspective
Estrogen is a potent mitogen that stimulates cell proliferation and prevents cell death in breast cancer cells through activation of the ER. Paradoxically, estrogen is also capable of inducing tumor regression of hormone-dependent breast cancer in post-menopausal women who have been treated exhaustively with anti-hormones. Pre-clinical studies suggest that the evolution of anti-hormone resistance over years of therapy reconfigures the survival mechanism of the breast cancer cell so that estrogen no longer functions as a survival factor but as a death signal. It is this reconfiguration that helps to explain the 'two faces' of estrogen: the ability to stimulate growth and to induce death. Interestingly, estrogen also induces tumor regression in raloxifene-resistant endometrial tumors (G Balaburski and VC Jordan, personal communication) and it prevents bone loss by inducing apoptosis in osteoclasts, suggesting a universal principle.
Pre-clinical data [34] clearly show that it is possible to enhance the apoptotic effect of low-dose E2 by combining it with BSO. Hence, the combination of BSO and E2 could be used to improve the efficacy of E2 as an apoptotic agent if GSH depletion is fundamental to tumor cell survival. Phase I clinical trials of BSO at doses resulting in both peripheral and tumor GSH depletion show that BSO can be safely administered to patients with refractory disease. BSO was administered intravenously twice daily either alone or together with chemotherapy to cancer patients whose disease had progressed despite multiple lines of previous chemotherapy [78].
Inhibitors of survival pathways also have the ability to enhance the apoptotic/growth-inhibitory effects of E2. Several groups have developed small-molecule inhibitors of Bcl-2 as anti-tumor agents [87]. These inhibitors encompass various drugs that bind the anti-apoptotic Bcl-2 family members with more or less efficacy. Oblimersen (Genasense; G3139) is an anti-Bcl-2 antisense oligonucleotide that has reached phase III clinical trials in combination therapy [88]. There are also natural inhibitors of Bcl-2, which include tea polyphenols such as catechins and theaflavins [89].
Conclusion
The discovery of a new biology of E2-induced apoptosis provides a unique signal transduction pathway to exploit in the treatment of metastatic breast cancer that has become refractory to exhaustive anti-hormone therapy. The clinical clues with the use of high-dose estrogen therapy have now been supported by a wealth of laboratory data defining apoptotic mechanisms. It is plausible to consider that the methodical evaluation of monoclonal antibodies and small-molecule tyrosine kinase inhibitors to prevent breast cancer survival could amplify the apoptotic actions of estradiol in a select group of patients. Indeed, if a study of the molecular biology of estrogen-induced apoptosis can define the mechanism precisely, then the molecules involved will become the target for a new drug group. These new drugs may be able to precipitate apoptosis in ER-negative breast tumors or indeed be used universally to treat cancer types other than breast cancer.
Abbreviations
AI: aromatase inhibitor; AP-1: activator protein 1; Bad: Bcl-2/Bcl-XL-associated death domain protein; Bak: Bcl-2 homologous antagonist-killer protein; Bax: Bcl-2-associated X protein; Bcl-2: B-cell lymphoma-2; Bcl-XL: Bcl-2-related gene, long form; BH: Bcl-2 (B-cell lymphoma-2) homology; Bid: Bcl-2-interacting domain; Bim: Bcl-2-interacting mediator of cell death; BSO: L-buthionine sulfoximine; CDK: cyclin-dependent kinase; CR: complete response; DES: diethylstilbestrol; E2: 17β-estradiol; ER: estrogen receptor; FasL: Fas ligand; GSH: glu-tathione; IL: interleukin; JNK: c-jun N-terminal kinase; LTED: long-term estrogen-deprived; MAPK: mitogen-activated protein kinase; NF-κB: nuclear factor-kappa-B; OPG: osteoprotegerin; p53: 53 kDa protein; PARP: poly(ADP-ribose)polymerase; PI3K: phosphoinositide 3-kinase; PKB/Akt: protein kinase B; PKC-α: protein kinase C-alpha; Puma: p53-upregulated modulator of apoptosis; RANK-L: ligand of the receptor activator of nuclear factor-kappa-B; SERM: selective estrogen receptor modulator; siRNA: short interfering RNA; Sp-1: specificity protein 1; TNF: tumor necrosis factor.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
This work was supported by National Institutes of Health Career Development Grant K01CA120051-01A2 (to JSL-W), American Cancer Society grant IRG-92-027-14 (JSL-W), the Hollenbach Family Fund (JSL-W), the Department of Defence Breast Program under award number BC050277 (to VCJ), Center of Excellence SPORE (Specialized Program of Research Excellence) in Breast Cancer grant CA 89018 (VCJ), grant R01 GM067156 (VCJ), Fox Chase Cancer Center Core Grant NIH P30 CA006927 (VCJ), the Avon Foundation (VCJ), the Genuardi's Fund (VCJ), and the Weg Fund of the Fox Chase Cancer Center (VCJ).
Contributor Information
Joan S Lewis-Wambi, Email: joan.lewis@fccc.edu.
V Craig Jordan, Email: v.craig.jordan@fccc.edu.
References
- Russo IH, Russo J. Role of hormones in mammary cancer initiation and progression. J Mammary Gland Biol Neoplasia. 1998;3:49–61. doi: 10.1023/A:1018770218022. [DOI] [PubMed] [Google Scholar]
- Beatson CT. On the treatment of inoperable cases of carcinoma of the mamma: suggestions for a new method of treatment with illustrative cases. Lancet. 1896;2:101–111. [PMC free article] [PubMed] [Google Scholar]
- Beral V. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362:419–427. doi: 10.1016/S0140-6736(03)14596-5. [DOI] [PubMed] [Google Scholar]
- Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J, Writing Group for the Women's Health Initiative Investigators Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women's Health Initiative randomized controlled trial. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists' Collaborative Group. Lancet. 1998;351:1451–1467. doi: 10.1016/S0140-6736(97)11423-4. [DOI] [PubMed] [Google Scholar]
- Iwase H. Current topics and perspectives on the use of aromatase inhibitors in the treatment of breast cancer. Breast Cancer. 2008;15:278–290. doi: 10.1007/s12282-008-0071-y. [DOI] [PubMed] [Google Scholar]
- Altucci L, Addeo R, Cicatiello L, Dauvois S, Parker MG, Truss M, Beato M, Sica V, Bresciani F, Weisz A. 17beta-Estradiol induces cyclin D1 gene transcription, p36D1-p34cdk4 complex activation and p105Rb phosphorylation during mitogenic stimulation of G(1)-arrested human breast cancer cells. Oncogene. 1996;12:2315–2324. [PubMed] [Google Scholar]
- Foster JS, Wimalasena J. Estrogen regulates activity of cyclin-dependent kinases and retinoblastoma protein phosphorylation in breast cancer cells. Mol Endocrinol. 1996;10:488–498. doi: 10.1210/me.10.5.488. [DOI] [PubMed] [Google Scholar]
- Pietras RJ, Marquez-Garban DC. Membrane-associated estrogen receptor signaling pathways in human cancers. Clin Cancer Res. 2007;13:4672–4676. doi: 10.1158/1078-0432.CCR-07-1373. [DOI] [PubMed] [Google Scholar]
- Moriarty K, Kim KH, Bender JR. Minireview: estrogen receptor-mediated rapid signaling. Endocrinology. 2006;147:5557–5563. doi: 10.1210/en.2006-0729. [DOI] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol. 2006;20:1996–2009. doi: 10.1210/me.2005-0525. [DOI] [PubMed] [Google Scholar]
- Stoica GE, Franke TF, Wellstein A, Czubayko F, List HJ, Reiter R, Morgan E, Martin MB, Stoica A. Estradiol rapidly activates Akt via the ErbB2 signaling pathway. Mol Endocrinol. 2003;17:818–830. doi: 10.1210/me.2002-0330. [DOI] [PubMed] [Google Scholar]
- Arpino G, Wiechmann L, Osborne CK, Schiff R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev. 2008;29:217–233. doi: 10.1210/er.2006-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompel A, Somai S, Chaouat M, Kazem A, Kloosterboer HJ, Beusman I, Forgez P, Mimoun M, Rostene W. Hormonal regulation of apoptosis in breast cells and tissues. Steroids. 2000;65:593–598. doi: 10.1016/S0039-128X(00)00172-0. [DOI] [PubMed] [Google Scholar]
- Jordan VC. The 38th David A. Karnofsky lecture: the paradoxical actions of estrogen in breast cancer – survival or death? J Clin Oncol. 2008;26:3073–3082. doi: 10.1200/JCO.2008.17.5190. [DOI] [PubMed] [Google Scholar]
- Jordan VC, Lewis JS, Osipo C, Cheng D. The apoptotic action of estrogen following exhaustive antihormonal therapy: a new clinical treatment strategy. Breast. 2005;14:624–630. doi: 10.1016/j.breast.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Lewis JS, Cheng D, Jordan VC. Targeting oestrogen to kill the cancer but not the patient. Br J Cancer. 2004;90:944–949. doi: 10.1038/sj.bjc.6601627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JS, Meeke K, Osipo C, Ross EA, Kidawi N, Li T, Bell E, Chandel NS, Jordan VC. Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J Natl Cancer Inst. 2005;97:1746–1759. doi: 10.1093/jnci/dji400. [DOI] [PubMed] [Google Scholar]
- Santen R, Jeng MH, Wang JP, Song R, Masamura S, McPherson R, Santner S, Yue W, Shim WS. Adaptive hypersensitivity to estradiol: potential mechanism for secondary hormonal responses in breast cancer patients. J Steroid Biochem Mol Biol. 2001;79:115–125. doi: 10.1016/S0960-0760(01)00151-0. [DOI] [PubMed] [Google Scholar]
- Song RX, Mor G, Naftolin F, McPherson RA, Song J, Zhang Z, Yue W, Wang J, Santen RJ. Effect of long-term estrogen deprivation on apoptotic responses of breast cancer cells to 17beta-estradiol. J Natl Cancer Inst. 2001;93:1714–1723. doi: 10.1093/jnci/93.22.1714. [DOI] [PubMed] [Google Scholar]
- Swaby RF, Jordan VC. Low-dose estrogen therapy to reverse acquired antihormonal resistance in the treatment of breast cancer. Clin Breast Cancer. 2008;8:124–133. doi: 10.3816/CBC.2008.n.012. [DOI] [PubMed] [Google Scholar]
- Wolf DM, Jordan VC. A laboratory model to explain the survival advantage observed in patients taking adjuvant tamoxifen therapy. Recent Results Cancer Res. 1993;127:23–33. doi: 10.1007/978-3-642-84745-5_4. [DOI] [PubMed] [Google Scholar]
- Yao K, Lee ES, Bentrem DJ, England G, Schafer JI, O'Regan RM, Jordan VC. Antitumor action of physiological estradiol on tamoxifen-stimulated breast tumors grown in athymic mice. Clin Cancer Res. 2000;6:2028–2036. [PubMed] [Google Scholar]
- Robertson CN, Roberson KM, Padilla GM, O'Brien ET, Cook JM, Kim CS, Fine RL. Induction of apoptosis by diethylstilbestrol in hormone-insensitive prostate cancer cells. J Natl Cancer Inst. 1996;88:908–917. doi: 10.1093/jnci/88.13.908. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Mor G, Naftolin F. Estrogen-regulated developmental neuronal apoptosis is determined by estrogen receptor subtype and the Fas/Fas ligand system. J Neurobiol. 2000;43:64–78. doi: 10.1002/(SICI)1097-4695(200004)43:1<64::AID-NEU6>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Saintier D, Khanine V, Uzan B, Ea HK, de Vernejoul MC, Cohen-Solal ME. Estradiol inhibits adhesion and promotes apoptosis in murine osteoclasts in vitro. J Steroid Biochem Mol Biol. 2006;99:165–173. doi: 10.1016/j.jsbmb.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Okasha SA, Ryu S, Do Y, McKallip RJ, Nagarkatti M, Nagarkatti PS. Evidence for estradiol-induced apoptosis and dysregulated T cell maturation in the thymus. Toxicology. 2001;163:49–62. doi: 10.1016/S0300-483X(01)00374-2. [DOI] [PubMed] [Google Scholar]
- Jiang SY, Jordan VC. Growth regulation of estrogen receptor-negative breast cancer cells transfected with complementary DNAs for estrogen receptor. J Natl Cancer Inst. 1992;84:580–591. doi: 10.1093/jnci/84.8.580. [DOI] [PubMed] [Google Scholar]
- Levenson AS, Jordan VC. Transfection of human estrogen receptor (ER) cDNA into ER-negative mammalian cell lines. J Steroid Biochem Mol Biol. 1994;51:229–239. doi: 10.1016/0960-0760(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Ingle JN, Ahmann DL, Green SJ, Edmonson JH, Bisel HF, Kvols LK, Nichols WC, Creagan ET, Hahn RG, Rubin J, Frytak S. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. N Engl J Med. 1981;304:16–21. doi: 10.1056/NEJM198101013040104. [DOI] [PubMed] [Google Scholar]
- Lewis JS, Osipo C, Meeke K, Jordan VC. Estrogen-induced apoptosis in a breast cancer model resistant to long-term estrogen withdrawal. J Steroid Biochem Mol Biol. 2005;94:131–141. doi: 10.1016/j.jsbmb.2004.12.032. [DOI] [PubMed] [Google Scholar]
- Song RX, Zhang Z, Mor G, Santen RJ. Down-regulation of Bcl-2 enhances estrogen apoptotic action in long-term estradiol-depleted ER(+) breast cancer cells. Apoptosis. 2005;10:667–678. doi: 10.1007/s10495-005-1903-2. [DOI] [PubMed] [Google Scholar]
- Santen RJ, Song RX, Masamura S, Yue W, Fan P, Sogon T, Hayashi S, Nakachi K, Eguchi H. Adaptation to estradiol deprivation causes up-regulation of growth factor pathways and hypersensitivity to estradiol in breast cancer cells. Adv Exp Med Biol. 2008;630:19–34. doi: 10.1007/978-0-387-78818-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis-Wambi JS, Kim HR, Wambi C, Patel R, Pyle JR, Klein-Szanto AJ, Jordan VC. Buthionine sulfoximine sensitizes anti-hormone-resistant human breast cancer cells to estrogen-induced apoptosis. Breast Cancer Res. 2008;10:R104. doi: 10.1186/bcr2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipo C, Gajdos C, Liu H, Chen B, Jordan VC. Paradoxical action of fulvestrant in estradiol-induced regression of tamoxifen-stimulated breast cancer. J Natl Cancer Inst. 2003;95:1597–1608. doi: 10.1093/jnci/djg079. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhao H, Asztalos S, Chisamore M, Sitabkhan Y, Tonetti DA. Estradiol-induced regression in T47D:A18/PKCalpha tumors requires the estrogen receptor and interaction with the extracellular matrix. Mol Cancer Res. 2009;7:498–510. doi: 10.1158/1541-7786.MCR-08-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddow A, Watkinson JM, Paterson E. Influence of synthetic oestrogens upon advanced malignant disease. BMJ. 1944;2:393–398. doi: 10.1136/bmj.2.4368.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AC, Sedransk N, Kelley RM, Ansfield FJ, Ravdin RG, Talley RW, Potter NR. Diethylstilbestrol: recommended dosages for different categories of breast cancer patients. Report of the Cooperative Breast Cancer Group. JAMA. 1977;237:2079–2078. doi: 10.1001/jama.237.19.2079. [DOI] [PubMed] [Google Scholar]
- Cole MP, Jones CT, Todd ID. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br J Cancer. 1971;25:270–275. doi: 10.1038/bjc.1971.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonning PE, Taylor PD, Anker G, Iddon J, Wie L, Jorgensen LM, Mella O, Howell A. High-dose estrogen treatment in post-menopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res Treat. 2001;67:111–116. doi: 10.1023/A:1010619225209. [DOI] [PubMed] [Google Scholar]
- Peethambaram PP, Ingle JN, Suman VJ, Hartmann LC, Loprinzi CL. Randomized trial of diethylstilbestrol vs. tamoxifen in postmenopausal women with metastatic breast cancer. An updated analysis. Breast Cancer Res Treat. 1999;54:117–122. doi: 10.1023/A:1006185805079. [DOI] [PubMed] [Google Scholar]
- Ellis MJ, Dehdahti F, Kommareddy A, Jamalabadi-Majidi S, Crowder R, Jeffe DB, Gao F, Fleming G, Silverman P, Dickler M, L Carey, Marcom PK. A randomized phase 2 trial of low dose (6 mg daily) versus high dose (30 mg daily) estradiol for patients with estrogen receptor positive aromatase inhibitor resistant advanced breast cancer [abstract] Cancer Res. 2009;69:s67. doi: 10.1001/jama.2009.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–8567. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Ziegler DS, Kung AL. Therapeutic targeting of apoptosis pathways in cancer. Curr Opin Oncol. 2008;20:97–103. doi: 10.1097/CCO.0b013e3282f310f6. [DOI] [PubMed] [Google Scholar]
- Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y. Bcl-2 family of proteins: life-or-death switch in mitochondria. Biosci Rep. 2002;22:47–58. doi: 10.1023/A:1016061006256. [DOI] [PubMed] [Google Scholar]
- Talapatra S, Thompson CB. Growth factor signaling in cell survival: implications for cancer treatment. J Pharmacol Exp Ther. 2001;298:873–878. [PubMed] [Google Scholar]
- Liu H, Lee ES, Gajdos C, Pearce ST, Chen B, Osipo C, Loweth J, McKian K, De Los Reyes A, Wing L, Jordan VC. Apoptotic action of 17beta-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst. 2003;95:1586–1597. doi: 10.1093/jnci/djg080. [DOI] [PubMed] [Google Scholar]
- Chisamore MJ, Ahmed Y, Bentrem DJ, Jordan VC, Tonetti DA. Novel antitumor effect of estradiol in athymic mice injected with a T47D breast cancer cell line overexpressing protein kinase Calpha. Clin Cancer Res. 2001;7:3156–3165. [PubMed] [Google Scholar]
- Tonetti DA, Chisamore MJ, Grdina W, Schurz H, Jordan VC. Stable transfection of protein kinase C alpha cDNA in hormone-dependent breast cancer cell lines. Br J Cancer. 2000;83:782–791. doi: 10.1054/bjoc.2000.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor G, Kohen F, Garcia-Velasco J, Nilsen J, Brown W, Song J, Naftolin F. Regulation of fas ligand expression in breast cancer cells by estrogen: functional differences between estradiol and tamoxifen. J Steroid Biochem Mol Biol. 2000;73:185–194. doi: 10.1016/S0960-0760(00)00081-9. [DOI] [PubMed] [Google Scholar]
- Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol Cell. 1998;1:543–551. doi: 10.1016/S1097-2765(00)80054-4. [DOI] [PubMed] [Google Scholar]
- Kavurma MM, Santiago FS, Bonfoco E, Khachigian LM. Sp1 phosphorylation regulates apoptosis via extracellular FasL-Fas engagement. J Biol Chem. 2001;276:4964–4971. doi: 10.1074/jbc.M009251200. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Wang X, Safe S. Estrogen receptor-Sp1 complexes mediate estrogen-induced cathepsin D gene expression in MCF-7 human breast cancer cells. J Biol Chem. 1994;269:15912–15917. [PubMed] [Google Scholar]
- Porter W, Saville B, Hoivik D, Safe S. Functional synergy between the transcription factor Sp1 and the estrogen receptor. Mol Endocrinol. 1997;11:1569–1580. doi: 10.1210/me.11.11.1569. [DOI] [PubMed] [Google Scholar]
- Duan R, Porter W, Safe S. Estrogen-induced c-fos protoonco-gene expression in MCF-7 human breast cancer cells: role of estrogen receptor Sp1 complex formation. Endocrinology. 1998;139:1981–1990. doi: 10.1210/en.139.4.1981. [DOI] [PubMed] [Google Scholar]
- Sun G, Porter W, Safe S. Estrogen-induced retinoic acid receptor alpha 1 gene expression: role of estrogen receptor-Sp1 complex. Mol Endocrinol. 1998;12:882–890. doi: 10.1210/me.12.6.882. [DOI] [PubMed] [Google Scholar]
- Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson JA, Safe S. Ligand-, cell-, and estrogen receptor subtype (alpha/beta)-dependent activation at GC-rich (Sp1) promoter elements. J Biol Chem. 2000;275:5379–5387. doi: 10.1074/jbc.275.8.5379. [DOI] [PubMed] [Google Scholar]
- Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–1132. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien S, Moore JO, Boyd TE, Larratt LM, Skotnicki A, Koziner B, Chanan-Khan AA, Seymour JF, Bociek RG, Pavletic S, Rai KR. Randomized phase III trial of fludarabine plus cyclophosphamide with or without oblimersen sodium (Bcl-2 antisense) in patients with relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol. 2007;25:1114–1120. doi: 10.1200/JCO.2006.07.1191. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.1833819. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/S0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- John S, Nayvelt I, Hsu HC, Yang P, Liu W, Das GM, Thomas T, Thomas TJ. Regulation of estrogenic effects by beclin 1 in breast cancer cells. Cancer Res. 2008;68:7855–7863. doi: 10.1158/0008-5472.CAN-07-5875. [DOI] [PubMed] [Google Scholar]
- Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27:5511–5526. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Dutta J, Fan Y, Gupta N, Fan G, Gelinas C. Current insights into the regulation of programmed cell death by NF-kappaB. Oncogene. 2006;25:6800–6816. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- Stoffel A, Levine AJ. Activation of NF-kappaB by the API2/MALT1 fusions inhibits p53 dependant but not FAS induced apoptosis: a directional link between NF-kappaB and p53. Cell Cycle. 2004;3:1017–1020. [PubMed] [Google Scholar]
- Krasil'nikov MA, Lobanova YS, Scherbakov AM, Shatskaya VA, Evteev VA. NF-kappaB suppression provokes the sensitization of hormone-resistant breast cancer cells to estrogen apoptosis. Mol Cell Biochem. 2009;324:65–71. doi: 10.1007/s11010-008-9985-0. [DOI] [PubMed] [Google Scholar]
- Voehringer DW. BCL-2 and glutathione: alterations in cellular redox state that regulate apoptosis sensitivity. Free Radic Biol Med. 1999;27:945–950. doi: 10.1016/S0891-5849(99)00174-4. [DOI] [PubMed] [Google Scholar]
- Ellerby LM, Ellerby HM, Park SM, Holleran AL, Murphy AN, Fiskum G, Kane DJ, Testa MP, Kayalar C, Bredesen DE. Shift of the cellular oxidation-reduction potential in neural cells expressing Bcl-2. J Neurochem. 1996;67:1259–1267. doi: 10.1046/j.1471-4159.1996.67031259.x. [DOI] [PubMed] [Google Scholar]
- Anderson CP, Tsai JM, Meek WE, Liu RM, Tang Y, Forman HJ, Reynolds CP. Depletion of glutathione by buthionine sulfoxine is cytotoxic for human neuroblastoma cell lines via apoptosis. Exp Cell Res. 1999;246:183–192. doi: 10.1006/excr.1998.4303. [DOI] [PubMed] [Google Scholar]
- Schroder CP, Godwin AK, O'Dwyer PJ, Tew KD, Hamilton TC, Ozols RF. Glutathione and drug resistance. Cancer Invest. 1996;14:158–168. doi: 10.3109/07357909609018891. [DOI] [PubMed] [Google Scholar]
- Schnelldorfer T, Gansauge S, Gansauge F, Schlosser S, Beger HG, Nussler AK. Glutathione depletion causes cell growth inhibition and enhanced apoptosis in pancreatic cancer cells. Cancer. 2000;89:1440–1447. doi: 10.1002/1097-0142(20001001)89:7<1440::AID-CNCR5>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Bailey HH. L-S,R-buthionine sulfoximine: historical development and clinical issues. Chem Biol Interact. 1998;111–112:239–254. doi: 10.1016/S0009-2797(97)00164-6. [DOI] [PubMed] [Google Scholar]
- Bailey HH, Mulcahy RT, Tutsch KD, Arzoomanian RZ, Alberti D, Tombes MB, Wilding G, Pomplun M, Spriggs DR. Phase I clinical trial of intravenous L-buthionine sulfoximine and melphalan: an attempt at modulation of glutathione. J Clin Oncol. 1994;12:194–205. doi: 10.1200/JCO.1994.12.1.194. [DOI] [PubMed] [Google Scholar]
- Bailey HH, Ripple G, Tutsch KD, Arzoomanian RZ, Alberti D, Feierabend C, Mahvi D, Schink J, Pomplun M, Mulcahy RT, Wilding G. Phase I study of continuous-infusion L-S,R-buthionine sulfoximine with intravenous melphalan. J Natl Cancer Inst. 1997;89:1789–1796. doi: 10.1093/jnci/89.23.1789. [DOI] [PubMed] [Google Scholar]
- Vasilevskaya I, O'Dwyer PJ. Role of Jun and Jun kinase in resistance of cancer cells to therapy. Drug Resist Updat. 2003;6:147–156. doi: 10.1016/S1368-7646(03)00043-8. [DOI] [PubMed] [Google Scholar]
- Altiok N, Koyuturk M, Altiok S. JNK pathway regulates estradiol-induced apoptosis in hormone-dependent human breast cancer cells. Breast Cancer Res Treat. 2007;105:247–254. doi: 10.1007/s10549-006-9451-1. [DOI] [PubMed] [Google Scholar]
- Park J, Kim I, Oh YJ, Lee K, Han PL, Choi EJ. Activation of c-Jun N-terminal kinase antagonizes an anti-apoptotic action of Bcl-2. J Biol Chem. 1997;272:16725–16728. doi: 10.1074/jbc.272.27.16725. [DOI] [PubMed] [Google Scholar]
- Riggs BL, Khosla S, Melton LJ., 3rd Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev. 2002;23:279–302. doi: 10.1210/er.23.3.279. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL. Osteoclasts: what do they do and how do they do it? Am J Pathol. 2007;170:427–435. doi: 10.2353/ajpath.2007.060834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas PD, Ensrud KE, Adachi JD, Harper KD, Sarkar S, Gennari C, Reginster JY, Pols HA, Recker RR, Harris ST, Wu W, Genant HK, Black DM, Eastell R, Mulitple Outcomes of Raloxifene Evaluation Investigators Efficacy of raloxifene on vertebral fracture risk reduction in postmenopausal women with osteoporosis: four-year results from a randomized clinical trial. J Clin Endocrinol Metab. 2002;87:3609–3617. doi: 10.1210/jc.87.8.3609. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Toraldo G, Weitzmann MN, Cenci S, Ross FP, Pacifici R. Estrogen decreases osteoclast formation by down-regulating receptor activator of NF-kappa B ligand (RANKL)-induced JNK activation. J Biol Chem. 2001;276:8836–8840. doi: 10.1074/jbc.M010764200. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y, Nishina H, Takeda S, Takayanagi H, Metzger D, Kanno J, Takaoka K, Martin TJ, Chambon P, Kato S. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130:811–823. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- Zeitlin BD, Zeitlin IJ, Nor JE. Expanding circle of inhibition: small-molecule inhibitors of Bcl-2 as anticancer cell and antiangiogenic agents. J Clin Oncol. 2008;26:4180–4188. doi: 10.1200/JCO.2007.15.7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CA, Benimetskaya L, Mani S. Antisense strategies for oncogene inactivation. Semin Oncol. 2005;32:563–572. doi: 10.1053/j.seminoncol.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Leone M, Zhai D, Sareth S, Kitada S, Reed JC, Pellecchia M. Cancer prevention by tea polyphenols is linked to their direct inhibition of antiapoptotic Bcl-2-family proteins. Cancer Res. 2003;63:8118–8121. [PubMed] [Google Scholar]