

Abstract
The amyloid-β (Aβ) peptide is a key aggregate species in Alzheimer's disease. Although important aspects of Aβ peptide aggregation are understood, the initial stage of aggregation from monomer to oligomer is still not clear. One potential mediator of this early aggregation process is interactions of Aβ with anionic cell membranes. We used unconstrained and umbrella sampling molecular dynamics simulations to investigate interactions between the 42-amino acid Aβ peptide and model bilayers of zwitterionic dipalmitoylphosphatidylcholine (DPPC) lipids and anionic dioleoylphosphatidylserine (DOPS) lipids. Using these methods, we determined that Aβ is attracted to the surface of DPPC and DOPS bilayers over the small length scales used in these simulations. We also found supporting evidence that the charge on both the bilayer surface and the peptide affects the free energy of binding of the peptide to the bilayer surface and the distribution of the peptide on the bilayer surface. Our work demonstrates that interactions between the Aβ peptide and lipid bilayer promotes a peptide distribution on the bilayer surface that is prone to peptide-peptide interactions, which can influence the propensity of Aβ to aggregate into higher-order structures.
Introduction
Neurodegenerative disorders, including Alzheimer's disease, share a similar mechanism of toxicity (1,2), namely, aggregation of unfolded peptides into amorphous oligomers that coalesce to form an ordered fibril. It is of great importance to understand both the exact steps behind fibril formation from the monomer state and the means of toxicity in these diseases. By further defining integral steps in the aggregation pathway for neurodegenerative disorders (in this work, Alzheimer's disease in particular), we can gain greater insight into the toxic mechanisms and potential therapeutic approaches for a host of fatal diseases.
One of the major aggregate species in Alzheimer's disease is the amyloid-β (Aβ) peptide (3–6). Aβ is a 38–42 amino acid cleavage product of the amyloid precursor protein, a large transmembrane protein of unknown function in the cell (3–5). Aβ contains two domains: a charged domain at the N-terminus and a hydrophobic domain situated at the C-terminus. NMR results (7,8) show that Aβ has a random coil structure in solution at pH 7. Upon onset of Alzheimer's disease, Aβ forms soluble oligomers that aggregate to form ordered fibrils with β-sheet morphology in the hydrophobic domain, as determined through solid-state NMR and electron microscopy (9,10). In this aggregation process, the steps involved in the initiation of aggregation from monomers to small oligomer structures are not well determined. There are many aspects of cellular function that may play a significant role in the early stages of Aβ aggregation, such as cellular pH (11), salt concentration (12), covalent attachments of Aβ due to oxidation, and interactions of Aβ with metal ions (13). However, one hypothesis (14–16) that shows promise for explaining both the early steps of aggregation and the effect of certain risk factors in Alzheimer's disease is the interaction between Aβ and cellular membranes. This hypothesis postulates that interactions between Aβ and lipids promote conversion of disordered Aβ into a partially folded intermediate that will aggregate under favorable conditions. The membrane can affect soluble proteins through a variety of ways: electrostatic interactions between amino acids and charged headgroups (14–18), new partially folded or unfolded free energy minima at the surface (14–18), increased aggregation due to faster diffusion over a two-dimensional (2D) surface (14–18), and a lower surface pH due to anionic lipid headgroups (17–19). In this work, we investigate these lipid-peptide interactions using molecular dynamics (MD) simulations and identify properties of lipid bilayers that may promote peptide-peptide interactions characteristic of aggregation.
Experimental investigations have been able to replicate the aggregation of Aβ peptides in vitro quite accurately. For the most part, the experimental conditions for in vivo and in vitro aggregation are similar; however, one significant difference is that in vitro aggregation requires a much higher peptide concentration (approximately micromolar concentration) to induce aggregation than in vivo aggregation (approximately submicromolar peptide concentration) (20–22). One potential hypothesis (14–16) to explain this discrepancy proposes that interactions with the cell membrane promote altered function and aggregation in vivo. This hypothesis is well founded in biology through signal peptide binding to bilayers during signaling cascades (23,24) and in peptide-lipid binding in toxin-related cell death (23,24). Early experiments that used circular dichroism (CD) spectroscopy to follow structural changes for Aβ incubated with lipid vesicles demonstrated that zwitterionic lipids headgroups (19–21), such as phosphatidylcholine (PC), did not significantly affect peptide structure. However, when Aβ was incubated with anionic lipid headgroups (19–21), such as phosphatidylserine (PS), a clear conversion from a random coil to β-structure was observed. Further, imaging experiments demonstrated that Aβ was aggregating into fibrils at concentrations near in vivo aggregation conditions in the presence of vesicles (25,26). 31P-NMR (27) and x-ray reflectivity (22) results have shown that Aβ peptides interact with anionic lipids and lead to significant alteration of the properties of the bilayer itself. These results provide a clear demonstration that lipids can fundamentally impact the aggregation pathway for Aβ; however, investigators have not been able to determine the exact interactions that are occurring on the bilayer surface that force this conformational change. Some controversies (15) also exist regarding the extent of interactions between Aβ and anionic lipids. In an experimental work (28), a claim was made that Aβ-anionic lipid interactions are weak or nonexistent under certain conditions. Therefore, a detailed understanding of the interactions between Aβ and lipids at the bilayer surface will be integral to resolving these controversies.
Although most experimental approaches do not have the necessary resolution to determine direct protein-lipid interactions on a single-molecule level, MD simulations provide an ideal approach to this system. MD with explicit (29–32) and implicit (30,33–35) solvent and free energy (31,33,35) calculations has been used to study peptide-lipid interactions with good agreement with experimental results. Further, MD has been used extensively with Aβ (36–47). Single-peptide MD simulations confirm a random coil structure for Aβ in solution; however, a transient β-hairpin structure is seen in longer-duration (36,39–42), replica-exchange (45,46), and low-pH (40) simulations. Previous studies used MD simulations of Aβ with lipid bilayers (42,47) to investigate the stability of a preinserted Aβ in a zwitterionic bilayer, but did not investigate the effect of headgroup charges and other bilayer properties on Aβ structure or stability near the bilayer surface. This previous computational work with Aβ further supports the use of MD for investigating the details of Aβ peptide-lipid interactions.
To examine peptide-lipid interactions in this system, we calculated the free energies of peptide binding to the bilayer surface for various lipid headgroup charges and peptide charges. The chosen lipids for these studies have zwitterionic PC and anionic PS headgroups. Lipids with PC headgroups are the most abundant lipids in neural membranes (48). Lipids with anionic PS headgroups play an integral role in localization to cell membranes and programmed cellular death mechanisms (49). Anionic lipids decrease local surface pH (17–19), so it is essential to vary both lipid charge and the peptide charge to understand the influence of electrostatics on the system. By investigating both electrostatic and hydrophobic aspects of the Aβ-bilayer interaction, we can obtain a more detailed picture of the influence of membranes on Aβ aggregation. The results from this work will help to determine the validity of the cell membrane as a catalytic element in Aβ aggregation, and, with knowledge of the toxic mechanism of this class of similar neurodegenerative diseases, will assist in the future treatment and prevention of such diseases.
Materials and Methods
Simulations in solution
Two structures were chosen for simulations of the 42 amino acid Aβ peptide both in solution and near the bilayer surface. By using two structures, one can eliminate some of the bias inherent to having an ordered starting structure. The first structure is Protein Database (PDB) code 1Z0Q and represents a random coil with some helix content as determined by NMR (8). The structure used in calculations was the first NMR structure (8) given in the file deposited in the PDB. The second structure is PDB code 2BEG, which is one peptide taken from the structure of an Aβ fibril as determined by solid-state NMR (9). Residues 1–16 in the N-terminal tail were unstructured and were not included in the PDB file. Therefore, these residues were added using the SYBYL software program (Tripos, St. Louis, MO). This β-hairpin structure is controversial because it is not universally accepted as the accurate monomer structure in fibrils (10). Recent results (10) have shown that the β-sheet structure detected in fibrils may be shared between two monomers. However, for this work, the β-hairpin structure is used because it represents the potential β-structure that can be formed from a monomer of Aβ. All structures from the PDB were edited using GROMACS software to convert the structures to a united atom format described by the GROMACS force field (50,51). Along with the two initial structures used for the simulations, three charge states of Aβ were used. At pH 7, Aβ has a −3 charge due to six aspartic and glutamic acid residues and three lysine and arginine residues, assuming uncharged termini. At pH ∼5, three histidine residues become protonated (19) to give Aβ a neutral charge. Then, at low pH values, the aspartic and glutamic acid residues are protonated to give Aβ a +6 charge. The termini were uncharged in these simulations so that the effect of peptide charge on the peptide-lipid interactions would be isolated to the amino acid side chains only. GROMACS utilities (50,51) were used to change the protonation state of relevant histidine, aspartic acid, and glutamic acid residues to give the appropriate charge for the peptide state. The combination of two initial structures and three pH states for each structure produced a set of six simulations performed in solution.
Each structure was solvated in a 6.4 nm × 6.4 nm × 8.1 nm box, with Na+ or Cl− counterions added to the system to balance the peptide charge, and NaCl salt added to produce a near-physiological concentration of 0.1 M NaCl (Table 1 a). The system was equilibrated with a 3 ns MD simulation, and then 80 ns MD simulations were performed for analysis. Temperature was held constant at 323K using a Nose-Hoover (52) scheme with a relaxation time of 0.5 ps under constant volume (NVT) conditions. All bonds in the system were constrained with the LINCS algorithm (53), which allowed a time step of 3.5 fs. Long-range electrostatics were handled using the SPME algorithm (54), and periodic boundary conditions were used in all three dimensions. The SPC/E model (55) of water was used for all simulations. Secondary structure was calculated using the DSSP package (56) in GROMACS.
Table 1.
Simulation contents for unconstrained simulations
| Starting structure | Aβ pH | SPC/E | Na+ | Cl− |
|---|---|---|---|---|
| a. Simulations in solution: 6.4 nm × 6.4 nm × 8.1 nm box | ||||
| Helix | pH 7 | 10534 | 22 | 19 |
| pH 5 | 10539 | 19 | 19 | |
| pH 3 | 10530 | 19 | 25 | |
| β-Hairpin | pH 7 | 10521 | 22 | 19 |
| pH 5 | 10523 | 19 | 19 | |
| pH 3 | 10515 | 19 | 25 | |
| b. Simulations with DPPC: 6.4 nm × 6.4 nm × 16.3 nm box | ||||
| Helix | pH 7 | 15587 | 27 | 24 |
| pH 5 | 15588 | 24 | 24 | |
| pH 3 | 15585 | 24 | 30 | |
| β-Hairpin | pH 7 | 15576 | 27 | 24 |
| pH 5 | 15580 | 24 | 24 | |
| pH 3 | 15569 | 24 | 30 | |
| c. Simulations with DOPS: 6.4 nm × 6.4 nm × 16.8 nm box | ||||
| Helix | pH 7 | 15906 | 155 | 24 |
| pH 5 | 15907 | 152 | 24 | |
| pH 3 | 15904 | 152 | 30 | |
| β-Hairpin | pH 7 | 15896 | 155 | 24 |
| pH 5 | 15899 | 152 | 24 | |
| pH 3 | 15888 | 152 | 30 | |
Unconstrained simulations on DPPC and DOPS
The six conditions used for Aβ simulations in solution were again used for simulations near a dipalmitoylphosphatidylcholine (DPPC) and dioleoylphosphatidylserine (DOPS) bilayer. Although direct biological considerations would promote the use of lipids such as palmitoyloleoylphosphatidylcholine (POPC) and dipalmitoylphosphatidylserine (DPPS) for our simulations, bilayers containing these lipids differ substantially in their area per headgroup. Since surface charge density is an important parameter for studying electrostatics, we decided to choose PC and PS lipids that have very close areas: DPPC and DOPS. Thus, our model lipid bilayers still contain biologically relevant headgroups.
Initially, both bilayers were brought to an equilibrated state before a peptide was placed near the bilayer. For both the DPPC and DOPS bilayers, a single lipid molecule was built using the SYBYL package, which was then used to create a symmetric 128 lipid bilayer. The DPPC bilayer was equilibrated for 30 ns with 3654 water molecules on the bilayer. The computational details for these simulations are similar to those used for previous simulations with Aβ in solution; however, a constant pressure ensemble (NPT) was used to allow the bilayer to reach an appropriate area per headgroup. The Parrinello-Rahman pressure coupling scheme (57) was used with a barostat relaxation time of 2.0 ps at a pressure of 1 atm. Further, the lipid force field parameters were taken from the work of Berger et al. (58). These simulations used a time step of 4 fs. The DPPC equilibration resulted in an area per headgroup value of 63.6 Å2, which is in agreement with previous experimental (59) and computational (60) results. The DOPS bilayer was also equilibrated for 30 ns with 128 Na+ counterions and 4102 water molecules on the bilayer. The DOPS bilayer equilibration resulted in an area per headgroup value of 63.9 Å2, which is also in agreement with experimental (61) and computational (62) results. Once the bilayers were equilibrated, simulations could be performed with Aβ near the bilayer surface. Both initial starting structures of Aβ at all three pH values were solvated with SPC/E water molecules, Na+ or Cl− counterions and NaCl salt in a 6.4 nm × 6.4 nm × 4.5 nm box. The solvated peptide box was then placed near the surface of the equilibrated bilayer. To ensure that the system was symmetric except for the peptide, a box of SPC/E water with similar ion concentrations was placed below the DPPC and DOPS bilayers to give one peptide with SPC/E water molecules, 128 lipid molecules, and Na+/Cl− ions in a 6.4 nm × 6.4 nm × 16.3 nm box for DPPC simulations (Table 1 b) and one peptide with SPC/E water molecules, 128 lipid molecules, and Na+/Cl− ions in a 6.4 nm × 6.4 nm × 16.8 nm box for DOPS simulations (Table 1 c). For these simulations on DPPC and DOPS bilayers, the peptide center of mass (COM) was placed at a distance of 6.0 nm and 6.2 nm, respectively, from the bilayer COM. This distance ensured that the peptide was completely surrounded by solvent and that no portion of the peptide would be influenced by short-range interactions with the bilayer surface due to the initial configuration of the simulation. The Aβ-bilayer system was then simulated, after energy minimization, for 80 ns. Simulation conditions were similar to the previously described Aβ simulations in solution. A constant volume (NVT) ensemble was used with a time step of 3 fs at a constant temperature of 323K with periodic boundary conditions along all three dimensions. All secondary structure analysis was performed using the DSSP package (56) in GROMACS.
Umbrella sampling simulations with Aβ near DPPC and DOPS
To calculate the free energies of binding of Aβ to the surface of DPPC and DOPS bilayers, umbrella sampling (63,64) was performed. Previous experimental evidence has demonstrated that Aβ has a random coil structure (7,8) in solution. Therefore, only one starting structure was used for umbrella sampling simulations because these calculations were set up to closely replicate an Aβ peptide approaching a bilayer from solution. The final structures from the simulations using the β-hairpin initial configuration of Aβ in solution were all predominantly random coil at the end of the 80 ns simulation and thus were ideal as starting structures for these umbrella sampling calculations. To improve sampling, three initial configurations were used for the Aβ-bilayer system. First, the peptide was placed so that it was parallel to the bilayer surface and neither the charged N-terminus nor the hydrophobic C-terminus was closer to the bilayer surface. Then the peptide underwent rigid-body rotation so that either the N-terminus or the C-terminus was close to the bilayer surface. Although these extra initial conditions cannot fully overcome sampling issues associated with limited timescales of MD simulations, the multiple free-energy calculations from the three initial conditions at each pH on DPPC and DOPS will improve the validity of the calculated free-energy profile. For each initial configuration, the random coil peptide was solvated with SPC/E water molecules, Na+ or Cl− counterions, and NaCl salt in a 6.4 nm × 6.4 nm × 4.2 nm box. This peptide box was then placed above the equilibrated DPPC or DOPS bilayers, and a similar box of SPC/E water and ions without the peptide was placed below the bilayer for symmetry purposes. This resulted in a system of 1 Aβ peptide above a bilayer of 128 lipids with SPC/E water molecules and Na+ or Cl− ions in a 6.3 nm × 6.3 nm × 15.8 nm box for simulations with DPPC (Table 2 a) and one peptide above a bilayer of 128 lipids with SPC/E water molecules and Na+ or Cl− ions in a 6.3 nm × 6.3 nm × 16.1 nm box for simulations with DOPS (Table 2 b). The COM separation for Aβ and the DPPC or DOPS bilayer in this initial configuration file was between 6.3 nm and 6.6 nm. For each initial configuration, a short 3 ns equilibration simulation was performed. In these simulations, the z-dimension of the peptide was constrained so that the peptide-bilayer COM separation would remain greater than 6.0 nm but the system would still be able to equilibrate. The simulation details of this short equilibration were the same as for the previous unconstrained simulations except that a 1 fs time step was used.
Table 2.
Simulation contents for umbrella sampling simulations
| Starting structure | Aβ pH | SPC/E | Na+ | Cl− |
|---|---|---|---|---|
| a. Simulations with DPPC: 6.4 nm × 6.4 nm × 15.8 nm box | ||||
| Random coil | pH 7 | 14764 | 27 | 24 |
| pH 5 | 14762 | 24 | 24 | |
| pH 3 | 14759 | 24 | 30 | |
| b. Simulations with DOPS: 6.4 nm × 6.4 nm × 16.1 nm box | ||||
| Random coil | pH 7 | 15083 | 155 | 24 |
| pH 5 | 15081 | 152 | 24 | |
| pH 3 | 15078 | 152 | 30 | |
For the umbrella sampling (63,64), 14 windows were chosen. These windows spanned the peptide-bilayer COM separations from 6.0 nm to 2.1 nm. Therefore, the spacing between each window was 0.3 nm, which would enable sufficient sampling. Further, this range of distance allows the peptide to be pulled from a full solvated, solution-like environment onto the surface of the bilayer and then into the interfacial region of the bilayer. The pulling was accomplished by applying a harmonic force with a force constant of 500 kJ/(mol∗nm2). For each window with the parallel initial configuration, an 80 ns MD simulation was performed. After error analysis was performed on the 80 ns MD simulations, it was determined that 40 ns simulations were sufficient to provide an error of ±4.1kcal/mol for the free energy of binding, which is adequate for the free energies calculated in this work. Error analysis was performed with the use of a block error analysis scheme. In this scheme, a 40 ns simulation was broken into smaller blocks and the average value and standard deviation for the potential of mean force at each COM separation was calculated. The maximum standard deviation converged to 4.1 kcal/mol after the simulation was split into blocks, for up to a total of 30 blocks. Thus, only 40 ns MD simulations were performed in each window for the N-terminus down and C-terminus down initial configurations. The computational details of the simulations performed in each window were exactly the same as in the previous unconstrained simulations except that a 3 fs time step was used. Again, the system used a constant volume (NVT) ensemble with periodic boundary conditions along all three dimensions with a constant temperature of 323K. For each window, secondary structure was calculated using the DSSP program (56) in GROMACS. Free energy was calculated for each configuration using the weighted histogram analysis method (WHAM) (65) adapted for in-house code. The COM fluctuations from the second 40 ns of the parallel initial configuration MD simulations were used for free-energy calculations to maintain a consistent 40 ns simulation time among all initial configurations, while the COM fluctuations from the full 40 ns MD simulations for the N-terminus down and C-terminus down initial configurations were used for free-energy calculations. To calculate an averaged free energy of binding for the three initial configurations of Aβ at each pH on either DPPC or DOPS, the COM fluctuations for all three initial configurations were combined and then analyzed using WHAM. If the free energies for each initial configuration are calculated with WHAM and then averaged by obtaining an unweighted average of the sum of the exponentials of individual potentials of mean force, as previously described (66), one can also get a potential of mean force if enough orientationally dependent potentials are included in the unweighted average, and if the peptide is a rigid body. Since both of these conditions are strongly violated in our case, we expect that this method will produce a large error. Thus, we calculated the free energies by combining the COM fluctuations and then analyzing them with WHAM. Nevertheless, we also calculated the free-energy curves for each initial orientation of the peptide to perform a contact value analysis. A contact value for Aβ binding to the bilayer was calculated for each umbrella sampling window throughout the umbrella sampling simulations. Contact was defined as a separation of less than 5Å in the Z-coordinate between any atom on a given amino acid and the average position of the phosphate atom in the lipid headgroup, which was calculated using the 64 phosphate atoms on the bilayer leaflet that Aβ was closest to. The residue was given a contact value of one if any atom in the amino acid was within 5Å of the average phosphate, and a contact value of zero if it was not. Contact values were calculated for all amino acids in Aβ and averaged over all time steps in the simulation, which gives contact values between zero (no contact) and 42 (full contact/binding) for a given umbrella sampling window. The contact values were then used to calculate a 2D free-energy surface using the unweighted probabilities obtained from the above-mentioned WHAM calculations on the COM separation coordinate and conditional probabilities calculated from the distribution of contact scores within each umbrella sampling window.
Results
Simulations in solution
Initial simulations were performed with the 42-amino acid Aβ peptide in solution. These simulations were used as a test of the protein force field to determine whether experimental solution structures would be obtained during simulations starting from ordered structures. Further, these simulations provide a baseline for comparison with the results from simulations of Aβ-lipid systems. Two initial structures were chosen for these simulations to eliminate any bias due to the starting configuration (Fig. 1; see Materials and Methods for further details of the structures). Both structures were fully solvated with appropriate counterions and with NaCl salt. The simulations were performed with a 1 ns equilibration followed by a full 80 ns simulation. The secondary structure of both the helix and β-hairpin starting structures changed drastically throughout the simulation. Both the helix and the β-hairpin structures lost the majority of the ordered structure content to become a full random coil (Table 3). These results agree with previous experimental NMR (7,8) and CD (8) results, as well as previous simulations (36,39–42,45), which showed that Aβ has a random coil structure in solution at pH 7.
Figure 1.
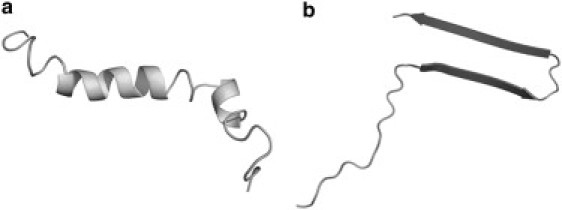
Initial configurations of Aβ used for simulations. (a) PDB code 1Z0Q is a coil-dominated structure (8). (b) PDB code 2BEG is a preformed β-hairpin (9).
Table 3.
Average structures for unconstrained Aβ simulations
| Initial conditions |
Final structure |
|||
|---|---|---|---|---|
| Starting structure∗ | Aβ pH† | In solution | On DPPC | On DOPS |
| helix | pH 7 | coil | coil/turn | helix |
| helix | pH 5 | coil | coil/turn | helix |
| helix | pH 3 | coil | coil/turn | helix |
| β-hairpin | pH 7 | coil | coil | β-hairpin |
| β-hairpin | pH 5 | coil | coil | β-hairpin |
| β-hairpin | pH 3 | coil/turn | coil | β-hairpin/turn |
Indicates whether the peptide was originally the 1Z0Q (helix) or 2BEG (β-hairpin) derived structure.
Refers to the charge on the peptide during the simulation.
In the studies of Aβ interactions with bilayers of various lipid headgroup charges, lipids with anionic headgroups were used as a model system. It has been shown that anionic lipids can lower the pH (17–19) of solution near the bilayer, which will in turn alter the protonation state of proteins near these bilayers. Calculations (19) fitting experimental data with Aβ bound to anionic palmitoyloleoylphosphatidylglycerol (POPG) lipids show that protonation of the three histidine residues upon binding does occur, which further supports the use of multiple pH states to study Aβ binding. Therefore, we also investigated various protonation states of Aβ near bilayers. Similarly to the previous simulations of Aβ in solution at pH7, simulations of Aβ at different protonation states in solution were performed for comparison with simulations of the Aβ-lipid bilayer systems. Aβ can undergo two major protonation events (termed pH 5 and pH 3 simulations) that result in a neutral and +6 net charge of the peptide (see Materials and Methods section for further details). Not only will studies with these three pH states provide insight into the effect of pH on Aβ structure in solution and near lipid bilayers, the use of an anionic, neutral, and cationic peptide will demonstrate the direct importance of electrostatics on peptide-charged bilayer interactions.
From our simulations, we observed that both initial peptide structures at pH 5 and the helical peptide at pH 3 essentially lost all secondary structure during the simulation, resulting in a random coil as the final structure (Table 3). For the β-hairpin starting structure at pH 3, the final structure was not completely random coil but had some transient turn content. Thus, this final configuration can be considered as having some transient order because a turn (67) is not purely random—it is a somewhat intermediate structure. Nevertheless, this amount of ordered structure at pH 3 is small, and it can be concluded that a random coil was the primary structure for Aβ in solution regardless of the starting structure used in the simulation and the total charge on the peptide.
Simulations near DPPC and DOPS bilayers
The results from the simulations of Aβ in solution were then extended to simulations of Aβ with a zwitterionic DPPC bilayer or an anionic DOPS bilayer. For simulations on the fluid DPPC and DOPS bilayers, both the helix starting structure and β-hairpin starting structure were again used. Also, simulations were performed at all three charge states for the Aβ peptide. Although a DPPC bilayer would not affect local pH (19) and thus not induce protonation state changes on Aβ, performing these simulations of Aβ near neutral lipids can provide insight into the role of the protonation state on the structure of Aβ near a surface with which it should not extensively interact. The results of the simulations in solution demonstrate that 80 ns is an adequate simulation time to allow for the peptide to undergo significant conformational flexibility, considering the computational restraints. Although it was possible for Aβ to pass through the upper periodic boundary and interact with the bottom leaflet of the bilayer in the chosen simulation setup, this did not occur during simulations because Aβ was clearly attracted to the surface of the DPPC bilayer and was near the upper leaflet surface for the majority of the simulation time. Near the DPPC bilayer, the helix starting structure at each peptide pH unfolded into a structure dominated by random coil and turns, whereas the β-hairpin starting structure unfolded into a full random coil, similar to the simulations performed in solution (Table 3). For these simulations, it is clear that, although Aβ was attracted to DPPC near the bilayer surface, the DPPC bilayer did not affect the overall secondary structure content of peptide. These results agree with previous experimental results (19–21) showing that vesicles composed of neutral lipids do not alter the secondary structure of Aβ when mixed. Near the DOPS bilayer, analogously to the simulations with DPPC, Aβ was attracted to the surface of the bilayer in all simulations, independent of peptide charge. For simulations involving the helix starting structure at all three pH values, the DOPS bilayer strongly enhanced the helical structure, especially near the N-terminus of the peptide (Table 3). For the β-hairpin starting structure, the β-hairpin configuration was mostly retained at pH 7 and pH 5, with some turn structure also developing. At pH 3, the β-hairpin unfolded slightly into a structure dominated by turns. Therefore, it appears that the DOPS bilayer influences the secondary structure of Aβ so that the random coil observed in solution or near a zwitterionic bilayer is not formed. These results agree to a certain extent with previous experimental measurements (19–21) that showed a significant secondary structure in Aβ near anionic lipids; however, the time restrictions inherent to MD simulations prevent observation of any significant secondary structure change on the surface of DOPS bilayers. These previous experimental measurements demonstrated that a random coil Aβ in solution will be converted to a β-sheet dominated structure upon addition of anionic vesicles (19–21,68), which can be converted to an α-helix upon further addition of anionic vesicles. Although the time constraints of these simulations limit the potential structural conversion for a single peptide, they also show that the anionic bilayer stabilizes both the β-structure and helix structure. The qualitative results of simulations with Aβ near DPPC and DOPS prompted us to further study this system using a more quantitative method to help elucidate why Aβ appeared to be attracted to the bilayer surface regardless of the peptide charge or bilayer charge.
Umbrella sampling simulations
To describe the Aβ-bilayer interactions by means of a quantitative method, umbrella sampling techniques were used. Umbrella sampling (63–65) determines a free energy of binding of Aβ to the surface of the lipid bilayer using a systematic routine. For these simulations, the initial Aβ structure was taken to be the final structure of the Aβ simulations in solution from starting β-hairpin structures. The final structures of the Aβ β-hairpin simulations in solution had very little ordered structure and were predominantly random coil. Therefore, the use of these random coil starting structures for umbrella sampling simulations will closely mimic the experimental conditions of an Aβ peptide in solution, which has a mostly random coil structure (7,8), approaching a cell membrane. Further, for each starting structure, three initial configurations of Aβ with respect to the bilayer surface were used: one with the N-terminus of Aβ close to the bilayer surface, one with the C-terminus of Aβ close to the bilayer surface, and one in which Aβ is parallel to the bilayer surface so that neither terminus is closer to the bilayer. The use of three initial configurations will improve the sampling of the free-energy calculations (see Materials and Methods section for further details). From these simulations, a free energy of binding for Aβ from solution to the bilayer surface can be calculated and compared with experimental predictions.
The calculated average free energies of binding are listed in Table 4 and presented as free-energy profiles in Fig. 2. As predicted from the unconstrained MD simulations, Aβ was attracted to the bilayer surface independently of the Aβ charge or bilayer headgroup charge. For calculations on the DPPC bilayer, Aβ at all pH values had ΔGbinding ≈ −16 kcal/mol to −19 kcal/mol (ΔA was actually obtained in our calculations, but, as is common for condensed systems, ΔG≈ΔA). This reduces to a ΔGbinding ≈−0.4 kcal/mol∗residue, which is close to previous experimental predictions for peptide-lipid binding interactions (68). For calculations on the DOPS bilayer, the free energy of binding depended significantly on the Aβ charge. The free energy of binding for the pH 5 and pH 3 Aβ were within error (±4.1 kcal/mol, as described in the Materials and Methods section) of free energies of binding for Aβ with DPPC. However, the free energy for binding of the anionic pH 7 peptide to DOPS was less then half of the binding free energy of the pH 5 and pH 3 peptide to DOPS. This discrepancy in binding free energies is likely due to the interplay of electrostatic interactions with lipid headgroups and interactions between the hydrophobic residues of Aβ and the interfacial region of the bilayer. For the highly negative free energies of binding on DPPC or DOPS, the majority of favorable interactions between the peptide and bilayer, which lead to the large, negative free energy of association, are derived from the interactions between the hydrophobic residues of Aβ and the interfacial region of the bilayer. However, for the pH7 peptide binding to DOPS, although the hydrophobic C-terminus of the peptide allows for a negative free energy of association for Aβ to DOPS, the anionic DOPS headgroups, even partially screened by Na+ counterions, interact strongly with the charged N-terminus of Aβ and prevent the full association of the peptide with the interfacial portions of DOPS.
Table 4.
Calculated free energies for binding of Aβ to the bilayer surface
| Bilayer type | Aβ pH state | Free energy (kcal/mol) |
|---|---|---|
| DPPC | pH 7 | −16.0 |
| pH 5 | −18.4 | |
| pH 3 | −18.9 | |
| DOPS | pH 7 | −6.6 |
| pH 5 | −14.1 | |
| pH 3 | −15.6 |
Figure 2.

Free-energy profiles for binding of Aβ to the surface of the (a) DPPC or (b) DOPS bilayer. The error associated with the minimum of these potentials is 4.1 kcal/mol.
Along with the magnitude of the free energy of binding, the free-energy profiles from these umbrella sampling calculations provide further information about the system (Fig. 2). The free-energy profiles supply some insight into the length scales for binding events. In the profiles, the free energy decreases smoothly as the peptide approaches the bilayer. For some of the Aβ-bilayer combinations, such as Aβ at pH 3 approaching a DOPS bilayer, small barriers are present in the free-energy profiles. These barriers have values in the range of 0.1–0.2 kcal/mol and are therefore insignificant at the considered temperature. Thus, the point in the free-energy curve in which the free energy begins to decrease marks the distance at which Aβ becomes significantly attracted to the bilayer surface. For Aβ binding to the DPPC bilayer and the pH 7 peptide binding to the DOPS bilayer, this distance is at a COM separation of 4.5 nm. For the pH 5 and pH 3 peptide binding to the DOPS bilayer, this distance is at a COM separation of 5.1 nm. Considering a bilayer leaflet thickness of ∼2–2.5 nm, the COM of the peptide is separated from the bilayer surface by over 2 nm at these COM distances, which is a significant length and is not appropriate for the interactions with the interfacial region of the bilayer that may be driving this binding. Thus, to better understand this binding and to demonstrate that this COM separation can be a deceiving coordinate, we calculated a contact value during the binding process (see Materials and Methods for details). In short, a value of one is assigned to any residue of Aβ that is bound to the interfacial region of the bilayer, and a value of zero is assigned to any residue that is not bound. This value is calculated for each residue and averaged over the full simulation. The contact value is calculated for every window in the umbrella sampling, and a 2D free-energy surface as a function of COM separation and number of contacts is determined (Fig. 3). The plot shown in Fig. 3 for the parallel initial configuration of the pH 7 Aβ peptide binding to DPPC is characteristic of most free-energy profiles for binding. At large COM separations, there is no contact between any amino acids and the bilayer surface. Then, at distances of ∼4 nm to 4.5 nm, the first amino acids of Aβ come in contact with the interfacial region of the bilayer, as shown in snapshot 1. As free energy begins to decrease significantly, more amino acids come into contact with the bilayer surface, as shown in snapshot 2. Finally, at the free-energy minimum, 90–95% of amino acids are in contact with the bilayer surface and the peptide is clearly bound to the interfacial region of the bilayer, as shown in snapshot 3. Snapshot 3 also demonstrates the parallel binding of the Aβ peptide to a DPPC bilayer, which was previously mentioned as a causative factor in the large, negative free energy of binding of Aβ to DPPC. Further, the free-energy surface shows that the most probable path taken for binding is that the Aβ peptide will approach the bilayer surface without making significant contact. Then, once the peptide is close to the bilayer surface at COM separations around 4 nm (snapshot 1), the peptide will begin to quickly make contact with the bilayer surface and the free energy will drop drastically as the peptide approaches the surface (snapshot 2) and tightly binds with the surface (snapshot 3). An alternate path in which the peptide creates contacts monotonically as it approaches the bilayer surface is not favored because it requires many more contacts at a given COM separation, which would force the peptide to extend and expose hydrophobic residues to solvent to ensure a free energy similar to that of the more favored binding path. Of interest, the pH 7 Aβ peptide has a lower number of total amino acids in contact with the DOPS bilayer surface at the free-energy minimum (only 36 of 42 amino acids are in contact, compared with 40 of 42 amino acids in contact in the other systems). This lower extent of contact between the anionic peptide and DOPS, due to electrostatic repulsion on the bilayer surface, helps to explain the smaller free energy of binding of pH 7 Aβ to DOPS even though the contact of 36 amino acids to the bilayer surface provides a favorable free energy of binding and drives the binding process. Further, the use of this contact score demonstrates that the large COM separations described by the free-energy profiles are still compatible with binding driven by association of the peptide with the interfacial region of the bilayer. Finally, the radius of gyration of Aβ was calculated as a function of COM separation (data not shown). For all peptide-bilayer combinations, the radius of gyration was constant until the peptide began to make contact with the bilayer surface. The radius of gyration then increased and peaked as the peptide made extensive contacts with the bilayer surface. Once the peptide had made a significant number of contacts with the bilayer surface, the radius of gyration decreased to a value slightly lower than the prebinding level and remained constant as the peptide finished the binding process. These radius-of-gyration calculations demonstrate that, similarly to the contact score calculations, the peptide alters its structure to make extensive contacts with the bilayer as it begins to interact with the bilayer surface.
Figure 3.
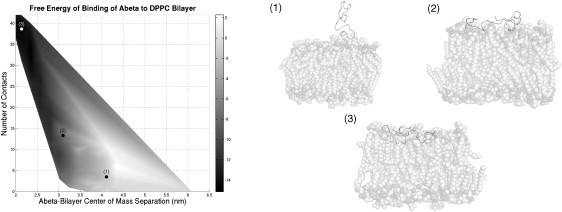
Free energies of binding of parallel pH 7 Aβ to the DPPC bilayer as a function of Aβ-bilayer COM separation and number of contacts. The surface shows (using the scale next to the figure) the relative free-energy change in units of kcal/mol as the peptide binds to the bilayer surface. Other peptide-bilayer combinations showed a similar free-energy surface. The snapshots represent points along the binding trajectory and show the extent of contact at 1), 4.2 nm; 2), 3.0 nm; and 3), 2.1 nm COM separations.
Along with inspecting quantitative aspects of Aβ binding to the bilayer surface by using umbrella sampling, we were able to analyze the secondary structure of Aβ throughout the process. Because each umbrella involved an MD simulation with a restrained COM separation (63–65), secondary structure analysis could be performed at each window for the entire simulation time. In these simulations, only the earliest stages of binding could be investigated due to the temporal limitations of simulations. In the Aβ-membrane binding process, we expect that the majority of conformational change will occur after significant binding has occurred. Therefore, the secondary structure analysis will provide insight into the earliest stages of conformational change and may help to predict any significant secondary structure change that occurs after binding. For all bilayer and Aβ combinations, the secondary structure was not greatly affected until the peptide came in full contact with the bilayer surface. Upon full contact with the bilayer surface, the secondary structure was influenced by the bilayer. For the simulations on a DPPC bilayer, the secondary structure remained a random coil. This is exactly as expected for the zwitterionic DPPC based on the unconstrained simulations mentioned above and in experimental results (19–21). For simulations on the DOPS bilayer, both turn and β-structure content increased upon contact with the bilayer for all Aβ pH regimes, similarly to the previous unconstrained simulations near the DOPS bilayer. However, these resultant transient β-structures were not nearly as well ordered as the β-hairpin (9) used in the initial unconstrained simulations and thus they only represent an intermediate Aβ structure. The DOPS bilayer was able to introduce some ordering of the Aβ peptide, but not enough to fully structure a single peptide. Similarly to the unconstrained simulations, the time restrictions imposed by all-atom MD simulations prevent observation of significant secondary structure changes on the timescales analyzed here. Other methods, such as parallel tempering, may be required to observe any structural change, or it may be that the structural changes observed in the experiments are due to protein-protein interactions formed in oligomers and are not stable on the single-peptide level.
Unconstrained simulations at free-energy minima
To study the effect of peptide-lipid interactions occurring on the bilayer surface of the Aβ-bilayer system, we performed the density-profiles analysis presented in Fig. 4. Density profiles for the system were calculated using GROMACS utilities (50,51). To ensure that the COM constraints did not influence the distribution of the peptide on the bilayer surface, unconstrained MD simulations were performed. For each of these simulations, the final structure from the umbrella sampling simulation in the window that was closest to the free-energy minima was chosen. For all three Aβ-DPPC simulations, the 2.1 nm COM separation window was closest to the free-energy minimum and was thus used for the initial structure of unconstrained simulations. For the Aβ-DOPS simulations, the 2.4 nm COM separation window final structures were used. The computational details of these simulations were exactly the same as for the previous umbrella sampling simulations except that the harmonic potential restraint was removed and each simulation was performed for 80 ns. The density profiles plotted in Fig. 4 were taken from the initial configuration with a free-energy profile closest to the average free-energy profile, which indicates that this initial configuration is the heaviest weighted initial configuration for the calculations. Thus, the density profiles plotted with DPPC were pH 7: parallel initial configuration, pH 5: N-terminus down initial configuration, and pH 3: C-terminus down initial configuration; with DOPS they were pH 7: C-terminus down initial configuration, pH 5: parallel initial configuration, and pH 3: parallel initial configuration.
Figure 4.

Density profiles of Aβ on DPPC and DOPS bilayers calculated from 80 ns simulation at the COM separation closest to the free-energy minima of profiles in Fig. 2. All plots on DPPC are taken at a COM separation of 2.1 nm. All plots on DOPS are taken from simulations at a COM separation of 2.4 nm. The “Aβ charged section” refers to residues 1–22 of the peptide, and the “Aβ hydrophobic section” refers to residues 23–42.
Aβ was separated into two segments for density calculations: residues 1–22, which are primarily charged and hydrophilic residues, and residues 23–42, which are primarily hydrophobic residues. In these density plots, both the charged and hydrophobic sections of Aβ on DPPC appear to be clearly bound to the bilayer where interactions with the interfacial regions of the bilayer dominate. At all pH values on DPPC, the charged section of Aβ and the hydrophobic section of Aβ overlap significantly with the interfacial portions of the DPPC density, creating an Aβ distribution wherein Aβ is parallel to the bilayer surface at the interface of the hydrophobic sections of the bilayer. Although these possible hydrophobic interactions with the bilayer may not involve hydrophobic insertion of the peptide into the bilayer core, binding to the interfacial region of the bilayer will lead to removal of water from the peptide and subsequent interactions with the interface of the hydrophobic core of the bilayer, which drives the binding. For Aβ on DOPS, it is clear that electrostatic interactions influence the distribution of the peptide on the bilayer surface due to different peptide density distributions concurrent with pH. At pH 7, the charged section of the peptide is repelled from the bilayer surface and remains outside of the bilayer density, whereas the hydrophobic section of Aβ is clearly distributed in the interfacial region of the bilayer. This creates a peptide distribution wherein Aβ, at pH 7, is situated almost perpendicular to the bilayer surface, with the hydrophobic region interacting with the bilayer interfacial region and the charged section repelled from the surface (Fig. 5). For the pH 5 and pH 3 Aβ on DOPS, the charged and hydrophobic sections of the peptide both clearly overlap with the DOPS density. However, overlap of the hydrophobic section of Aβ with the interfacial region of the bilayer is still more extensive then overlap of the charged section of Aβ with the interfacial region of the bilayer (Fig. 4). Also, in comparison to the distribution of Aβ on the DPPC bilayer, pH 5 and pH 3 Aβ is more solvent-exposed and less tightly bound to the interfacial surface of the bilayer, as indicated by both the lower overlap of either region of the peptide with the DOPS bilayer in comparison to the significant overlap of both regions of Aβ with the DPPC bilayer, and snapshots from the simulations (Fig. 5). These peptide and lipid charge-dependent density distributions of Aβ on the bilayer surface clearly demonstrate the effect of both electrostatic and interfacial interactions with this region of the bilayer and may play a role in the availability of Aβ for peptide-peptide interactions near the bilayer surface, which drives aggregation.
Figure 5.
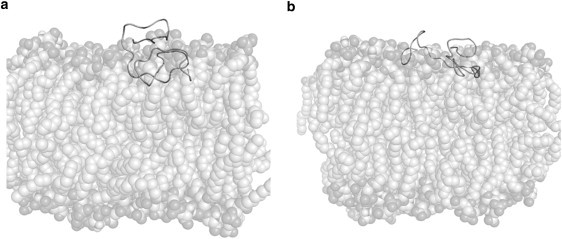
Comparison of simulation snapshots from 80 ns unconstrained MD simulations of (a) pH 7 Aβ and (b) pH 5 Aβ on DOPS at the free-energy minima COM separation.
Discussion
The results obtained from simulations with the 42 amino acid Aβ peptide provide insight into the detailed interactions that occur between Aβ and lipids on the surface of a pure lipid bilayer. The unconstrained simulations both in solution and near a DPPC or DOPS membrane demonstrate that the MD techniques used in this study can effectively replicate various experimental results. During the simulations in solution, Aβ unfolded into a random coil peptide from ordered starting structures. Near bilayers, Aβ was attracted to both the DPPC and DOPS bilayers over the short length scales used in these simulations. The DOPS bilayer stabilized the secondary structure to a greater extent than the DPPC bilayer. These results support previous experimental work using CD and NMR spectroscopy (19–21), which demonstrated that the addition of anionic vesicles to a solution of random coil Aβ peptides leads to a significant change in the secondary structure of the peptide, whereas the addition of zwitterionic vesicles does not affect the peptide structure.
In this work, the most insightful results were derived from the umbrella sampling simulations on DPPC and DOPS bilayers. Not only did these calculations provide quantitative details for the extent of attraction of Aβ to the bilayer surface through free energies of binding, the setup of these simulations allowed for detailed analysis of peptide structure and distribution as Aβ systematically approached the bilayer surface. This analysis revealed intriguing aspects of the Aβ-bilayer system. The umbrella sampling simulations provided insight into the distribution of Aβ on the bilayer surface dependent on peptide and lipid headgroup charges. From the density profiles in Fig. 4 and the simulation snapshots in Figs. 3 and 5, it is apparent that electrostatic interactions at the bilayer surface greatly influence peptide distribution. On the DPPC bilayer, Aβ, independently of peptide charge, sits essentially parallel to the bilayer surface near the interface between the headgroup and hydrophobic core regions of the bilayer. This orientation maximizes interactions with the interfacial region of the bilayer throughout the peptide without completely burying hydrophilic and charged residues found on the N-terminus of the peptide in the bilayer core. On DOPS, Aβ does not adopt this parallel arrangement and instead promotes a much more superficial interaction with the bilayer surface for the neutral (pH 5) and cationic (pH 3) Aβ peptides. Further, for the anionic pH 7 peptide, an almost perpendicular arrangement is observed wherein the hydrophobic C terminus of the peptide interacts with the hydrophobic core of the bilayer while the hydrophilic N-terminus becomes solvent-exposed. This configuration results from the interplay of interfacial association of C-terminal tail of the peptide with the hydrophobic core of the bilayer and electrostatic repulsion between the anionic N-terminal tail and anionic lipid headgroups. Thus, for the neutral and cationic Aβ bound to DOPS, the energetically favorable electrostatic interactions between the peptide and the lipid headgroups prevent the extreme solvent exposure of the N-terminus. However, these electrostatic attractions between the charged headgroups and the N-terminus amino acid side chains also prevent the tight association of the N-terminus with the interfacial region of DOPS, in contrast to the peptide distribution on DPPC. This attraction near the headgroup region of DOPS with the pH 5 and pH 3 peptides promotes a more solvent-exposed distribution of the N-terminus of the peptide, which forces the entire peptide to be bound less tightly to the bilayer interface and thus more exposed for protein-protein interactions that may drive oligomerization on the bilayer surface. Therefore, the charge on Aβ during binding to an anionic bilayer surface will significantly influence the distribution of the peptide upon nonspecific binding.
Further, secondary structure analysis during the binding process provides some insight into the computational approach to this system. Any peptide secondary structure change required that the peptide be in full contact with the bilayer, which occurred near the free-energy minima presented in Fig. 2. Even though the peptide began to make contact with the bilayer at large COM separations, secondary structure change was only seen when the peptide was in full contact with the bilayer at COM separations of 2.1–2.4 nm. Further, this secondary structure change was not very extensive. Only in the extreme case of a pH 3 peptide on a DOPS bilayer was any secondary structure change observed. For the more physiologically feasible pH 5 peptide on DOPS, there was some transient stabilization of β-structure, but not to the extent of formation of a distinct β-structure as in the predicted fibril structure of Aβ. Therefore, our results appear to support the hypothesis that the bilayer cannot fully order a single peptide into a fibril-like structure, but likely acts to stabilize an intermediate state that is aggregation-prone. Further, recent results indicate that the β-structure observed in Aβ fibrils is not formed from a single peptide but is a β-structure shared between two Aβ peptides (10). If these structural predictions hold true, we are unlikely to see any physiologically relevant formation of a β-hairpin in these simulations, as β-structure formation would be due to peptide-peptide interactions facilitated by the bilayer surface. It is also possible that the 40 ns timescales used in this simulation are not adequate for observing significant secondary structure changes. The 80 ns unconstrained MD simulations at the free-energy minima were also analyzed for secondary structure change, and very little structural change was observed. Throughout 120 ns of combined unconstrained and constrained MD simulations at the free-energy minima, secondary structure change was transient at best. Thus, approaches such as replica exchange, similar to some previously performed work (69), or coarse-grained MD will likely be required to adequately explore Aβ secondary structure formation on the bilayer surface.
The results of this work lead to a rough mechanism for elucidating how the detailed balance between electrostatic and hydrophobic forces on the bilayer surface may affect Aβ aggregation. Initially, the Aβ peptide is brought close to the surface of a bilayer due either to diffusion (through interaction with sugar groups on lipids, such as gangliosides) or to cleavage from the amyloid precursor protein. Once the peptide is close enough to the surface, it will favorably bind with the lipids. If this binding occurs on a mostly zwitterionic bilayer, the peptide will strongly interact with the interface at the hydrophobic core of the bilayer, as seen in the density profiles of Fig. 4, thus precluding extensive interactions with other nearby peptides and preventing any secondary structure change, in agreement with previous experiments (19–21). However, if this binding occurs on an anionic bilayer, the peptide will not be as strongly associated with the bilayer core and will be more exposed to the solvent and other bound peptides. If the anionic headgroups on lipids are able to lower the local pH by one to two units, the hydrophobic portion of the peptide will become exposed, as demonstrated in density profiles in Fig. 4, and more likely to interact with other nearby peptides, thus driving oligomerization. Also, previous research (11) has shown that fibrilization occurs more rapidly in solution at a lower pH (≈5). Therefore, lowering pH near the anionic lipid surface may also promote aggregation by intrinsically increasing protein-protein interactions through a reduction in the electrostatic repulsion between peptides, which, along with altering peptide distribution on the bilayer, will promote oligomer formation. On the basis of previous structural determinations (10), it is likely that the resulting peptide-peptide interactions on the bilayer surface will drive the secondary structure changes observed in experiment (19–21) and promote fibrilization. Therefore, an anionic lipid membrane appears to promote aggregation by 1), increasing peptide diffusion by altering diffusion from a 3D to a 2D process; 2), locally increasing Aβ concentration on the bilayer surface due to the highly favorable free energy of binding; and 3), decreasing the local pH on the bilayer surface to promote an Aβ configuration that would be amenable to protein-protein interactions that can drive oligomerization.
Many aspects of this system remain to be elucidated by future MD simulations. As mentioned above, it would be very interesting to employ replica-exchange MD to analyze Aβ secondary structure changes and determine the direct role of the bilayer on peptide secondary structure near the bilayer surface. Further, simulations using multiple peptides on the bilayer may provide insight into the role of peptide-peptide interactions on early oligomer formation near the bilayer surface. Finally, a study similar to a previous replica-exchange MD investigation (69) using the WALP peptide on the DPPC bilayer, in which both bilayer surface binding and peptide insertion into the bilayer core were simulated with subsequent calculation of a 2D free-energy surface, would be very informative for this system. For the study presented here, which examines only Aβ binding to the bilayer surface, a 2D free-energy surface calculation using a second reaction coordinate similar to the extent of helix formation used in the WALP-DPPC study is not applicable. However, Aβ binding and insertion could be studied using a similar order parameter, and a free-energy surface for the full process could be calculated. Performing such a study on the full insertion process would provide great insight into a full range of Aβ-bilayer interactions that would only be available on the detailed scale of MD simulations. Thus, future experimental and computational endeavors with Aβ on the bilayer surface will be integral to confirming that the structural change observed in experiment is due to protein-protein interactions that occur during the early stages of oligomerization, and essential for further characterizing the influence of anionic membranes on Aβ aggregation in Alzheimer's disease.
Acknowledgments
The authors thank Professor O. Andersen and Dr. G. Hummer for useful suggestions, Z. Zhang for technical assistance with the MD simulations, and V. Williams and the UNC Research Computing Group for providing and maintaining the computing resources used in this work. This work was supported by the National Science Foundation under grant number MCB-0615469.
Contributor Information
Charles H. Davis, Email: cdavis@email.unc.edu.
Max L. Berkowitz, Email: maxb@unc.edu.
References
- 1.Thirumalai D., Klimov D.K., Dima R.I. Emerging ideas on the molecular basis of protein and peptide aggregation. Curr. Opin. Struct. Biol. 2003;13:146–159. doi: 10.1016/s0959-440x(03)00032-0. [DOI] [PubMed] [Google Scholar]
- 2.Stefani M. Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world. Biochim. Biophys. Acta. 2004;1739:5–25. doi: 10.1016/j.bbadis.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Kang J., Lemaire H.-G., Unterbeck A., Salbaum J.M., Masters C.L. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 4.Miller D.L., Papayannopoulos I.A., Styles J., Bobin S.A., Lin Y.Y. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer's disease. Arch. Biochem. Biophys. 1993;301:41–52. doi: 10.1006/abbi.1993.1112. [DOI] [PubMed] [Google Scholar]
- 5.Selkoe D.J. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 6.Shankar G.M., Li S., Mehta T.H., Garcia-Munoz A., Shepardson N.E. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hou L., Shao H., Zhang Y., Li H., Menon N.K. Solution NMR studies of the Aβ (1–40) and Aβ (1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 8.Tomaselli S., Esposito V., Vangone P., van Nuland N.A.J., Bonvin A.M.J.J. The α-to-β conformational transition of Alzheimer's Aβ-(1–42) peptide in aqueous media is reversible: a step-by-step conformational analysis suggests the location of β conformational seeding. Chembiochem. 2006;7:257–267. doi: 10.1002/cbic.200500223. [DOI] [PubMed] [Google Scholar]
- 9.Lührs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B. 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sasche C., Fandrich M., Grigorieff N. Paired β-sheet structure of an Aβ(1–40) amyloid fibril revealed by electron microscopy. Proc. Natl. Acad. Sci. USA. 2008;105:7462–7466. doi: 10.1073/pnas.0712290105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirkitadze M.D., Condron M.M., Teplow D.B. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J. Mol. Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 12.Klement K., Wieligmann K., Meinhardt J., Hortschansky P., Richter W. Effect of different salt ions on the propensity of aggregation and on the structure of Alzheimer's Aβ(1–40) amyloid fibrils. J. Mol. Biol. 2007;373:1321–1333. doi: 10.1016/j.jmb.2007.08.068. [DOI] [PubMed] [Google Scholar]
- 13.Smith D.G., Cappai R., Barnham K.J. The redox chemistry of the Alzheimer's disease amyloid β peptide. Biochim. Biophys. Acta. 2007;1768:1976–1990. doi: 10.1016/j.bbamem.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Gorbenko G.P., Kinnunen P.K.J. The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids. 2006;141:72–82. doi: 10.1016/j.chemphyslip.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 15.Matsuzaki K. Physicochemical interactions of amyloid β-peptide with lipid bilayers. Biochim. Biophys. Acta. 2007;1768:1935–1942. doi: 10.1016/j.bbamem.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 16.Aisenbrey C., Borowik T., Bystrom R., Bokvist M., Lindstrom F. How is protein aggregation in amyloidogenic diseases modulated by biological membranes? Eur. Biophys. J. 2008;37:247–255. doi: 10.1007/s00249-007-0237-0. [DOI] [PubMed] [Google Scholar]
- 17.Krishtalik L.I., Cramer W.A. On the physical basis for the cis-positive rule describing protein orientation in biological membranes. FEBS Lett. 1995;369:140–143. doi: 10.1016/0014-5793(95)00756-y. [DOI] [PubMed] [Google Scholar]
- 18.Van Klompenburg W., de Kruijff B. The role of anionic lipids in protein insertion and translocation in bacterial membranes. J. Membr. Biol. 1998;162:1–7. doi: 10.1007/s002329900336. [DOI] [PubMed] [Google Scholar]
- 19.Terzi E., Hölzemann G., Seelig J. Self-association of β-amyloid peptide (1–40) in solution and binding to lipid membranes. J. Mol. Biol. 1995;252:633–642. doi: 10.1006/jmbi.1995.0525. [DOI] [PubMed] [Google Scholar]
- 20.Terzi E., Hölzemann G., Seelig J. Interaction of Alzheimer β-amyloid peptide(1–40) with lipid membranes. Biochemistry. 1997;36:14845–14852. doi: 10.1021/bi971843e. [DOI] [PubMed] [Google Scholar]
- 21.McLaurin J., Chakrabartty A. Characterization of the interactions of Alzheimer β-amyloid peptides with phospholipid membranes. Eur. J. Biochem. 1997;245:355–363. doi: 10.1111/j.1432-1033.1997.t01-2-00355.x. [DOI] [PubMed] [Google Scholar]
- 22.Ege C., Majewski J., Wu G., Kjaer K., Lee K.Y.C. Templating effect of lipid membrane on Alzheimer's amyloid β peptide. Chemphyschem. 2005;6:226–229. doi: 10.1002/cphc.200400468. [DOI] [PubMed] [Google Scholar]
- 23.Kinnunen P.K.J., Köiv A., Lehtonen J.Y.A., Rytömaa M., Mustonen P. Lipid dynamics and peripheral interactions of proteins with membrane surfaces. Chem. Phys. Lipids. 1994;73:181–207. doi: 10.1016/0009-3084(94)90181-3. [DOI] [PubMed] [Google Scholar]
- 24.Lee A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 25.Yip C.M., Darabie A.A., McLaurin J. Aβ42-peptide assembly on lipid bilayers. J. Mol. Biol. 2002;318:97–107. doi: 10.1016/S0022-2836(02)00028-1. [DOI] [PubMed] [Google Scholar]
- 26.Ambroggio E.E., Kim D.H., Separovic F., Barrow C.J., Barrow C.J. Surface behavior and lipid interaction of Alzheimer β-amyloid peptide 1–42: a membrane-disrupting peptide. Biophys. J. 2005;88:2706–2713. doi: 10.1529/biophysj.104.055582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bokvist M., Lindström F., Watts A., Gröbner G. Two types of Alzheimer's β-amyloid (1–40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 2004;335:1039–1049. doi: 10.1016/j.jmb.2003.11.046. [DOI] [PubMed] [Google Scholar]
- 28.Reference deleted in proof.
- 29.Appelt C., Eisenmenger F., Kuhne R., Schmieder P., Soderhall J.A. Interaction of the antimicrobial peptide cyclo(RRWWRF) with membranes by molecular dynamics simulations. Biophys. J. 2005;89:2296–2306. doi: 10.1529/biophysj.105.063040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gumbart J., Wang Y., Aksimentiev A., Tajkhorshid E., Schulten K. Molecular dynamics simulations of proteins in lipid bilayers. Curr. Opin. Struct. Biol. 2005;15:423–431. doi: 10.1016/j.sbi.2005.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hénin J., Pohorille A., Chipot C. Insights into the recognition and association of transmembrane α-helices. The free energy of α-helix dimerization in glycophorin A. J. Am. Chem. Soc. 2005;127:8478–8484. doi: 10.1021/ja050581y. [DOI] [PubMed] [Google Scholar]
- 32.Freitas M.S., Gaspar L.P., Lorenzoni M., Almelda F.C.L., Tinoco L.W. Structure of the Ebola fusion peptide in a membrane-mimetic environment and the interaction with lipid rafts. J. Biol. Chem. 2007;282:27306–27314. doi: 10.1074/jbc.M611864200. [DOI] [PubMed] [Google Scholar]
- 33.Kessel A., Shental-Bechor D., Haliloglu T., Ben-Tal N. Interactions of hydrophobic peptides with lipid bilayers: Monte Carlo simulations with M2δ. Biophys. J. 2003;85:3431–3444. doi: 10.1016/S0006-3495(03)74765-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lazaridis T. Implicit solvent simulations of peptide interactions with anionic lipid membranes. Proteins: Struct. Funct. Bioinform. 2005;58:518–527. doi: 10.1002/prot.20358. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J., Lazaridis T. Calculating the free energy of association of transmembrane helices. Biophys. J. 2006;91:1710–1723. doi: 10.1529/biophysj.106.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mager P.P. Molecular simulation of the primary and secondary structure of the Aβ(1–42)-peptide of Alzheimer's disease. Med. Res. Rev. 1998;18:403–420. doi: 10.1002/(sici)1098-1128(199811)18:6<403::aid-med4>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 37.Simona F., Tiana G., Broglia R.A., Colombo G. Modeling the α-helix to β-hairpin transition mechanism and the formation of oligomeric aggregates of the fibrillogenic peptide Aβ(12–28): insights from all-atom molecular dynamics simulations. J. Mol. Graph. Model. 2004;23:263–273. doi: 10.1016/j.jmgm.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 38.Buchete N.-V., Tycko R., Hummer G. Molecular dynamics simulations of Alzheimer's β-amyloid protofilaments. J. Mol. Biol. 2005;353:804–821. doi: 10.1016/j.jmb.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 39.Cruz L., Urbanc B., Borreguero J.M., Lazo N.D., Teplow D.B. Solvent and mutation effects on the nucleation of amyloid β-protein folding. Proc. Natl. Acad. Sci. USA. 2005;102:18258–18263. doi: 10.1073/pnas.0509276102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han W., Wu Y.-D. A strand-loop-strand structure is a possible intermediate in fibril elongation: long time simulations of amyloid-β peptide (10–35) J. Am. Chem. Soc. 2005;127:15408–15416. doi: 10.1021/ja051699h. [DOI] [PubMed] [Google Scholar]
- 41.Urbanc B., Cruz L., Ding F., Sammond D., Khare S. Molecular dynamics simulation of amyloid β dimer formation. Biophys. J. 2005;87:2310–2321. doi: 10.1529/biophysj.104.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Y., Shen J., Luo X., Zhu W., Chen K. Conformational transition of amyloid β-peptide. Proc. Natl. Acad. Sci. USA. 2005;102:5403–5407. doi: 10.1073/pnas.0501218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jang S., Shin S. Amyloid β-peptide oligomerization in silico: dimer and trimer. J. Phys. Chem. B. 2006;110:1955–1958. doi: 10.1021/jp055568e. [DOI] [PubMed] [Google Scholar]
- 44.Wei G., Shea J.-E. Effects of solvent on the structures of the Alzheimer amyloid-β(25–35) peptide. Biophys. J. 2006;91:1638–1647. doi: 10.1529/biophysj.105.079186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meinke J.H., Hansmann U.H.E. Aggregation of β-amyloid fragments. J. Chem. Phys. 2007;126:014706. doi: 10.1063/1.2423013. [DOI] [PubMed] [Google Scholar]
- 46.Sgourakis N.K., Yan Y., McCallum S.A., Wang C., Garcia A.E. The Alzheimer's peptides Aβ40 and 42 adopt distinct conformations in water: a combined MD/NMR study. J. Mol. Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lemkul J.A., Bevan D.R. A comparative molecular dynamics analysis of the amyloid β-peptide in a lipid bilayer. Arch. Biochem. Biophys. 2008;470:54–63. doi: 10.1016/j.abb.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 48.Norton W.T., Abe T., Poduslo S.E., DeVries G.H. The lipid composition of isolated brain cells and axons. J. Neurosci. Res. 1975;1:57–75. doi: 10.1002/jnr.490010106. [DOI] [PubMed] [Google Scholar]
- 49.Elliott J.I., Surprenant A., Marelli-Berg F.M., Cooper J.C., Cassady-Cain R.L. Membrane phosphatidylserine distribution as a non-apoptotic signaling mechanism in lymphocytes. Nat. Cell Biol. 2005;7:808–816. doi: 10.1038/ncb1279. [DOI] [PubMed] [Google Scholar]
- 50.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. [Google Scholar]
- 51.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 52.Nosé S.J. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984;81:511–519. [Google Scholar]
- 53.Hess B., Bekker H., Berendsen H.J.C., Fraaige J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 54.Essman U., Perera L., Berkowitz M.L., Darden T., Lee H. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 55.Berendsen H.J.C., Grigera J.R., Straatsma T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987;91:6269–6271. [Google Scholar]
- 56.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 57.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- 58.Berger O., Edholm O., Jahnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure and constant temperature. Biophys. J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagle J.F., Zhang R.T., Tristram-Nagle S., Sun W.J., Petrache H.I. X-ray structure determination of fully hydrated L(α) phase dipalmitoylphosphatidylcholine bilayers. Biophys. J. 1996;70:1419–1431. doi: 10.1016/S0006-3495(96)79701-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smondyrev A.M., Berkowitz M.L. United atom force field for phospholipid membranes: constant pressure molecular dynamics simulation of dipalmitoylphosphatidicholine/water system. J. Comput. Chem. 1999;50:531–545. [Google Scholar]
- 61.Petrache H.I., Tristram-Nagle S., Gawrisch K., Harries D., Parsegian V.A. Structure and fluctuations of charged phosphatidylserine bilayers in the absense of salt. Biophys. J. 2004;86:1574–1586. doi: 10.1016/S0006-3495(04)74225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhide S.Y., Berkowitz M.L. Structure and dynamics of water at the interface with phospholipids bilayers. J. Chem. Phys. 2005;123:224702. doi: 10.1063/1.2132277. [DOI] [PubMed] [Google Scholar]
- 63.Patey G.N., Valleau J.P. Free-energy of spheres with dipoles – Monte-Carlo with multistage sampling. Chem. Phys. Lett. 1973;21:297–300. [Google Scholar]
- 64.Torrie G.M., Valleau J.P. Non-physical sampling distributions in Monte-Carlo free-energy estimation – umbrella sampling. J. Comput. Phys. 1977;23:187–199. [Google Scholar]
- 65.Kumar S., Rosenberg J.M., Bouzida D., Swendsen R.H., Kollman P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. 1. The method. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 66.Vivcharuk V., Tomberli B., Tolokh I.S., Gray C.G. Prediction of binding free energy for adsorption of antimicrobial peptide lactoferricin on a POPC membrane. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2008;77:031913. doi: 10.1103/PhysRevE.77.031913. [DOI] [PubMed] [Google Scholar]
- 67.Chou P.Y., Fasman G.D. β-turns in proteins. J. Mol. Biol. 1977;115:135–175. doi: 10.1016/0022-2836(77)90094-8. [DOI] [PubMed] [Google Scholar]
- 68.Meier M., Seelig J. Thermodynamics of the coil ↔ β-sheet transition in a membrane environment. J. Mol. Biol. 2007;369:277–289. doi: 10.1016/j.jmb.2007.02.082. [DOI] [PubMed] [Google Scholar]
- 69.Nymeyer H., Woolf T.B., Garcia A.E. Folding is not required for bilayer insertion: replica exchange simulations of an α-helical peptide with an explicit lipid bilayer. Proteins: Struct. Funct. Bioinform. 2005;59:783–790. doi: 10.1002/prot.20460. [DOI] [PubMed] [Google Scholar]
- 70.Kirkitadze M.D., Condron M.M., Teplow D.B. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J. Mol. Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]