

Abstract
Liver disorders with a likely autoimmune pathogenesis in childhood include autoimmune hepatitis (AIH), autoimmune sclerosing cholangitis (ASC), and de novo AIH after liver transplantation. AIH is divided into two subtypes according to seropositivity for smooth muscle and/or antinuclear antibody (SMA/ANA, type 1) or liver kidney microsomal antibody (LKM1, type 2). There is a female predominance in both. LKM1 positive patients tend to present more acutely, at a younger age, and commonly have partial IgA deficiency, while duration of symptoms before diagnosis, clinical signs, family history of autoimmunity, presence of associated autoimmune disorders, response to treatment, and long-term prognosis are similar in both groups. The most common type of paediatric sclerosing cholangitis is ASC. The clinical, biochemical, immunological, and histological presentation of ASC is often indistinguishable from that of AIH type 1. In both, there are high IgG, non-organ specific autoantibodies, and interface hepatitis. Diagnosis is made by cholangiography. Children with ASC respond to immunosuppression satisfactorily and similarly to AIH in respect to remission and relapse rates, times to normalization of biochemical parameters, and decreased inflammatory activity on follow up liver biopsies. However, the cholangiopathy can progress. There may be evolution from AIH to ASC over the years, despite treatment. De novo AIH after liver transplantation affects patients not transplanted for autoimmune disorders and is strikingly reminiscent of classical AIH, including elevated titres of serum antibodies, hypergammaglobulinaemia, and histological findings of interface hepatitis, bridging fibrosis, and collapse. Like classical AIH, it responds to treatment with prednisolone and azathioprine. De novo AIH post liver transplantation may derive from interference by calcineurin inhibitors with the intrathymic physiological mechanisms of T-cell maturation and selection. Whether this condition is a distinct entity or a form of atypical rejection in individuals susceptible to the development of autoimmune phenomena is unclear. Whatever its etiology, the recognition of this potentially life-threatening syndrome is important since its management differs from that of standard anti-rejection therapy.
Keywords: Autoimmune hepatitis, Autoimmune sclerosing cholangitis, Liver transplant, Children
INTRODUCTION
Autoimmune liver disorders of childhood are inflammatory liver diseases characterized histologically by a dense mononuclear cell infiltrate in the portal tract and serologically by the presence of non-organ and liver specific autoantibodies and increased levels of transaminases and IgG, in the absence of a known etiology. They usually respond to immunosuppressive treatment, which should be instituted as soon as diagnosis is made. In children, as well as in young adults, autoimmune hepatitis (AIH) often presents acutely and has a more aggressive course than in older patients. The previously accepted requirement of 6 mo duration of symptoms before diagnosis can be made has been abandoned. In children, there are two liver disorders in which the liver damage is likely to arise from an autoimmune attack: classical AIH and AIH/sclerosing cholangitis overlap syndrome (autoimmune sclerosing cholangitis, ASC). A possible autoimmune pathogenesis has also been postulated for the so called post liver transplantation “de novo AIH”, a condition originally described in children and later confirmed in adults. According to data collected at the Kings College Hospital tertiary center, there is an increase in the yearly prevalence of AIH and ASC in childhood, although referral bias may play a role. Thus, in the 1990s these conditions were diagnosed in 2.3% of about 400 children older than 4 mo referred during one year, while in the 2000s their incidence increased to 12%.
AUTOIMMUNE HEPATITIS (AIH)
Clinical features
Two types of childhood AIH are recognized: AIH type 1 is characterized by the presence of smooth muscle (SMA) and/or antinuclear (ANA) antibodies; AIH type 2 is positive for anti liver kidney microsomal type 1 (anti-LKM-1) antibody[1]. Type 1 AIH represents two thirds of the cases and is a disease of children and adults, while type 2 AIH is mainly described in children. Severity of disease is similar in the two types of AIH[1]. In both, there is a predominance of girls (75%-80%). Anti-LKM-1 positive patients are younger and have a greater tendency to present with acute liver failure, but the duration of symptoms before diagnosis and the frequency of hepatosplenomegaly are similar in both groups. Both have a high frequency of associated autoimmune disorders (about 20%) and a family history of autoimmune disease (40%). Associated autoimmune disorders include thyroiditis, inflammatory bowel disease, vitiligo, insulin-dependent diabetes, nephrotic syndrome in both types[1]. Type 2 AIH can be associated to autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), an autosomal recessive genetic disorder in which the liver disease is reportedly present in about 20% of the cases[2].
There are three clinical patterns of disease onset[1]: (1) in at least 40% of patients, the presentation is indi-stinguishable from that of an acute viral hepatitis (non-specific symptoms of malaise, nausea/vomiting, anorexia, and abdominal pain, followed by jaundice, dark urine, and pale stools), some children, particularly anti-LKM-1 positive, develop acute hepatic failure with grade II to IV encephalopathy 2-8 wk from onset of symptoms; (2) in 25%-40% of patients, the onset is insidious, with an illness characterized by progressive fatigue, relapsing jaundice, headache, anorexia, and weight loss, lasting from several months and even years before diagnosis; (3) in about 10% of patients, there is no history of jaundice, and the diagnosis follows presentation with complications of portal hypertension, such as splenomegaly, hematemesis from esophageal varices, bleeding diathesis, chronic diarrhea, and weight loss. The mode of presentation of AIH in childhood is therefore variable, and the disease should be suspected and excluded in all children presenting with symptoms and signs of prolonged or severe liver disease. The course of the disease can be fluctuating, with flares and spontaneous remissions, a pattern which may result in delayed referral and diagnosis. The majority of children, however, even those presenting with acute hepatitis, on physical examination reveal clinical signs of an underlying chronic liver disease, i.e. cutaneous stigmata (spider nevi, palmar erythema, leukonychia, striae), firm liver and splenomegaly; at ultrasound the liver parenchyma is often nodular and heterogeneous.
Diagnosis and laboratory findings
Diagnosis of AIH is based on a series of positive and negative criteria defined by the International AIH Group (IAIHG)[3,4]. Though these criteria have been produced mainly for research purposes, they have been validated also in the clinical practice. Liver biopsy is necessary to establish the diagnosis of AIH, the typical histological picture include: a dense mononuclear and plasma cell infiltration of the portal areas, which expands into the liver lobule; destruction of the hepatocytes at the periphery of the lobule with erosion of the limiting plate (“interface hepatitis”); connective tissue collapse resulting from hepatocyte death and expanding from the portal area into the lobule (“bridging collapse”); hepatic regeneration with “rosette” formation (Figure 1). In addition to the typical histology, other positive criteria include elevated serum transaminase and IgG/gammaglobulin levels, and presence of ANA, SMA, or anti-LKM-1. Negative criteria relevant to the paediatric age are evidence of infection with hepatitis B or C virus, Wilson disease, and drug or alcohol consumption.
Figure 1.
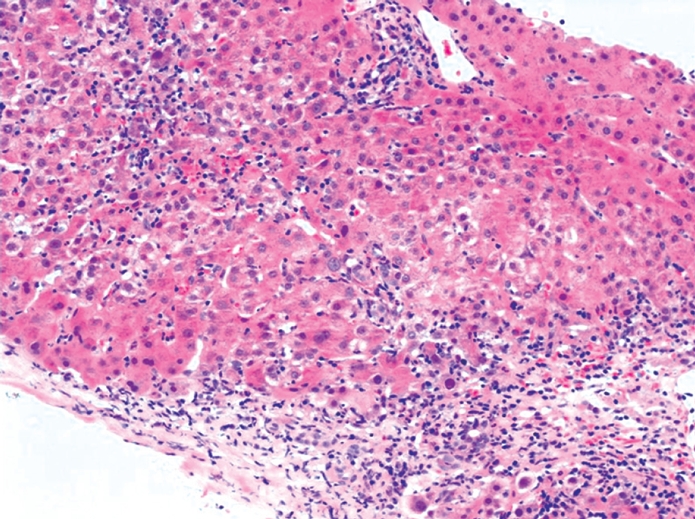
Portal and periportal lymphocyte and plasma cell infiltrate, extending to and disrupting the parenchymal limiting plate (interface hepatitis). Swollen hepatocytes, pyknotic necroses, and acinar inflammation are present. HE staining (Picture kindly provided by Dr. Alberto Quaglia).
Autoantibodies: A key criterion for the diagnosis of AIH is the detection of ANA, SMA, and anti-LKM-1 by indirect immunofluorescence. Autoantibody detection not only assists in the diagnosis but also allows, as mentioned above, differentiation of AIH in type 1 and type 2. ANA and SMA and anti-LKM-1 are practically mutually exclusive[5]; in those rare instances when they are present simultaneously, the child is classified as having AIH type 2. Recognition and interpretation of the immunofluorescence patterns is not always straightforward[5]. The operator dependency of the technique and the relative rarity of AIH explain the non-infrequent occurrence of errors in reporting, particularly of less frequent specificities such as anti-LKM-1. Problems do exist between laboratory reporting and clinical interpretation of the results that are partly dependent on clinicians’ unfamiliarity with the disease spectrum of AIH, but also partly dependent on insufficient standardization of the tests. This problem is being addressed by the autoimmune serology committee of the IAIHG[5].
The basic technique for the routine testing of autoantibodies relevant to AIH is indirect immuno-fluorescence on a freshly prepared rodent substrate that should include kidney, liver, and stomach to allow the detection of ANA, SMA, anti-LKM-1 as well as anti liver cytosol type 1 (anti-LC-1, see below), but also of anti-mitochondrial antibody (AMA), the serological hallmark of primary biliary cirrhosis, a disease affecting almost exclusively adults. Since a high proportion of healthy adults may show ANA or SMA reactivity at the conventional starting serum dilution of 1/10, the arbitrary dilution of 1/40 is considered clinically significant by the IAIHG in the adult population. In contrast, in healthy children autoantibody reactivity is infrequent, so that titers of 1/20 for ANA and SMA and 1/10 for anti-LKM-1 are clinically relevant. Hence, the laboratory should report any level of positivity from 1/10, and the attending physician should interpret the result within the clinical context and the age of the patient.
ANA is detectable as a nuclear staining in kidney, stomach, and liver. Its pattern can be homogeneous, or coarsely or finely speckled. In most cases of AIH, but not in all, the pattern is homogeneous. For a clearer and easier definition of the nuclear pattern, HEp2 cells that have prominent nuclei can be used. These cells, however, should not be used for screening purposes, because nuclear reactivity to HEp2 cells is frequent at low serum dilution (1/40) in the normal population[6]. ANA reactivity is not specific to AIH, being detectable in chronic viral hepatitis B and especially C, though at lower titre, and in non-hepatic autoimmune disorders.
SMA is detected on kidney, stomach, and liver. On the renal substrate, it is possible to visualize a V (vessels), G (glomeruli), and T (tubules) staining[7]. VG and VGT patterns are the most frequently detected in AIH[8]. The VGT pattern corresponds to the so called “F actin” or microfilament (MF) pattern observed using cultured fibroblasts as substrate. Though “anti-actin” reactivity is present in the majority of patients with AIH type 1, some 20% of SMA positive AIH type 1 patients do not have the F-actin/VGT pattern[8]. The absence, therefore, of anti-actin SMA does not exclude the diagnosis of AIH[8]. As for ANA, SMA can be found in chronic viral hepatitis B or C and extrahepatic autoimmune disorders.
Anti-LKM-1 stains brightly the liver cell cytoplasm and the P3 portion of the renal tubules, but does not stain gastric parietal cells. Anti-LKM-1 is often confused with AMA, since both autoantibodies stain liver and kidney, though AMA stains the liver more faintly and the renal tubules more diffusely with an accentuation of the small distal ones and, in contrast to anti-LKM-1, it also stains the gastric parietal cells. In the context of childhood AIH, patients reported to be AMA positive are almost invariably positive for anti-LKM-1, since AMA positive AIH in children is extremely rare[9] and PBC even rarer, only two cases having been documented histologically and immunoserologically, both being teenage girls[10]. The identification of the molecular targets of anti-LKM-1, i.e. cytochrome P4502D6, and of AMA, i.e. enzymes of the 2-oxo-acid dehydrogenase complexes, has led to the establishment of commercial immunoassays based on the use of the recombinant or purified antigens[11] that can resolve any doubts remaining after immunofluorescence examination. Anti-LKM-1 is highly specific for AIH type 2, being found outside this condition in a small proportion (-5%) of patients chronically infected by the hepatitis C virus, that usually possess the human leukocyte antigen (HLA) allele DRB1*07.
Other autoantibodies less commonly tested but of diagnostic importance in paediatric AIH include those to liver cytosol type 1 (LC-1), anti-neutrophil cytoplasm (ANCA) and soluble liver antigen (SLA). Anti-LC-1, which can be present on its own, but frequently occurs in association with anti-LKM-1, is an additional marker for AIH type 2 and targets formimino-transferase cyclodeaminase (FTCD)[12]. In AIH type 1, as well as in inflammatory bowel disease and sclerosing cholangitis, ANCA is frequently found and targets a peripheral nuclear perinuclear antigen (hence the suggested name of pANNA, i.e. peripheral anti nuclear neutrophil antibody). pANNA is virtually absent in type 2 AIH[7]. Anti-SLA that was originally described as the hallmark of a third type of AIH[13], is also found in some 50% of paediatric patients with type 1 and type 2 AIH, where it defines a more severe course[14].
There are a small proportion of children with AIH without detectable autoantibodies. The prevalence and the clinical characteristics of this rare seronegative form of AIH, which responds to immunosuppression similarly to the seropositive forms, remain to be defined.
Comparison between type 1 and type 2 AIH: Clinical, laboratory and histological features of type 1 and 2 AIH are summarized in Table 1. In Northern Europe, type 1 AIH is associated with the presence of human leukocyte antigen (HLA) DRB1*03[1,11], while type 2 AIH is associated with the presence of DRB1*07 and, less frequently, with DRB1*03[15]. In South America, the HLA DRB1*1301 allele is reported to predispose to paediatric AIH type 1 and is also associated with persistent infection with the endemic hepatitis A virus[16,17]. Interestingly, in Northern European children HLA DRB1*1301 is associated with autoimmune sclerosing cholangitis (see below). It is conceivable that some South American children diagnosed as having AIH type 1, but in whom routine cholangiograms were not performed, indeed had ASC.
Table 1.
Clinical, laboratory, and histological features at presentation of autoimmune hepatitis type 1, autoimmune hepatitis type 2, and autoimmune sclerosing cholangitis[1,20]
| Type 1 AIH | Type 2 AIH | ASC | |
| Median age in year | 11 | 7 | 12 |
| Females (%) | 75 | 75 | 55 |
| Mode of presentation (%) | |||
| Acute hepatitis | 47 | 40 | 37 |
| Acute liver failure | 3 | 25 | 0 |
| Insidious onset | 38 | 25 | 37 |
| Complication of chronic liver disease | 12 | 10 | 26 |
| Associated autoimmune diseases (%) | 22 | 20 | 48 |
| Inflammatory bowel disease (%) | 20 | 12 | 44 |
| Family history of autoimmune disease (%) | 43 | 40 | 37 |
| Abnormal cholangiogram (%) | 0 | 0 | 100 |
| ANA/SMA (%) | 100 | 25 | 96 |
| Anti LKM1 (%) | 0 | 100 | 4 |
| pANCA (%) | 45 | 11 | 74 |
| Anti SLA (%)1 | 58 | 58 | 41 |
| Increased IgG level (%) | 84 | 75 | 89 |
| Partial IgA deficiency (%) | 9 | 45 | 5 |
| Low C4 level (%) | 89 | 83 | 70 |
| Increased frequency of HLA DR*0301 | Yes | No2 | No |
| Increased frequency of HLA DR*0701 | No | Yes | No |
| Increased frequency of HLA DR*1301 | No | No | Yes |
| Interface hepatitis (%) | 66 | 72 | 35 |
| Biliary features (%) | 28 | 6 | 31 |
| Cirrhosis (%) | 69 | 38 | 15 |
| Remission after immunosuppressive Treatment (%) | 97 | 87 | 89 |
AIH: Autoimmune hepatitis; ASC: Autoimmune sclerosing cholangitis; ANA: Anti-nuclear antibodies; SMA: Anti-smooth muscle antibody; LKM1: Liver kidney microsomal type 1 antibody; pANCA: Perinuclear anti-neutrophil cytoplasmic antibody; SLA: Soluble liver antigen; C4: C4 component of complement; HLA: Human leukocyte antigen.
Measured by radioligand assay;
Increased in HLA DR*0701 negative patients.
Paediatric patients with AIH, whether anti-LKM-1 or ANA/SMA positive, have isolated partial deficiency of the HLA class III complement component C4, which is genetically determined[18].
Anti-LKM-1-positive patients have higher levels of bilirubin and transaminases at presentation than those who are ANA/SMA positive and present significantly more frequently with fulminant hepatic failure[1]. Excluding children with the fulminant presentation, a severely impaired hepatic synthetic function, as assessed by the presence of both prolonged prothrombin time and hypoalbuminemia, is more common in ANA/SMA-positive than in anti-LKM-1 positive patients. The vast majority of patients have increased levels of IgG, but some 20% do not[1], indicating that normal IgG values do not exclude the diagnosis of AIH. Partial IgA deficiency is significantly more common in LKM1-positive than in ANA/SMA-positive patients[1,19].
The severity of interface hepatitis at diagnosis is similar in both types, but cirrhosis on initial biopsy is more frequent in type 1 than in type 2 AIH, suggesting a more chronic course of disease in the former. Of note is that most patients already cirrhotic at diagnosis present with a clinical picture reminiscent of that of prolonged acute viral-like hepatitis. Multiacinar or panacinar collapse, which suggests an acute liver injury, is more frequently seen in type 2 AIH. The question as to whether the acute presentation in these patients represents a sudden deterioration of an underlying unrecognized chronic process or a genuinely acute liver damage remains open. Progression to cirrhosis during treatment is more frequent in type 1 AIH. As mentioned above, in both a more severe disease and a higher tendency to relapse is associated to the possession of antibodies to soluble liver antigen (SLA), which are present in about half of the patients with AIH type 1 or 2 at diagnosis[11].
Differential diagnosis: Since positive autoimmune serology can be present in conditions other than AIH, in particular ASC[20], chronic hepatitis B[21] or C[22] virus infections, and Wilson disease[23], all these disorders must be considered in the differential diagnosis and excluded. ASC, described below, shares the same serological profile as type 1 AIH, but has typical bile duct lesions on cholangiography. Up to 50% of children with hepatitis B and C are positive for ANA and/or SMA, usually at low titres[21,22], and some 5% of patients with chronic hepatitis C have anti-LKM-1 antibodies. In these patients the histology can also mimic AIH, though usually the degree of inflammation is milder. Detection of the typical viral markers allows a correct diagnosis. ANA, and at times SMA, can be present in Wilson disease, in association with high IgG and an inflammatory liver histology, which can make the differential diagnosis with AIH type 1 difficult. Urinary, serum, and liver tissue copper studies and search for Kayser Fleischer rings should be performed in all cases.
APECED: APECED is a monogenic disorder[24,25] with a variable phenotype. About 20% of the cases develop AIH that resembles AIH type 2[2]. This condition, also known as autoimmune polyendocrine syndrome 1 is an autosomal recessive disorder caused by homozygous mutations in the AIRE1 gene and characterized by a variety of organ-specific autoimmune diseases, the most common of which are hypoparathyroidism and primary adrenocortical failure, accompanied by chronic mucocutaneous candidiasis.
Etiology and pathogenesis
The etiology of AIH is unknown, though both genetic and environmental factors are involved in its expression. Etiological hypotheses and possible mechanisms leading to the liver autoimmune attack are described under “Etiopathogenesis of AIH” in this issue.
Management and prognosis
AIH is exquisitely responsive to immunosuppression. The rapidity and degree of response depends on the disease severity at presentation. All types of present-ations, apart from fulminant hepatic failure with encephalopathy, respond to standard treatment with prednisolone with or without azathioprine.
Standard treatment for AIH consists of prednisolone 2 mg/kg per day (maximum 60 mg/d), which is gradually decreased over a period of 4 to 8 wk with progressive normalization of the transaminases, and then the patient is maintained on the minimal dose able to sustain normal transaminase levels, usually 2.5 mg/d or 5 mg/d depending on age[1,26]. During the first 6 to 8 wk of treatment, liver function tests should be checked weekly to allow a frequent fine-tuning, avoiding severe steroid side effects. If progressive normalization of the liver function tests is not obtained over this period of time or if too high a dose of prednisolone is required to maintain normal transaminases, azathioprine is added at a starting dose of 0.5 mg/kg per day, which, in the absence of signs of toxicity, is increased up to a maximum of 2-2.5 mg/kg per day until biochemical control is achieved. Azathioprine is not recommended as first-line treatment because of its hepatotoxicity in severely jaundiced patients, but 85% of the patients will eventually require azathioprine addition. A preliminary report in a cohort of 30 children with AIH suggests that the measurements of the azathioprine metabolites 6-thioguanine and 6-methylmercaptopurine are useful in identifying drug toxicity and non-adherence and in achieving a level of 6-thioguanine considered therapeutic for inflammatory bowel disease[27], though what is an ideal therapeutic level for AIH has not been determined. Although an 80% decrease of initial transaminase activity is obtained within 6 wk from starting treatment in most patients, complete normalization of liver function may take several months. In the King’s series, normalization of serum transaminase activity occurred at median of 6 mo in ANA/SMA positive children and 9 mo in LKM-1 positive children[1]. Relapse while on treatment is common, occurring in about 40% of the patients and requiring a temporary increase of the steroid dose. An important role in relapse is played by non-adherence that is common, particularly in adolescents[28]. Moreover, the risk of relapse is higher if steroids are administered on an alternate-day schedule, often instituted in the belief that it has a less negative effect on the child’s growth. Small daily doses are more effective in maintaining disease control and minimize the need for high-dose steroid pulses during relapses, with consequent more severe side effects[1].
A question frequently asked by the parents of teenaged girls is whether treatment can be safely continued during pregnancy. Although the experience is limited, there does not appear to be adverse events for mother and baby[29]. In particular, no teratogenic effects have been described with azathioprine in humans, though for women concerned about its use, treatment with steroids alone can be used.
Cessation of treatment is considered if a liver biopsy shows minimal or no inflammatory changes after at least one year of normal liver function tests. However, it is advisable not to attempt to withdraw treatment within three years from diagnosis or during or immediately before puberty, when relapses are more common. The reasons for this are unclear, though an important role may be played by non-adherence, as mentioned above. In the Kings experience, successful long-term withdrawal of treatment was achieved in 20% of patients with AIH type 1, but in none with AIH type 2[1].
In paediatrics, an important role in monitoring the response to treatment is the measurement of autoantibody titers and IgG levels, the fluctuation of which is correlated with disease activity[30].
Despite the efficacy of standard immunosuppressive treatment, severe hepatic decompensation may develop even after many years of apparently good biochemical control, leading to transplantation 10-15 years after diagnosis in 10% of the patients. In the Kings College Hospital series[1], over 97% of the patients treated with standard immunosuppression were alive between 0.3 and 19 years (median 5 years) after diagnosis, including 8% after liver transplantation. Side effects of steroid treatment were mild, the only serious complication being psychosis during induction of remission in 4%, which resolved after prednisolone withdrawal. All patients developed a transient increase in appetite and mild cushingoid features during the first few weeks of treatment. After five years of treatment, 56% of the patients maintained the baseline centile for height or went up across a centile line, 38% dropped across one centile line, and only 6% dropped across two centile lines[31]. Moreover, it has recently been shown that long-term daily treatment with prednisolone in children with autoimmune liver disease does not affect their expected final adult height according to parental stature[32].
Sustained remission, achieved with prednisolone and azathioprine, can be maintained with azathioprine alone in some children with AIH type 1, akin to the experience in adults[33], but not in AIH type 2.
In those patients (up to 10%) in whom standard immunosuppression is unable to induce stable remission or who are intolerant to azathioprine, mycophenolate mofetil at a dose of 20 mg/kg twice daily can be successfully used[31]. In case of persistent no response or of intolerance to mycophenolate mofetil (headache, diarrhea, nausea, dizziness, hair loss, or neutropenia), the use of calcineurin inhibitors (cyclosporine A or tacrolimus) should be considered.
Children who present with acute hepatic failure pose a particularly difficult therapeutic problem. If not encephalopathic, they usually benefit from conventional immunosuppressive therapy, but only one of the six children with acute liver failure and encepaholpathy in the Kings series responded to immunosuppression and survived without transplantation[1].
AUTOIMMUNE SCLEROSING CHOLANGITIS
ASC has the same prevalence as AIH type 1 in child-hood[18]. This has been shown in a prospective study conducted over a period of 16 years, in which all children with serological (i.e. positive autoantibodies, high IgG levels) and histological (i.e. interface hepatitis) features of autoimmune liver disease underwent a cholangiogram at the time of presentation. Approximately 50% of these patients had alterations of the bile ducts characteristic of sclerosing cholangitis, though generally less advanced than those observed in adult primary sclerosing cholangitis (Figure 2). A quarter of the children with ASC, despite abnormal cholangiograms, had no histological features suggesting bile duct involvement and the diagnosis of sclerosing cholangitis was only possible because of the cholangiographic studies. Virtually all patients were seropositive for ANA and/or SMA. Fifty-five percent were girls, and the mode of presentation was similar to that of typical AIH. Inflammatory bowel disease was present in about 45% of children with ASC compared to about 20% of those with AIH, and 90% of children with ASC had greatly increased serum IgG levels. At the time of presentation, standard liver function tests did not help in discriminating between AIH and ASC (Table 2), though the alkaline phosphatase/aspartate aminotransferase ratio was significantly higher in ASC. pANNA were present in 74% of patients with ASC compared to 45% of patients with AIH type 1 and 11% of those with AIH type 2. Susceptibility to ASC in children is conferred by the presence of HLA DRB1*1301[32]. Clinical, laboratory, and histological features of type 1 and 2 AIH and ASC are compared in Table 1.
Figure 2.
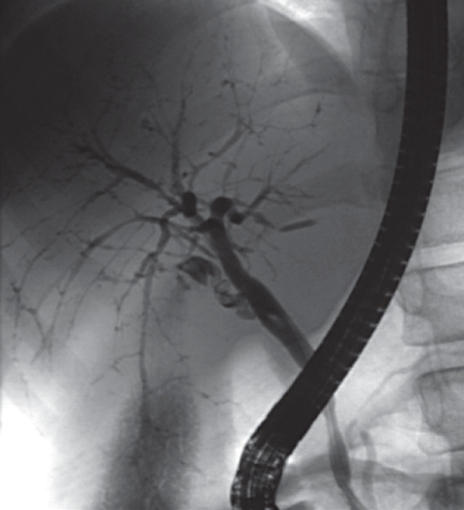
Retrograde cholangiogram of a child with autoimmune sclerosing cholangitis showing widespread bile duct strictures and dilatations (Picture kindly provided by Dr. Maria Sellars).
Table 2.
Laboratory parameters at presentation in children with autoimmune hepatitis and autoimmune sclerosing cholangitis[20]
| AIH | ASC | |
| Bilirubin (nv < 20 micromol/L) | 35 (4-306) | 20 (4-179) |
| Albumin (nv > 35 g/L) | 35 (25-47) | 39 (27-54) |
| AST (nv < 50 IU/L) | 333 (24-4830) | 102 (18-1215) |
| INR (< 1.2) | 1.2 (0.96-2.5) | 1.1 (0.9-1.6) |
| GGT (nv < 50 IU/L) | 76 (29-383) | 129 (13-948) |
| AP (nv < 350 IU/L) | 356 (131-878) | 303 (104-1710) |
AST: Aspartate aminotransferase; INR: International normalized prothrombin ratio; GGT: Gamma glutamyl transpeptidase; AP: Alkaline phosphatase; nv: Normal values.
Children with ASC respond to the same immuno-suppressive schedule described above for AIH[18], liver test abnormalities resolving within a few months after starting treatment in most patients. Steroids and azathioprine, however, though beneficial in abating the parenchymal inflammatory lesions, appear to be less effective in controlling the bile duct disease. Following favorable reports in adult primary sclerosing cholangitis[33,34], ursode-oxycholic acid is added at the dose of 20-30 mg/kg per day, though there is no information as to whether it is helpful in arresting the progression of ASC. Akin to AIH, measurement of autoantibody titers and IgG levels is useful in monitoring disease activity and response to treatment[20]. The medium-term prognosis is good, with a reported 7-year survival of 100%, though 15% of the patients required liver transplantation during this period of follow-up[18]. Evolution from AIH to ASC has been documented suggesting that AIH and ASC are part of the same pathogenic process[18].
The prospective study conducted at Kings College Hospital shows that in childhood ASC and AIH have a similar prevalence[20]. It also shows that ASC is more frequent than sclerosing cholangitis without autoimmune features[20], autoantibody negative sclerosing cholangitis having been observed in only 9 children referred over the 16-year study period[20].
Whether childhood ASC and adult PSC belong to the same disease spectrum remains to be established, since no prospective study in a large patient cohort has investigated at the time of presentation the presence of bile duct damage in adults with features of autoimmune liver disease. Interestingly, in a retrospective study, a high proportion of adult patients originally diagnosed as having AIH type 1 were found to have sclerosing cholangitis on magnetic resonance cholangiography[34]. The long term follow up of the Kings paediatric ASC series will provide important information about the possible links between ASC and PSC.
DE NOVO AIH AFTER LIVER TRANSPLANTATION
In the late 1990s, it was observed that AIH can arise de novo after liver transplantation in children who had not been transplanted for autoimmune liver disease[35]. Characteristic of this condition is a histological picture of interface hepatitis and multilobular collapse associated with increased IgG levels and positive autoantibodies. These include ANA, SMA, and classical anti-LKM-1, but also atypical anti-LKM-1, staining the renal tubules but not the liver. After this original report, de novo AIH after liver transplantation has been confirmed by several studies both in adult and paediatric patients[36,37]. Importantly, treatment with prednisolone and azathioprine using the same schedule for classical AIH, concomitant with reduction of the calcineurin inhibitor dose, is highly effective in de novo AIH, leading to excellent graft and patient survival. It is of interest that these patients do not respond satisfactorily to standard anti-rejection treatment, making it essential to reach an early diagnosis to avoid graft loss.
Whether the liver damage observed in these patients is a form of rejection or the consequence of an “auto-immune” injury, possibly triggered by drugs or viral infection, remains to be established. The administration of cyclosporin A or tacrolimus to rodents after bone marrow transplantation can result in a “paradoxical” autoimmune syndrome in which the immunosuppressive drugs interfere with maturation of T lymphocytes and favor the emergence of autoaggressive T-cell clones[35–37]. This experience in animals may explain, in part, the development of this disorder in immunosuppressed children after liver transplantation.
The manifestations of the autoimmune condition in rodents vary in different strains and depend on genetic factors possibly encoded by the major histocompatibility complex[35]. Analysis of the HLA phenotypes of the recipients and donors in the original report did not show an association between the development of autoimmune features, the presence of either HLA DRB1*03 or -DRB1*04, or the degree of donor-recipient HLA mismatch[28]. Five of the seven patients, however, had received livers from donors with HLA markers known to be associated with susceptibility to AIH, including two with DRB1*04, one with DRB1*03, and two with both DRB1*03 and DRB1*04[38].
Footnotes
S- Editor Li DL L- Editor Mihm S E- Editor Yin DH
References
- 1.Gregorio GV, Portmann B, Reid F, Donaldson PT, Doherty DG, McCartney M, Mowat AP, Vergani D, Mieli-Vergani G. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology. 1997;25:541–547. doi: 10.1002/hep.510250308. [DOI] [PubMed] [Google Scholar]
- 2.Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829–1836. doi: 10.1056/NEJM199006283222601. [DOI] [PubMed] [Google Scholar]
- 3.Johnson PJ, McFarlane IG. Meeting report: International Autoimmune Hepatitis Group. Hepatology. 1993;18:998–1005. doi: 10.1002/hep.1840180435. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, Chapman RW, Cooksley WG, Czaja AJ, Desmet VJ, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929–938. doi: 10.1016/s0168-8278(99)80297-9. [DOI] [PubMed] [Google Scholar]
- 5.Vergani D, Alvarez F, Bianchi FB, Cançado EL, Mackay IR, Manns MP, Nishioka M, Penner E. Liver autoimmune serology: a consensus statement from the committee for autoimmune serology of the International Autoimmune Hepatitis Group. J Hepatol. 2004;41:677–683. doi: 10.1016/j.jhep.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Tan EM, Feltkamp TE, Smolen JS, Butcher B, Dawkins R, Fritzler MJ, Gordon T, Hardin JA, Kalden JR, Lahita RG, et al. Range of antinuclear antibodies in "healthy" individuals. Arthritis Rheum. 1997;40:1601–1611. doi: 10.1002/art.1780400909. [DOI] [PubMed] [Google Scholar]
- 7.Bottazzo GF, Florin-Christensen A, Fairfax A, Swana G, Doniach D, Groeschel-Stewart U. Classification of smooth muscle autoantibodies detected by immunofluorescence. J Clin Pathol. 1976;29:403–410. doi: 10.1136/jcp.29.5.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muratori P, Muratori L, Agostinelli D, Pappas G, Veronesi L, Granito A, Cassani F, Terlizzi P, Lenzi M, Bianchi FB. Smooth muscle antibodies and type 1 autoimmune hepatitis. Autoimmunity. 2002;35:497–500. doi: 10.1080/0891693021000054066. [DOI] [PubMed] [Google Scholar]
- 9.Gregorio GV, Portmann B, Mowat AP, Vergani D, Mieli-Vergani G. A 12-year-old girl with antimitochondrial antibody-positive autoimmune hepatitis. J Hepatol. 1997;27:751–754. doi: 10.1016/s0168-8278(97)80093-1. [DOI] [PubMed] [Google Scholar]
- 10.Dahlan Y, Smith L, Simmonds D, Jewell LD, Wanless I, Heathcote EJ, Bain VG. Pediatric-onset primary biliary cirrhosis. Gastroenterology. 2003;125:1476–1479. doi: 10.1016/j.gastro.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 11.Donaldson PT. Genetics in autoimmune hepatitis. Semin Liver Dis. 2002;22:353–364. doi: 10.1055/s-2002-35705. [DOI] [PubMed] [Google Scholar]
- 12.Lapierre P, Hajoui O, Homberg JC, Alvarez F. Formimino-transferase cyclodeaminase is an organ-specific autoantigen recognized by sera of patients with autoimmune hepatitis. Gastroenterology. 1999;116:643–649. doi: 10.1016/s0016-5085(99)70186-1. [DOI] [PubMed] [Google Scholar]
- 13.Manns M, Gerken G, Kyriatsoulis A, Staritz M, Meyer zum Büschenfelde KH. Characterisation of a new subgroup of autoimmune chronic active hepatitis by autoantibodies against a soluble liver antigen. Lancet. 1987;1:292–294. doi: 10.1016/s0140-6736(87)92024-1. [DOI] [PubMed] [Google Scholar]
- 14.Ma Y, Okamoto M, Thomas MG, Bogdanos DP, Lopes AR, Portmann B, Underhill J, Dürr R, Mieli-Vergani G, Vergani D. Antibodies to conformational epitopes of soluble liver antigen define a severe form of autoimmune liver disease. Hepatology. 2002;35:658–664. doi: 10.1053/jhep.2002.32092. [DOI] [PubMed] [Google Scholar]
- 15.Ma Y, Bogdanos DP, Hussain MJ, Underhill J, Bansal S, Longhi MS, Cheeseman P, Mieli-Vergani G, Vergani D. Polyclonal T-cell responses to cytochrome P450IID6 are associated with disease activity in autoimmune hepatitis type 2. Gastroenterology. 2006;130:868–882. doi: 10.1053/j.gastro.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 16.Fainboim L, Canero Velasco MC, Marcos CY, Ciocca M, Roy A, Theiler G, Capucchio M, Nuncifora S, Sala L, Zelazko M. Protracted, but not acute, hepatitis A virus infection is strongly associated with HLA-DRB*1301, a marker for pediatric autoimmune hepatitis. Hepatology. 2001;33:1512–1517. doi: 10.1053/jhep.2001.24562. [DOI] [PubMed] [Google Scholar]
- 17.Pando M, Larriba J, Fernandez GC, Fainboim H, Ciocca M, Ramonet M, Badia I, Daruich J, Findor J, Tanno H, et al. Pediatric and adult forms of type I autoimmune hepatitis in Argentina: evidence for differential genetic predisposition. Hepatology. 1999;30:1374–1380. doi: 10.1002/hep.510300611. [DOI] [PubMed] [Google Scholar]
- 18.Vergani D, Wells L, Larcher VF, Nasaruddin BA, Davies ET, Mieli-Vergani G, Mowat AP. Genetically determined low C4: a predisposing factor to autoimmune chronic active hepatitis. Lancet. 1985;2:294–298. doi: 10.1016/s0140-6736(85)90348-4. [DOI] [PubMed] [Google Scholar]
- 19.Homberg JC, Abuaf N, Bernard O, Islam S, Alvarez F, Khalil SH, Poupon R, Darnis F, Lévy VG, Grippon P. Chronic active hepatitis associated with antiliver/kidney microsome antibody type 1: a second type of "autoimmune" hepatitis. Hepatology. 1987;7:1333–1339. doi: 10.1002/hep.1840070626. [DOI] [PubMed] [Google Scholar]
- 20.Gregorio GV, Portmann B, Karani J, Harrison P, Donaldson PT, Vergani D, Mieli-Vergani G. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: a 16-year prospective study. Hepatology. 2001;33:544–553. doi: 10.1053/jhep.2001.22131. [DOI] [PubMed] [Google Scholar]
- 21.Gregorio GV, Jones H, Choudhuri K, Vegnente A, Bortolotti F, Mieli-Vergani G, Vergani D. Autoantibody prevalence in chronic hepatitis B virus infection: effect in interferon alfa. Hepatology. 1996;24:520–523. doi: 10.1002/hep.510240309. [DOI] [PubMed] [Google Scholar]
- 22.Gregorio GV, Pensati P, Iorio R, Vegnente A, Mieli-Vergani G, Vergani D. Autoantibody prevalence in children with liver disease due to chronic hepatitis C virus (HCV) infection. Clin Exp Immunol. 1998;112:471–476. doi: 10.1046/j.1365-2249.1998.00574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dhawan A, Taylor RM, Cheeseman P, De Silva P, Katsiyiannakis L, Mieli-Vergani G. Wilson's disease in children: 37-year experience and revised King's score for liver transplantation. Liver Transpl. 2005;11:441–448. doi: 10.1002/lt.20352. [DOI] [PubMed] [Google Scholar]
- 24.Liston A, Lesage S, Gray DH, Boyd RL, Goodnow CC. Genetic lesions in T-cell tolerance and thresholds for autoimmunity. Immunol Rev. 2005;204:87–101. doi: 10.1111/j.0105-2896.2005.00253.x. [DOI] [PubMed] [Google Scholar]
- 25.Simmonds MJ, Gough SC. Genetic insights into disease mechanisms of autoimmunity. Br Med Bull. 2004;71:93–113. doi: 10.1093/bmb/ldh032. [DOI] [PubMed] [Google Scholar]
- 26.Mieli-Vergani G, Vergani D. Autoimmune hepatitis in children. Clin Liver Dis. 2002;6:623–634. doi: 10.1016/s1089-3261(02)00020-x. [DOI] [PubMed] [Google Scholar]
- 27.Rumbo C, Emerick KM, Emre S, Shneider BL. Azathioprine metabolite measurements in the treatment of autoimmune hepatitis in pediatric patients: a preliminary report. J Pediatr Gastroenterol Nutr. 2002;35:391–398. doi: 10.1097/00005176-200209000-00032. [DOI] [PubMed] [Google Scholar]
- 28.Kerkar N, Annunziato RA, Foley L, Schmeidler J, Rumbo C, Emre S, Shneider B, Shemesh E. Prospective analysis of nonadherence in autoimmune hepatitis: a common problem. J Pediatr Gastroenterol Nutr. 2006;43:629–634. doi: 10.1097/01.mpg.0000239735.87111.ba. [DOI] [PubMed] [Google Scholar]
- 29.Heneghan MA, Norris SM, O'Grady JG, Harrison PM, McFarlane IG. Management and outcome of pregnancy in autoimmune hepatitis. Gut. 2001;48:97–102. doi: 10.1136/gut.48.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregorio GV, McFarlane B, Bracken P, Vergani D, Mieli-Vergani G. Organ and non-organ specific autoantibody titres and IgG levels as markers of disease activity: a longitudinal study in childhood autoimmune liver disease. Autoimmunity. 2002;35:515–519. doi: 10.1080/0891693021000056721. [DOI] [PubMed] [Google Scholar]
- 31.Mieli-Vergani G, Bargiota K, Samyn M, Vergani D. Therapeutic aspects of autoimmune liver disease in children. In: Dienes HP, Leuschner U, Lohse AW, Manns MP, et al., editors. Autoimmune Liver Diseases-Falk Symposium Dordrecht: Springer; 2005. pp. 278–282. [Google Scholar]
- 32.Samaroo B, Samyn M, Buchanan C, Mieli-Vergani G. Long-term daily oral treatment with prednisolone in children with autoimmune liver disease does not affect final adult height. Hepatology. 2006;44:438A. [Google Scholar]
- 33.Johnson PJ, McFarlane IG, Williams R. Azathioprine for long-term maintenance of remission in autoimmune hepatitis. N Engl J Med. 1995;333:958–963. doi: 10.1056/NEJM199510123331502. [DOI] [PubMed] [Google Scholar]
- 34.Abdalian R, Dhar P, Jhaveri K, Haider M, Guindi M, Heathcote EJ. Prevalence of sclerosing cholangitis in adults with autoimmune hepatitis: evaluating the role of routine magnetic resonance imaging. Hepatology. 2008;47:949–957. doi: 10.1002/hep.22073. [DOI] [PubMed] [Google Scholar]
- 35.Bucy PB, Yan Xu X, Li J, Huang GQ. Cyclosporin A-induced autoimmune disease in mice. J Immunol. 1993;151:1039–1050. [PubMed] [Google Scholar]
- 36.Cooper MH, Hartman GG, Starzl TE, Fung JJ. The induction of pseudo-graft-versus-host disease following syngeneic bone marrow transplantation using FK 506. Transplant Proc. 1991;23:3234–3235. [PMC free article] [PubMed] [Google Scholar]
- 37.Hess AD, Fischer AC, Horwitz LR, Laulis MK. Cyclosporine-induced autoimmunity: critical role of autoregulation in the prevention of major histocompatibility class II-dependent autoaggression. Transplant Proc. 1993;25:2811–2813. [PubMed] [Google Scholar]
- 38.Donaldson PT, Doherty DG, Hayllar KM, McFarlane IG, Johnson PJ, Williams R. Susceptibility to autoimmune chronic active hepatitis: human leukocyte antigens DR4 and A1-B8-DR3 are independent risk factors. Hepatology. 1991;13:701–706. [PubMed] [Google Scholar]