

Abstract
Many biological processes require proteins to undergo conformational changes at the surface of membranes. For example, some precursor proteins unfold at the surface of mitochondria and chloroplasts before translocation into the organelles, and toxins such as colicin A unfold to the molten globule state at bacterial surfaces before inserting into the cell membrane. It is commonly thought that the membrane surfaces and the associated protein machinery destabilize the substrate proteins and that this effect is required for membrane insertion or translocation. One of the best characterized translocation processes is protein import into mitochondria. By measuring the contributions of individual interactions within a model protein to its stability at the mitochondrial surface and in free solution, we show here that the mitochondrial surface neither induces the molten globule state in this protein nor preferentially destabilizes any type of interaction (e.g., hydrogen bonds, nonpolar, etc.) within the protein. Because it is not possible to measure absolute protein stability at the surface of mitochondria, we determined the stability of a tightly associated protein–protein complex at the mitochondrial import site as a model of the stability of a protein. We found the binding constants of the protein–protein complex at the mitochondrial surface and in free solution to be identical. Our results demonstrate that the mitochondrial surface does not destabilize importing precursor proteins in its vicinity.
Regulated unfolding is critically important in the life cycle of many proteins, such as those translocated across mitochondrial and chloroplast membranes, as well as those degraded by ATP-dependent proteases such as the proteasome. About half of all proteins synthesized in the eukaryotic cell are transported into or across a membrane (1). The protein translocation machineries are well defined biologically, but the means by which they transport and unfold proteins are not well understood at the biochemical and biophysical level.
One of the best characterized translocation processes is protein import into mitochondria. Precursor proteins are transported into mitochondria by a macromolecular machine that spans two membranes and contains at least nine different polypeptides. Import requires ATP and an electrochemical potential across the mitochondrial inner membrane (1). Protein unfolding is intimately associated with translocation. Although precursor proteins normally lack all structure during import into mitochondria (2–4), some proteins are fully folded before translocation (5, 6). The import of these proteins can be many hundred times faster than their spontaneous unfolding, indicating that mitochondria can actively unfold proteins (7, 8). Protein unfolding occurs at the mitochondrial surface, and it has been found that the import machinery in the outer membrane by itself unfolds (9) or destabilizes (10–13) precursor proteins. This unfolding activity is thought to be required for the translocation of proteins into mitochondria.
More generally, it has been found that various membranes destabilize proteins in their vicinity and induce the molten globule state in these proteins (14–18). The molten globule state is an in vitro folding intermediate observed in the refolding pathways of most proteins and is defined by the presence of native-like secondary structure elements, but the absence of tertiary interactions (19). Specifically, it has been suggested that membrane surfaces induce the molten globule state in the toxin colicin A, which then undergoes conformational changes and inserts into the membranes where it is active (20). Molten globule formation also occurs during membrane insertion of diphtheria toxin (18). Synthesizing these observations, it has been suggested that biological membranes have a general destabilizing effect on proteins in their vicinity and that this destabilizing effect is biologically important (19, 20).
We investigated the structure and energetics of a model precursor protein at the mitochondrial surface immediately before import and compared the findings to the situation in free solution. The precursor consisted of the ribonuclease barnase, whose folding in vitro has been studied extensively (21), fused to a mitochondrial targeting sequence derived from cytochrome b2 (7). To investigate the interactions of specific amino acids within barnase, we removed these individually by site-directed mutagenesis and compared the stability of wild-type and mutant proteins at the import site and in free solution. We found that each interaction tested contributed approximately the same energy to protein stability at the import site as in free solution. This result was obtained even for interactions that are broken in the molten globule state of barnase. Therefore, barnase precursors are not in the molten globule state at the mitochondrial import site before translocation. Furthermore, the mitochondrial surface does not specifically destabilize interactions of a particular chemical nature (e.g., nonpolar interactions, hydrogen bonds, salt bridges, etc.). To determine whether the mitochondrial surface destabilizes barnase globally, by affecting all interactions to a similar extent, it would be necessary to measure the absolute stability of the protein at the mitochondrial surface. This measurement is technically impossible. Instead, we investigated the tight interactions between barnase and its inhibitor protein barstar as a model for the intramolecular interactions in a folded protein (22–24). We found that the dissociation constants, and therefore the stability, of the barnase–barstar complex at the mitochondrial protein import site and in free solution coincide at physiological ionic strength. We conclude that precursors are not destabilized by the mitochondrial surface at the import site during translocation.
Materials and Methods
Proteins and Mitochondria.
Precursor proteins, consisting of a mitochondrial targeting sequence fused to the N terminus of the passenger protein, were constructed in pGEM-3Zf(+) vectors (Promega; ref. 7). The targeting sequence was derived from the first 35 amino acids of cytochrome b2, starting at the initiator methionine, and contained an Arg30 → Gly mutation to prevent processing by the mitochondrial matrix processing protease (25). The passenger protein was barnase, a ribonuclease from Bacillus amyloliquefaciens (26). Barnase contained a mutation changing His102 to Ala to inactivate its ribonuclease activity (27), as well as a mutation changing Gln2 to Met to allow radioactive labeling (7). In addition, barnase was destabilized by the mutations Ile76 → Val + Ile88 → Val + Ile96 → Val to allow efficient import (8). The mutations that probe protein structure were introduced by subcloning from the original vectors (28) or by using the QuickChange kit (Stratagene). All mutations were verified by DNA sequencing.
To purify chemical amounts of precursor protein, each precursor was subcloned into an expression vector derived from pQE60 (Qiagen, Chatsworth, CA), where the protein is under the control of a tac promoter. Lac repressor was co-expressed from the plasmid pREP4 (Qiagen). Escherichia coli M15 cells harboring both plasmids were induced in late log phase with 1 mM isopropyl β-d-thiogalactoside (IPTG) for 4 h before collection. Cells were broken open by sonication, inclusion bodies were collected and washed, and protein was extracted from the inclusion bodies with 8 M guanidine hydrochloride (U.S. Biological, Swampcott, MA). Protein was then dialyzed against 1.5 M guanidine hydrochloride, diluted 100 times into 50 mM sodium acetate (pH 5.0), and concentrated in a stirred cell.
Barstar is produced by B. amyloliquefaciens to protect the bacteria from any barnase that may accumulate in the cytosol. It inhibits barnase by forming a 1:1 complex with barnase. We used the Cys40 → Ala + Cys82 → Ala double mutant of barstar to avoid dimer formation (29). Barstar was expressed in E. coli strain JM109 from the vector pMT643 (24) by inducing the tac promoter controlling the barstar gene with 1 mM IPTG for 4 h after the cells reached late log phase. Cells were broken open by sonication, and the cell debris was removed by centrifugation. Barstar was purified by precipitation from the supernatant with 80% ammonium sulfate, followed by size exclusion (Sephadex 75, Pharmacia) and anion exchange (Mono Q, Pharmacia) column chromatography.
Mitochondria were purified from Saccharomyces cerevisiae strain D273-10B (MATα, American Type Culture Collection 25657) and purified by centrifugation through a Nycodenz (Nycomed, Westbury, NY) gradient (30).
Import Experiments.
Import rates were measured as described (3). Briefly, radioactive proteins were expressed from a T7 promoter by in vitro transcription and translation in rabbit reticulocyte lysate (Promega) supplemented with [35S]methionine and partially purified by high-speed centrifugation and ammonium sulfate precipitation (7). Precursors were then incubated with mitochondria at 0.5 mg mitochondrial protein per ml in import buffer [0.6 M sorbitol/50 mM Hepes–KOH (pH 7.4)/50 mM KCl/10 mM MgCl2/2 mM KH2PO4/1 mg⋅ml−1 fatty acid free BSA/5 mM methionine] containing 4 mM ATP, 10 mM creatine phosphate, and 0.15 mg⋅ml−1 creatine kinase. At designated times, aliquots were transferred to ice-cold stop buffer [0.6 M sorbitol/20 mM Hepes–KOH (pH 7.4)/2 μM valinomycin/0.2 mg⋅ml−1 proteinase K). After 10 min, the protease was inhibited with 1 mM PMSF, and mitochondria were reisolated by centrifugation. Samples were separated by SDS/PAGE, and the amount of imported protein was quantified by electronic autoradiography. The extent of import was plotted as a percentage of the total amount of precursor in the import reaction, and import kinetics were analyzed by using the software package KALEIDAGRAPH (Synergy Software, Reading, PA) assuming first-order kinetics.
Measurement of Dissociation Constants at the Mitochondrial Surface.
Ligand binding generally stabilizes proteins against unfolding (31). Because protein import into mitochondria requires protein unfolding, ligand binding to precursors inhibits their import (2, 8). Therefore, the dissociation constant of the precursor–ligand complex at the mitochondrial surface can be measured through the effect of ligand concentrations on import rates when unfolding is rate determining for import. For the barnase–barstar complex, the free and ligand-bound precursors are in rapid equilibrium compared with unfolding and import (compare the association and dissociation rate constants in Table 3 with the import rate constants for free precursor of 0.3 min−1). The measured import rate constant in the presence of ligand is then given by the sum of the products of the fractions of free and bound precursor and the respective import rate constants (compare ref. 32):
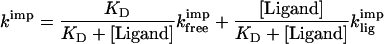 |
1 |
where kimp is the experimentally measured import rate constant, KD is the dissociation constant of the barnase–barstar complex, [Ligand] is the concentration of free barstar ligand, and kfreeimp and kligimp are the import rate constants of free and bound precursor. We found that barnase precursors bound to barstar are imported much slower than free barnase precursors; therefore, equation 1 simplifies to:
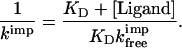 |
2 |
Thus, by measuring the import rate constants at different barstar concentrations, we can determine the dissociation constant of the barnase–barstar complex at the site of import (Table 3, Fig. 2).
Table 3.
Barnase-barstar dissociation constants at different ionic strengths*
| I, M | Off rate (s−1) in free solution† | On rate (107 M−1⋅s−1) in free solution† | KD, nM in free solution | KD, nM at the mitochondrial surface‡ |
|---|---|---|---|---|
| 0.05 | 1.2 ± 0.1 | 14 ± 1 | 9 ± 1 | 18 ± 1 |
| 0.07 | 1.1 ± 0.3 | 6.8 ± 0.6 | 16 ± 5 | 23 ± 2 |
| 0.17 | 1.0 ± 0.2 | 2.3 ± 0.2 | 43 ± 8 | 51 ± 3 |
All measurements were with barnase precursors with a Thr6 → Ala + Ile76 → Val + Ile88 → Val + Ile96 → Val mutant background. All errors are SE.
Determined at 35°C in import buffer lacking BSA.
Determined at 35°C in import buffer.
Figure 2.

Inhibition of import by ligand binding. The graph shows the inverse of the measured initial import rate of barnase precursors, standardized to the initial import rate of the unliganded precursors, vs. the barstar concentration.
In the ligand-binding experiments, import rates were measured as described above, but barnase precursors and import mix were preincubated with barstar at the indicated concentrations for 5 min. Equilibrium dissociation constants of the barnase–barstar complex, KD, were calculated by fitting the inverse of initial import rates, normalized to the initial import rate in the absence of barstar, against barstar concentrations.
To determine the ionic strength dependence of the dissociation constant, import rates were also measured in two additional import buffers: buffer A [0.6 M sorbitol/10 mM Hepes–KOH (pH 7.4)/0.5 mM KCl/5 mM MgCl2/0.25 mM KH2PO4/1 mg⋅ml−1 fatty acid free BSA) with 1 mM ATP, 10 mM creatine phosphate, and 0.15 mg⋅ml−1 creatine kinase and buffer B, which was the same as buffer A, but contained 20 mM KCl.
In Vitro Stability and Dissociation Constant Measurements.
The in vitro equilibrium stability of the precursor proteins purified from E. coli was measured as described (8, 33). Briefly, 100 μl of approximately 9 μM protein solution in 0.5 mM DTT, import buffer lacking BSA (pH 7.4), was added to 900 μl of 40 different urea solutions in import buffer of concentrations equally spaced between 0 and 10 M urea. After equilibration for 2–4 h at 25°C, the intrinsic protein fluorescence was measured for all samples in an ISS (Champaign, IL) PC1 fluorometer (excitation wavelength 290 nm, emission wavelength 320 nm, slits 8 nm). The fluorescence data were fitted assuming a two-state transition by using the published equations (28). Destabilization energies were calculated by multiplying the difference in urea concentrations at which wild-type and mutant proteins are 50% unfolded by the dependence of the free energy of unfolding on urea concentrations (m-value; ref. 28). Stability measurements were also performed at 30°C and 35°C for several of the mutants and yielded the same destabilization energies as at 25°C, within experimental error.
The in vitro dissociation constants of the complexes formed by barnase precursors with barstar were determined by measuring the association and dissociation rate constants as described (22, 23). Association rates were measured under second order conditions, with equal concentrations of both barnase precursor and barstar (5 μM each) in import buffer, which lacked BSA. The intrinsic protein fluorescence was monitored with an SX.18MV Applied Photophysics (Surrey, U.K.) stopped-flow apparatus at 35°C by using an excitation wavelength of 230 or 280 nm, 2-mm slit width, and a 305-nm glass cut-on filter. The second order rate constant for association was obtained by fitting fluorescence traces to the appropriate equation as described (22). Dissociation rates were measured by first forming the complex with 2 μM of barnase precursor and barstar and then competing off the barnase precursor with a large excess of a Trp35 → Phe mutant barnase. The Trp35 → Phe mutation in barnase changes the fluorescence spectrum of the barnase–barstar complex, which allows spectroscopic monitoring of the dissociation process (22). The intrinsic protein fluorescence was again followed, with the stopped flow apparatus at 35°C, and the fluorescence traces were fitted to first order kinetics as described (22). The dissociation constant (KD) was then calculated by dividing the dissociation rate constant by the association rate constant. Similar dissociation constants were measured for barnase with and without a targeting sequence (data not shown).
Results
Stability of Precursors in Free Solution.
The contributions of many amino acid side chains to the stability of barnase against unfolding in free solution have been measured (28). In these experiments, specific amino acid side chains were removed by site-directed mutagenesis, and the stabilities of both wild-type and mutant proteins were determined by urea denaturation while observing unfolding by monitoring the intrinsic protein fluorescence. The difference in stability between wild type and mutant reflects the contribution of the mutated interactions to the stability of wild-type protein. We have attached a mitochondrial targeting sequence to the N terminus of barnase and measured the effects of a subset of previously analyzed mutations on the stability of precursor proteins under conditions mimicking those of import experiments (Table 1). The absolute stabilities of wild-type barnase and barnase precursors with targeting sequences of different lengths were identical within experimental error (8). The effects of mutations on barnase with and without a targeting sequence were similar (compare Table 1 with table 3 in ref. 28).
Table 1.
Apparent destabilization energies (ΔΔG) at the mitochondrial surface and in free solution in kcal/mol
| Mutation in barnase | Destabilization at the mitochondrial surface*† | Destabilization of native state in free solution*‡ | Acceleration of unfolding in free solution§¶ | Destabilization of the molten globule state in free solution¶ |
|---|---|---|---|---|
| Ile4 → Ala | 0.6 ± 0.1 | 0.9 ± 0.1 | 1.5 | 0.0 |
| His18 → Gln | 0.7 ± 0.1 | 1.2 ± 0.1 | 0.2 | 1.2 |
| Asn23 → Ala | 1.4 ± 0.1 | 1.8 ± 0.1 | 2.6 | −0.1 |
| Ile25 → Val | 0.7 ± 0.1 | 1.1 ± 0.1 | 1.5 | −0.3 |
| Thr26 → Ala | 1.0 ± 0.2 | 1.9 ± 0.1 | 2.0 | 0.0 |
| Val36 → Ala | 0.6 ± 0.1 | 1.1 ± 0.1 | 1.5 | −0.3 |
| Ile51 → Val | 1.1 ± 0.2 | 1.7 ± 0.1 | 2.2 | −0.3 |
| Asp54 → Ala | 1.6 ± 0.2 | 2.6 ± 0.1 | 3.6 | −0.5 |
| Asp54 → Asn | 1.4 ± 0.2 | 2.2 ± 0.1 | 3.0 | −0.5 |
| Asn58 → Ala | 1.5 ± 0.1 | 2.1 ± 0.1 | 0.1 | 1.9 |
| Asn77 → Ala | 1.3 ± 0.2 | 1.8 ± 0.1 | 1.9 | 0.0 |
Mutations in barnase precursors. Mutations were generated in an Ile76 → Val + Ile88 → Val + Ile96 → Val mutant background. Errors are SE calculated from at least three repeat measurements.
The effects of mutations on protein stability at the mitochondrial surface were determined as the effects of mutations on import under the conditions used in these experiments. The apparent destabilization ΔΔG is calculated according to equation 4. Import experiments were performed at 35°C.
Determined at 25°C in the import buffer lacking BSA. Results are the same at 35°C, within error.
We define acceleration of unfolding in free solution as ΔΔGunfolding = RTln(kmutantunf/kwild typeunf).
Determined at 25°C in MES buffer (pH 6.3), data calculated from ref. 38. SE are smaller than 0.2 kcal/mol. Measurements were performed with barnase lacking a targeting sequence and lacking the mutant background.
Interactions at the Mitochondrial Surface.
The targeting sequence of the precursor proteins investigated in this study was 35 amino acids long, which is too short to allow the folded passenger protein at the mitochondrial surface to engage the unfolding and import machinery associated with the inner mitochondrial membrane (7, 8). Instead, the precursor can interact only with the import site in the outer membrane before the passenger protein unfolds spontaneously and then is translocated into the matrix. Rate constants for import of barnase precursors into purified yeast mitochondria do not depend on mitochondrial or precursor concentrations (data not shown). Therefore, import is described by the following process:
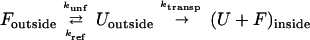 |
3 |
where Foutside is the folded precursor at the mitochondrial surface, Uoutside is the unfolded precursor at the mitochondrial surface, (U + F)inside is all forms of the precursor inside the mitochondria, kunf is the rate constant of protein unfolding, kref is the rate constant of protein refolding, and ktransp is the rate constant of the transport steps. The import experiments measure changes in (U + F)inside with time and spontaneous unfolding is the rate-determining step (8). Our approach to analysis of this mechanism is analogous to that of hydrogen exchange in proteins (34). Two limiting kinetic schemes could describe import: (i) the measured import rate constant could be equal to the unfolding rate constant, kunf, or (ii) the measured import rate constant could be equal to the equilibrium constant for unfolding, K = kunf/kref, multiplied by the rate constant for the transport steps, ktransp. Our data show that the second mechanism applies, because mutations in barnase affect import rates in the way that they affect the stability of barnase in free solution and not the unfolding rates (compare His18 → Gln and Asn58 → Ala to the other mutations in columns two, three, and four of Table 1). Mutations in barnase affect only the stability of the protein and not the intrinsic translocation rate constant of the import machinery (8). Therefore, we can calculate the energy apparently contributed by the mutated groups to protein stability (28) at the mitochondrial surface (ΔΔGimport) from the import rate constants of mutant and wild-type proteins as follows:
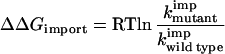 |
4 |
where kwild typeimp is the import rate constant of wild-type precursor, kmutantimp is the import rate of mutant precursor, R is the gas constant, and T is the absolute temperature. The results are shown in Table 1. Unfolding is slow compared with precursor binding to mitochondria as is determined by the observation that import rates do not depend on mitochondrial or precursor concentrations (data not shown). Therefore, the apparent interaction energies measured through the import kinetics reflect the state of the protein at the mitochondrial surface before import. Identical destabilization energies were measured when precursors were prebound to mitochondria and import was initiated by the addition of ATP (data not shown).
Precursors Are Not in the Molten Globule State at the Protein Import Site.
The folding pathway of barnase involves an intermediate that accumulates to different degrees depending on solution conditions (35–38). This intermediate is equivalent to the molten globule state (19) and is characterized by the presence of most secondary structure elements and the absence of many tertiary interactions. We know quantitatively how interactions in various parts of barnase contribute to the stability of both the native state and the molten globule state (38). A large number of the tertiary interactions that stabilize the native state do not contribute to the stability of the molten globule state. Here, we measured the contribution of a subset of these tertiary interactions to protein stability at the mitochondrial surface (Table 1). Clearly, removing interactions in barnase affects the stability of precursors at the mitochondrial surface in the same way it affects the stability of the native state in free solution and not the molten globule state (Table 1, Fig. 1). In other words, mutations that do not destabilize the molten globule state in vitro still accelerate import. The molten globule state of barnase is not very stable in free solution. However, the experiments described here probe the relative stabilities of native and molten globule states and detect any stabilization of the molten globule state independently of the extent to which it accumulates originally. In summary, the molten globule state of barnase is not preferentially stabilized at the mitochondrial surface, and barnase does not accumulate in the molten globule state before import.
Figure 1.

Comparison of the effects of mutations on protein stability at the mitochondrial surface (A) and in the molten globule state in vitro (B). To simplify the comparison, destabilization energies are standardized, in A by dividing the destabilization energy of mutations at the mitochondrial surface (ΔΔGimport) by the destabilization energy of the same mutations measured for the native state in vitro, and in B by dividing the destabilization energy of mutations in the molten globule state by the destabilization energy of the same mutations in the native protein. Data for B were calculated from ref. 38.
No Specific Type of Interactions is Preferentially Destabilized at the Mitochondrial Surface.
If interactions of a particular type (e.g., hydrogen bonds or salt bridges) were destabilized by the mitochondrial surface compared with other interactions (e.g., nonpolar), their contributions to protein stability at the mitochondrial surface would be smaller than their contributions in free solution. We find that no interaction is preferentially destabilized, regarding either the nature of the interactions or their location within the barnase structure (Table 1, Fig. 1).
The effect of membranes on protein stability has been related to the local environment near the membrane surface caused by negatively charged phospholipids (39, 40). Therefore, a destabilizing effect may become more pronounced at lower ionic strengths. We measured the import rates of barnase precursors at the lowest ionic strength experimentally accessible (I = 0.05 M compared with I = 0.17 M for import buffer at physiological ionic strength). Decreasing the ionic strength of the import buffer did not unmask any destabilizing effects of the mitochondrial surface (Table 2).
Table 2.
Apparent destabilization energies (ΔΔG) at the mitochondrial surface at two different ionic strengths in kcal/mol
| Mutation in barnase* | I = 0.17 M†§ | I = 0.05 M‡§ |
|---|---|---|
| Ile4 → Ala | 0.6 ± 0.1 | 0.7 ± 0.2 |
| His18 → Gln | 0.7 ± 0.1 | 0.7 ± 0.2 |
| Asn23 → Ala | 1.4 ± 0.1 | 1.0 ± 0.2 |
| Ile25 → Val | 0.7 ± 0.1 | 0.8 ± 0.2 |
| Thr26 → Ala | 1.0 ± 0.2 | 1.2 ± 0.2 |
| Val36 → Ala | 0.6 ± 0.1 | 0.8 ± 0.1 |
| Ile51 → Val | 1.1 ± 0.2 | 1.1 ± 0.2 |
| Asp54 → Ala | 1.6 ± 0.2 | 1.2 ± 0.2 |
| Asp54 → Asn | 1.4 ± 0.2 | 1.2 ± 0.2 |
| Asn58 → Ala | 1.5 ± 0.1 | 1.1 ± 0.2 |
| Asn77 → Ala | 1.3 ± 0.2 | 1.4 ± 0.2 |
Mutations were generated in an Ile76 → Val + Ile88 → Val + Ile96 → Val mutant background.
Determined in normal import buffer. Errors are SE.
Determined in low ionic strength import buffer A. Errors are SE.
I is the ionic strength of import buffers.
Destabilization energies measured at the mitochondrial surface are on average approximately 0.5 kcal/mol lower than those measured in free solution (Table 1). The deviation appears to be somewhat larger for the most destabilizing mutation (e.g., Asp54 → Ala) and therefore may be due to systematic measurement errors. For strongly destabilizing mutations, the actual import rates approach the upper limit of the turnover of the import machinery so that steps other than unfolding become rate limiting. Therefore, the calculated destabilization energies will underestimate the mutational effects.
The Mitochondrial Surface Does Not Globally Destabilize Proteins.
The mitochondrial surface could act by stabilizing the unfolded state, just as denaturants do. Interestingly, a global destabilization of a protein at the mitochondrial surface is not expected to lead to a reduction in the individual mutational destabilization energies (ΔΔGs) because both wild type and mutant would be affected in similar ways. (For illustration, consider conventional urea denaturation experiments: mutational destabilization energies are measured at urea concentrations at which the protein unfolds and extrapolated to the absence of denaturant. Yet, the fact that these energies are independent of urea concentration does not lead to the conclusion that urea does not destabilize proteins.) Therefore, to detect a global destabilization of barnase it would be necessary to measure the absolute stability of the protein both at the mitochondrial surface and in free solution. Unfortunately, this measurement cannot be made experimentally. However, it is straightforward to measure absolute dissociation constants both in vitro and at the mitochondrial surface. The strong interactions between two proteins are a good model for the intramolecular interactions within a protein. In addition, the complex between two proteins will be weakened when the participating proteins are destabilized. Barnase binds to its inhibitor protein barstar with a nanomolar dissociation constant (22, 29). The interaction interface contains nonpolar, hydrogen bonding, and salt bridge interactions (41), with the latter two dominating energetically (22–24).
The dissociation constants at the mitochondrial surface and in free solution coincide within experimental error at physiological ionic strengths (Table 3, Fig. 2). Therefore, the stability of the barnase–barstar complex is not affected by the mitochondrial surface under physiological conditions. This observation rules out the possibility that the mitochondrial protein import site has a global destabilizing effect on this protein. At the lowest ionic strength tested, there appears to be a small but significant destabilization of barnase–barstar complex. The fact that this destabilization is eliminated by physiological ionic strength suggests that the effect is electrostatic in nature. However, the magnitude of this effect is too small to be interpreted with confidence, and the minimally required components of the import buffer rule out the exploration of lower ionic strengths.
Discussion
Most mitochondrial proteins are synthesized in the cytosol and imported into mitochondria posttranslationally. Precursor proteins are normally in a fully unfolded conformation during import into mitochondria (2–4), and some proteins fold before translocation (5, 6). What makes protein import possible? A series of earlier studies suggested that the import machinery at the mitochondrial surface (9–13) induces protein unfolding or the accumulation of the molten globule state in precursor proteins. In contrast, our results show that a precursor protein is not destabilized by the mitochondrial surface during translocation and does not accumulate in the molten globule state. How can the two sets of observations be reconciled?
We have investigated the stability of a precursor protein at the site of import. Most earlier studies have been performed with phospholipid vesicles. These vesicles may differ from the mitochondrial outer membrane in their lipid composition and always differ in their protein composition. The mitochondrial outer membrane has a relatively high protein content [20% (wt/wt); ref. 42], whereas lipid vesicles lack proteins. During import, precursor proteins will most likely be closest to the proteins making up the import site rather than the lipid components of the membrane because their targeting sequences interact specifically with the import receptors (e.g., 9, 43). In this case, our results report on the effect of the environment specific to the import site rather than the lipid membranes. Regardless, the protein import assay measures the environment that is most relevant to protein import.
A potential cause for misunderstandings is the fact that membranes can trap unfolded proteins. Proteins spontaneously unfold and refold under all relevant experimental conditions. The unfolded form of some proteins binds to membrane surfaces efficiently so that refolding is inhibited. When these proteins are incubated with membranes, the amount of folded protein decreases as spontaneously unfolding protein is trapped by the membrane. Depending on how one defines the protein folding equilibrium, the trapping effect can be viewed as a destabilization of the protein (9, 44). However, because unfolded proteins that are productively bound to the import machinery are immediately imported and refold after translocation, this definition appears somewhat misleading.
How good a model is barnase for other mitochondrial precursors? The folding of barnase has been studied extensively in vitro (21) and appears ordinary when compared with the behavior of other proteins. Where we investigate features of barnase that are common to all soluble proteins, such as the contributions of hydrogen bonds, salt bridges, and nonpolar interactions to protein stability, our results will be applicable to all proteins. Conversely, the relative stabilities of molten globule intermediates and folded state are specific to each protein. Our experiments probe the relative stabilities of native and molten globule state, and we would detect a stabilization of the molten globule state, relative to the native state, independently of the extent to which the molten globule state accumulates. Because we do not detect any stabilization of the molten globule state, our results regarding this question are unambiguous: even proteins with greater tendencies to form the molten globule state than barnase will not accumulate in this folding intermediate during import.
Another question is whether the interaction between two proteins is a relevant model for the stability of a protein. Barnase and barstar interact through an extended interface that is stabilized through the same types of nonpolar, hydrogen bonding and salt-bridge interactions that stabilize proteins. Electrostatic interactions probably play a greater role in stabilizing the barnase–barstar complex than they do in the stability of some proteins (23). Because we find no destabilization of the complex under physiological conditions, the relative contributions of different types of interactions are irrelevant. In addition, any effect of the membrane caused by charged lipids will have a strong impact on electrostatic interactions, making the barnase–barstar complex a particularly sensitive probe. At the lowest ionic strength accessible, the barnase–barstar binding constant is lowered by a factor of approximately two (equivalent to a destabilization of 0.4 kcal/mol) at the mitochondrial surface, compared with the situation in free solution. Although apparently above the error of our measurements, this effect is too small to interpret with confidence.
In summary, we conclude that precursor proteins are not significantly destabilized by the environment at import sites in the outer mitochondrial membrane. Therefore, the import machinery in inner membrane is responsible for the unfolding of precursor proteins.
Acknowledgments
We thank Drs. Cheolju Lee and R. MacDonald for helpful discussions and acknowledge the use of instruments of the Keck Biophysics Facility at Northwestern University. This work was funded by a Research Project Grant from the American Cancer Society. S.H. gratefully acknowledges the support by a Gramm Travel Fellowship Award from the Robert H. Lurie Comprehensive Cancer Center of Northwestern University.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.230243097.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.230243097
References
- 1.Schatz G, Dobberstein B. Science. 1996;271:1519–1526. doi: 10.1126/science.271.5255.1519. [DOI] [PubMed] [Google Scholar]
- 2.Eilers M, Schatz G. Nature (London) 1986;322:228–232. doi: 10.1038/322228a0. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz M P, Huang S, Matouschek A. J Biol Chem. 1999;274:12759–12764. doi: 10.1074/jbc.274.18.12759. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz M P, Matouschek A. Proc Natl Acad Sci USA. 1999;96:13086–13090. doi: 10.1073/pnas.96.23.13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bomer U, Meijer M, Guiard B, Dietmeier K, Pfanner N, Rassow J. J Biol Chem. 1997;272:30439–30446. doi: 10.1074/jbc.272.48.30439. [DOI] [PubMed] [Google Scholar]
- 6.Wienhues U, Becker K, Schleyer M, Guiard B, Tropschug M, Horwich A L, Pfanner N, Neupert W. J Cell Biol. 1991;115:1601–1609. doi: 10.1083/jcb.115.6.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matouschek A, Azem A, Ratliff K, Glick B S, Schmid K, Schatz G. EMBO J. 1997;16:6727–6736. doi: 10.1093/emboj/16.22.6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang S, Ratliff K S, Schwartz M P, Spenner J M, Matouschek A. Nat Struct Biol. 1999;6:1132–1138. doi: 10.1038/70073. [DOI] [PubMed] [Google Scholar]
- 9.Mayer A, Neupert W, Lill R. Cell. 1995;80:127–137. doi: 10.1016/0092-8674(95)90457-3. [DOI] [PubMed] [Google Scholar]
- 10.Eilers M, Hwang S, Schatz G. EMBO J. 1988;7:1139–1145. doi: 10.1002/j.1460-2075.1988.tb02923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Endo T, Schatz G. EMBO J. 1988;7:1153–1158. doi: 10.1002/j.1460-2075.1988.tb02925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Endo T, Eilers M, Schatz G. J Biol Chem. 1989;264:2951–2956. [PubMed] [Google Scholar]
- 13.Hartmann C M, Gehring J, Christen P. Eur J Biochem. 1993;218:905–910. doi: 10.1111/j.1432-1033.1993.tb18446.x. [DOI] [PubMed] [Google Scholar]
- 14.Muga A, Mantsch H H, Surewicz W K. Biochemistry. 1991;30:7219–7224. doi: 10.1021/bi00243a025. [DOI] [PubMed] [Google Scholar]
- 15.van der Goot F G, Gonzalez-Manas J M, Lakey J H, Pattus F. Nature (London) 1991;354:408–410. doi: 10.1038/354408a0. [DOI] [PubMed] [Google Scholar]
- 16.de Jongh H H, Killian J A, de Kruijff B. Biochemistry. 1992;31:1636–1643. doi: 10.1021/bi00121a008. [DOI] [PubMed] [Google Scholar]
- 17.Bychkova V E, Dujsekina A E, Klenin S I, Tiktopulo E I, Uversky V N, Ptitsyn O B. Biochemistry. 1996;35:6058–6063. doi: 10.1021/bi9522460. [DOI] [PubMed] [Google Scholar]
- 18.Ren J, Kachel K, Kim H, Malenbaum S E, Collier R J, London E. Science. 1999;284:955–957. doi: 10.1126/science.284.5416.955. [DOI] [PubMed] [Google Scholar]
- 19.Ptitsyn O B. Adv Protein Chem. 1995;47:83–229. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
- 20.van der Goot F G, Lakey J H, Pattus F. Trends Cell Biol. 1992;2:343–348. [PubMed] [Google Scholar]
- 21.Fersht A R. FEBS Lett. 1993;325:5–16. doi: 10.1016/0014-5793(93)81405-o. [DOI] [PubMed] [Google Scholar]
- 22.Schreiber G, Fersht A R. Biochemistry. 1993;32:5145–5150. doi: 10.1021/bi00070a025. [DOI] [PubMed] [Google Scholar]
- 23.Schreiber G, Fersht A R. J Mol Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- 24.Jucovic M, Hartley R W. Proc Natl Acad Sci USA. 1996;93:2343–2347. doi: 10.1073/pnas.93.6.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arretz M, Schneider H, Guiard B, Brunner M, Neupert W. J Biol Chem. 1994;269:4959–4967. [PubMed] [Google Scholar]
- 26.Hartley R W. Biochemistry. 1975;14:2367–2370. doi: 10.1021/bi00682a015. [DOI] [PubMed] [Google Scholar]
- 27.Mossakowska D E, Nyberg K, Fersht A R. Biochemistry. 1989;28:3843–3850. doi: 10.1021/bi00435a033. [DOI] [PubMed] [Google Scholar]
- 28.Serrano L, Kellis J T, Jr, Cann P, Matouschek A, Fersht A R. J Mol Biol. 1992;224:783–804. doi: 10.1016/0022-2836(92)90562-x. [DOI] [PubMed] [Google Scholar]
- 29.Hartley R W. Biochemistry. 1993;32:5978–5984. doi: 10.1021/bi00074a008. [DOI] [PubMed] [Google Scholar]
- 30.Glick B S, Pon L A. Methods Enzymol. 1995;260:213–223. doi: 10.1016/0076-6879(95)60139-2. [DOI] [PubMed] [Google Scholar]
- 31.Makarov A A, Protasevich I I, Lobachov V M, Kirpichnikov M P, Yakovlev G I, Gilli R M, Briand C M, Hartley R W. FEBS Lett. 1994;354:251–254. doi: 10.1016/0014-5793(94)01127-3. [DOI] [PubMed] [Google Scholar]
- 32.Sancho J, Meiering E, Fersht A R. J Mol Biol. 1991;221:1007–1014. doi: 10.1016/0022-2836(91)80188-z. [DOI] [PubMed] [Google Scholar]
- 33.Matouschek A, Fersht A R. Methods Enzymol. 1991;202:82–112. doi: 10.1016/0076-6879(91)02008-w. [DOI] [PubMed] [Google Scholar]
- 34.Hvidt A, Nielsen S O. Adv Protein Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 35.Oliveberg M, Fersht A R. Biochemistry. 1996;35:2738–2749. doi: 10.1021/bi950967t. [DOI] [PubMed] [Google Scholar]
- 36.Sanz J M, Johnson C M, Fersht A R. Biochemistry. 1994;33:11189–11199. doi: 10.1021/bi00203a015. [DOI] [PubMed] [Google Scholar]
- 37.Sanz J M, Fersht A R. Biochemistry. 1993;32:13584–13592. doi: 10.1021/bi00212a026. [DOI] [PubMed] [Google Scholar]
- 38.Matouschek A, Serrano L, Fersht A R. J Mol Biol. 1992;224:819–835. doi: 10.1016/0022-2836(92)90564-z. [DOI] [PubMed] [Google Scholar]
- 39.McLaughlin S. Curr Top Membranes Transport. 1977;9:71–144. [Google Scholar]
- 40.Eisenberg M, Gresalfi T, Riccio T, McLaughlin S. Biochemistry. 1979;18:5213–5223. doi: 10.1021/bi00590a028. [DOI] [PubMed] [Google Scholar]
- 41.Buckle A M, Schreiber G, Fersht A R. Biochemistry. 1994;33:8878–8889. doi: 10.1021/bi00196a004. [DOI] [PubMed] [Google Scholar]
- 42.Tzagoloff A. Mitochondria. New York: Plenum; 1982. [Google Scholar]
- 43.Haucke V, Lithgow T, Rospert S, Hahne K, Schatz G. J Biol Chem. 1995;270:5565–5570. doi: 10.1074/jbc.270.10.5565. [DOI] [PubMed] [Google Scholar]
- 44.Rapaport D, Mayer A, Neupert W, Lill R. J Biol Chem. 1998;273:8806–8813. doi: 10.1074/jbc.273.15.8806. [DOI] [PubMed] [Google Scholar]